


Abstract
This study was undertaken to test the hypothesis that P-glycoprotein (P-gp) modulates opioid peptide pharmacodynamics. [d-Penicillamine2,5]enkephalin (DPDPE) (10 mg/kg i.v.) was administered to mdr1a(−/−) and wild-type mice to assess systemic disposition and antinociception. A subsequent dose-response experiment examined the impact of P-gp on DPDPE antinociception. In addition, the time course of antinociception was determined after a 0.9-mg/kg [mdr1a(−/−) mice] or 24-mg/kg (FVB mice) i.v. dose. Data were fit with a series of pharmacokinetic-pharmacodynamic models to compare the disposition and action of DPDPE in the two mouse strains. A 10-mg/kg dose produced >80% maximum possible response at all time points inmdr1a(−/−) mice; peak antinociception was <20% maximum possible response in FVB mice. DPDPE systemic disposition did not differ between the two mouse strains. Although brain tissue concentrations were 2- to 4-fold higher in mdr1a(−/−)compared to FVB mice, the dose required to elicit comparable antinociception was nearly 30-fold lower in mdr1a(−/−)mice; brain tissue EC50 differed by an order of magnitude in the two mouse strains. Pharmacokinetic-pharmacodynamic modeling indicated that the difference in antinociception betweenmdr1a(−/−) and FVB mice was a function of DPDPE distribution within brain, as well as between blood and brain, and not due to differences in intrinsic response. The results of this study suggest that DPDPE is a substrate of P-gp, and that P-gp is responsible, in part, for the low penetration of DPDPE into brain. The substantial difference in brain tissue EC50 in the absence vs. presence of P-gp suggests that P-gp modulates DPDPE-associated antinociception at sites other than the blood-brain interface.
DPDPE is an opioid pentapeptide developed as a potential drug for treatment of pain (Williams et al., 1996). DPDPE has been used extensively in receptor-binding studies due to selectivity for δ-opioid receptors (Mosberg et al., 1983; Toth et al., 1990). Extensive in vitro (Weber et al., 1991; Chen and Pollack, 1997a;b) and in vivo(Weber et al., 1991, 1992; Chen and Pollack, 1997a;b) studies have shown that biotransformation of DPDPE is minimal. Despite favorable stability due to conformational restriction from a disulfide bond-linked cyclic structure (Weber et al., 1992), the residence of DPDPE in vivo is short (half-life ∼14 min in rats; Chen and Pollack, 1996); extensive biliary excretion is responsible for the short sojourn in vivo (Chen and Pollack, 1997a). This rapid removal of DPDPE from blood is one reason for the short duration of antinociceptive action (Chen and Pollack, 1997b).
Substantial antinociception, with rapid onset after i.v. administration, indicates that DPDPE penetrates the blood-brain barrier (Chen and Pollack, 1997b). Previous studies in CD-1 mice suggested that brain penetration of DPDPE was low, with a brain:blood concentration ratio of ∼0.1 (Chen and Pollack, 1997b), despite relatively high (5 × 10−3 cm/min) membrane permeability in vitro (Shah et al., 1989). Although in vitromodels of the blood-brain barrier may be leakier than the in vivo barrier (Pardridge et al., 1990), the low brain partitioning in vivo, compared to membrane permeabilityin vitro, could be due to an active efflux pump that extrudes DPDPE from brain. Indeed, Chen and Pollack (1997b) showed that the brain:blood concentration ratio increased with increasing dose of DPDPE; the relationship between brain tissue partitioning and blood concentration was best described by a kinetic model incorporating active efflux from brain.
P-gp, a product of the multidrug-resistance (mdr) gene, is overexpressed in tumor cells and is present in normal tissues (e.g., brain capillary endothelium, bile canaliculi; Lum and Gosland, 1995). Substrate specificity for P-gp is limited. For example, vincristine, a vinca alkaloid and cyclosporine A, an immunosuppressive decapeptide, are substrates of this export protein (Foxwell et al., 1989; Lum et al., 1993). Although most studies of P-gp have focused on chemotherapeutic substrates, several studies have demonstrated that opioids such as morphine (Callaghan and Riordan 1993;Letrent et al., 1997) and loperamide (Schinkel et al., 1996), and peptides such as cyclosporine A (Foxwell et al., 1989; Saeki et al., 1993) and N-acetyl-leucyl-norleucinal (Sharma et al., 1992), interact with P-gp. It has been postulated that extruding peptides from cells is one of the physiological functions of P-gp (Sharma et al., 1992; Saeki et al., 1993). Although these data indicate that some peptides are substrates for P-gp, the possible role of P-gp in DPDPE disposition has not been addressed.
The availability of mice that lack the gene for production of the drug-transporting mdr1a P-gp (Schinkel et al., 1994) has facilitated investigations of the role of P-gp in the disposition, and consequently the pharmacological activity, of several agents. Absence of mdr1a P-gp, which is localized predominantly in brain, intestine, liver and testis (Teeter et al., 1990), has been shown to alter the systemic disposition and/or increase the brain penetration of several substrates (Schinkelet al., 1994, 1995a, b, 1996), including opioids. Brain tissue concentrations of the mu-opioid receptor agonist morphine was ∼2 fold higher in mdr1a(−/−) mice as compared to control mice (Schinkel et al., 1996). Loperamide, a non-peptide opioid that is devoid of central nervous system activity in wild-type mice, displayed central opiate effects inmdr1a(−/−) mice (Schinkel et al., 1996).
Taken together, the evidence to date suggests that penetration of DPDPE into the brain may be limited by a saturable efflux process, and that P-gp may mediate, at least in part, efflux of DPDPE from the brain. To address this hypothesis, the present study was undertaken to examine the pharmacokinetics and pharmacodynamics of DPDPE inmdr1a(−/−) mice, in comparison with wild-type mice, to assess the impact of P-gp on disposition and pharmacodynamics of DPDPE, and therefore to gain insight into the factors that limit pharmacologic effect of metabolically stable opioid peptides.
Methods
Materials.
DPDPE was a gift from NIDA and was used without further purification. [Tyr2,6-3H]-DPDPE (1332 Gbq/mmol) was obtained from Du Pont New England Nuclear (Boston, MA). All other reagents used in this study were of the highest grade available.
Animals.
Male FVB and mdr1a(−/−) mice, 4 to 5 wk of age (Taconic, Germantown, NY), were housed individually in wire-mesh cages with free access to food and water, and were maintained on a 12-hr light/dark cycle. Mice were anesthetized with i.p. ketamine (85 mg/kg) and xylazine (0.3 mg/kg), and a silicone rubber cannula (0.015 in o.d.) was implanted (∼1 cm) in the right jugular vein as described previously (Chen and Pollack, 1997b) 24 hr before the experiment. All procedures were approved by the Institutional Animal Care and Use Committee of The University of North Carolina at Chapel Hill.
DPDPE disposition and antinociception.
Based on previous studies in CD-1 mice (Chen and Pollack, 1997b), a 10-mg/kg dose was selected to produce ∼15% MPR in the hotplate test. Mice were randomized into five groups corresponding to times at which brain tissue and gallbladder would be obtained. Baseline hotplate latency was determined before administration of DPDPE as described elsewhere (Chen and Pollack, 1997b). Latency was defined as the time interval between placement on the hotplate (55°C) and licking the hind paws or jumping. Mice with control latencies (determined after cannulation) ≤25 sec received an i.v. bolus dose of 3H-DPDPE (10 mg/kg; 4 μCi) through the cannula. Hotplate latency was determined, and blood samples were obtained, at 5, 10, 15, 20, 30, 40 min; tissue was harvested at 5, 10, 20, 30 or 40 min (n = 5/group). A cut-off test latency of 60 sec was used to avoid tissue damage. Antinociception was calculated as:
Dose-response experiment.
The 10-mg/kg dose experiment, which produced >80% MPR in mdr1a(−/−) mice for up to 24 hr (see “Results”), indicated that a lower dose can elicit maximum response in this strain. Thus, a dose-response experiment was conducted to investigate the relationship between antinociception and DPDPE concentrations in blood and brain in the presence and absence of P-gp.3H-DPDPE (0.4, 0.6, 0.8, 1.0 mg/kg formdr1a(−/−); 20, 30, 40, 80 mg/kg for FVB; 4 μCi) was administered as described above. Antinociception was determined at 5 or 10 min after administration of DPDPE for FVB andmdr1a(−/−) mice, respectively, representing the time of peak effect in each strain as determined in preliminary experiments. Immediately after testing, blood samples were obtained from the jugular vein cannula and the mice were sacrificed by decapitation for collection of brain and gallbladder.
Time course of antinociception.
The time course of antinociception was examined for mdr1a(−/−) and FVB mice after a 0.9- or 24-mg/kg (containing 4 μCi 3H-DPDPE) i.v. dose, representing the approximate ED50 values for DPDPE in each mouse strain. Mice from each strain were divided into two groups (n = 5). In one group, blood was obtained at 5, 15, 30, 40, 60 min, and antinociception at 2, 10, 20, 40, 60 min; in the other group, blood was obtained at 2, 10, 20, 40, 60 min and antinociception at 5, 15, 30, 40, 60 min. All samples were stored at −20°C.
Quantitation of DPDPE.
Blood samples (10 μl) were mixed with scintillation cocktail (5 ml; Bio-Safe II, RPI Corp., Mount Prospect, IL) for determination of radioactivity with liquid scintillation spectrometry. Whole brain was isolated, blotted dry, weighed and homogenized (1:2 w/v in phosphate buffered saline). Aliquots of homogenate (100 μl) were mixed with scintillation cocktail. Brain tissue concentrations were corrected for residual blood content as described previously (Chen and Pollack, 1997b). The whole gallbladder was removed and added to 5 ml cocktail. Previous studies in this laboratory have shown that DPDPE-derived radioactivity represents the unchanged parent peptide after administration of3H-DPDPE.
Estimation of systemic pharmacokinetic parameters.
Initial pharmacokinetic analysis was based on non-compartmental methods. In addition, blood concentrations from both FVB andmdr1a(−/−) mice after a 10-mg/kg dose were fit simultaneously with a two-compartment model (WinNonlin, SCI, Apex, NC) to recover a single set of pharmacokinetic parameters for the two mouse strains. Data from the 24-mg/kg dose in FVB mice were fit separately, as DPDPE disposition appeared to differ from the lower dose.
Pharmacokinetic-pharmacodynamic analysis.
Stepwise nonlinear regression was used to develop a PK-PD model for DPDPE inmdr1a(−/−) and FVB mice. This effort addressed the hypothesis that P-gp transport results in apparent sequestration of DPDPE at a brain tissue site separate from the pharmacological receptor. The general approach was to fit portions of the model to specific data sets, allowing resolution of parameter subsets. These subsets then were used to develop increasingly complex models, culminating in the models depicted in figure1. The steps taken in this analysis were as follows: 1) A two-compartment model was fit simultaneously to blood concentration-time data from both strains (10-mg/kg dose), allowing resolution of systemic pharmacokinetic parameters (k12, k21, k10 and Vc). 2) Brain tissue concentration and effect data from the dose-response experiment inmdr1a(−/−) mice (i.e., in the absence of P-gp-mediated transport) were used to obtain the pharmacodynamic parameters EC50 and γ. 3) Parameters obtained in steps 1) and 2), together with blood, brain tissue, and effect data from the 10-mg/kg dose in mdr1a(−/−) mice, were used to estimate non-P-gp-mediated flux between blood and brain (k13 and k31). 4) Each of these parameters, together with the blood, brain tissue and effect data from the 10-mg/kg dose in FVB mice, were used to estimate the remaining rate constants (k31a, representing P-gp-mediated flux at the blood-brain interface, and k34, k43, k45, k54, representing flux within the brain compartment). The final models were used to simulate the effect vs. time profile after a 0.9-mg/kg dose in mdr1a(−/−) mice and a 24-mg/kg dose in FVB mice. For the 24-mg/kg dose, saturable brain uptake of DPDPE was required, consistent with previous reports in rats (Thomas et al., 1997).

Scheme for the PK-PD model of DPDPE inmdr1a(−/−) (top) and FVB (bottom) mice. The volume of the central compartment (Vc) and the rate constants k12, k21 and k10, were obtained by fitting a two-compartment model to the blood concentration-time data inmdr1a(−/−) and FVB mice after a 10-mg/kg dose. The effect parameters EC50 and γ were obtained from the dose-response experiment in mdr1a(−/−) mice, and the rate constants k13 and k31 were obtained from blood, brain tissue and effect data after the 10-mg/kg dose inmdr1a(−/−) mice. The remaining rate constants (k31a, representing the contribution of P-gp at the blood-brain interface, and k34, k43, k45, k54) were obtained from blood, brain tissue and effect data after the 10-mg/kg dose in FVB mice. See “Methods” for further details.
Statistical analysis.
Data are presented as mean ± S.E. ANOVA and Student’s t test, where appropriate, were used to assess the significance of differences in antinociception, DPDPE blood and brain tissue concentrations, and mass of DPDPE in the gallbladder between mdr1a(−/−) and FVB mice. In all cases, P = .05 was used as the criterion of significance.
Results
Disposition and antinociception after a 10-mg/kg i.v. dose.
No difference was observed in the systemic disposition of DPDPE betweenmdr1a(−/−) and FVB mice; the time course of concentrations in blood was almost superimposable between the two groups of mice (fig.2). Pharmacokinetic parameters recovered by non-compartmental analysis are presented in table1. No statistical difference was observed for any of the parameters examined; it was possible to fit the combined data from both strains with a single set of kinetic parameters (solid line, fig. 2).

DPDPE blood concentration-time profiles inmdr1a(−/−) (○) and FVB (•) mice after a 10-mg/kg i.v. bolus dose. Data are presented as mean ± S.E. (n= 4 per group). Solid line indicates fit of a two-compartment model to the data (see table 2 for parameters).
Noncompartmental pharmacokinetic parameters for DPDPE inmr1a(−/−) and FVB mice1-a
In both mdr1a(−/−) and FVB mice, brain tissue concentrations were maximal at 20 min postadministration, with only a marginal decline through 40 min (fig. 3). However, brain tissue concentrations in mdr1a(−/−) mice were 2- to 4-fold higher than those in FVB mice at each time point examined. Two-way ANOVA revealed a statistically significant difference (P < .0001) in brain tissue concentration between the two mouse strains across time.

Brain tissue concentration-time profiles inmdr1a(−/−) (○) and FVB (•) mice after a 10-mg/kg i.v. bolus dose of DPDPE. Data are presented as mean ± S.E. (n = 4 per group). Two-way ANOVA indicated that there was a significant difference (P < .0001) in brain tissue concentrations across time between the two mouse strains.
Although there was a trend toward a higher mass of DPDPE in the gallbladder of mdr1a(−/−) mice as compared to FVB mice, no statistical difference was observed between the two strains of mice due to substantial variability in the data (data not shown).
DPDPE (10 mg/kg) elicited an antinociceptive response within 5 min in FVB mice; response remained measurable and constant for 15 min (14.1 ± 5.1% MPR across the 5-, 10- and 15-min time points). Effect was not measurable beyond 15 min. In contrast, the 10-mg/kg dose elicited 100% MPR within 5 min in mdr1a(−/−) mice; significant effect (89.9 ± 10.1% MPR) remained through the final sampling point (40 min postdose). In a separate group ofmdr1a(−/−) mice, hotplate latency did not return to baseline by 24 hr (data not shown). At the 10-mg/kg dosemdr1a(−/−) mice evidenced substantial behavioral effects not observed in wild-type animals, including apparent sedation, uncoordinated locomotion and inability to jump or to lick the hind paw (the end-point of hotplate latency test; Loh et al., 1976).
Dose-response experiment.
With the exception of the highest dose in each mouse strain, the dose-response relationship was log-linear (fig. 4). Significant differences were observed in the degree of antinociception produced by DPDPE between the two strains. Response per unit dose was >10-fold higher in mdr1a(−/−) as compared to FVB mice; based on a log-linear dose-response model, the ED50 was 0.9 and 24 mg/kg, respectively.

Antinociceptive effect vs. dose of DPDPE in mr1a(−/−) (○) and FVB (•) mice. Data are expressed as mean ± S.E. (n ≥ 4 per group). A significant difference (ANOVA; P < .05) in antinociception was observed among dose groups in both control and knockout mice.
As was the case for the dose-response profile, the relationship between antinociception and DPDPE concentration in mdr1a(−/−) mice also was shifted leftward as compared to FVB mice (fig.5). Application of the sigmoidal Emax model to the data from mdr1a(−/−) mice yielded a brain tissue EC50 of 12.3 ± 0.2 ng/g and γ = 7.44 ± 0.55 (compared to an apparent EC50 of 160 ± 14 ng/g and γ = 4.24 ± 2.02 in FVB mice). The parameters obtained from mdr1a(−/−) mice were used in further PK-PD model development to address the hypothesis that the difference in EC50 between the two mouse strains was not due to differences in intrinsic sensitivity to the opioid peptide, but rather to differences in compartmentation of the peptide within the brain (see below).
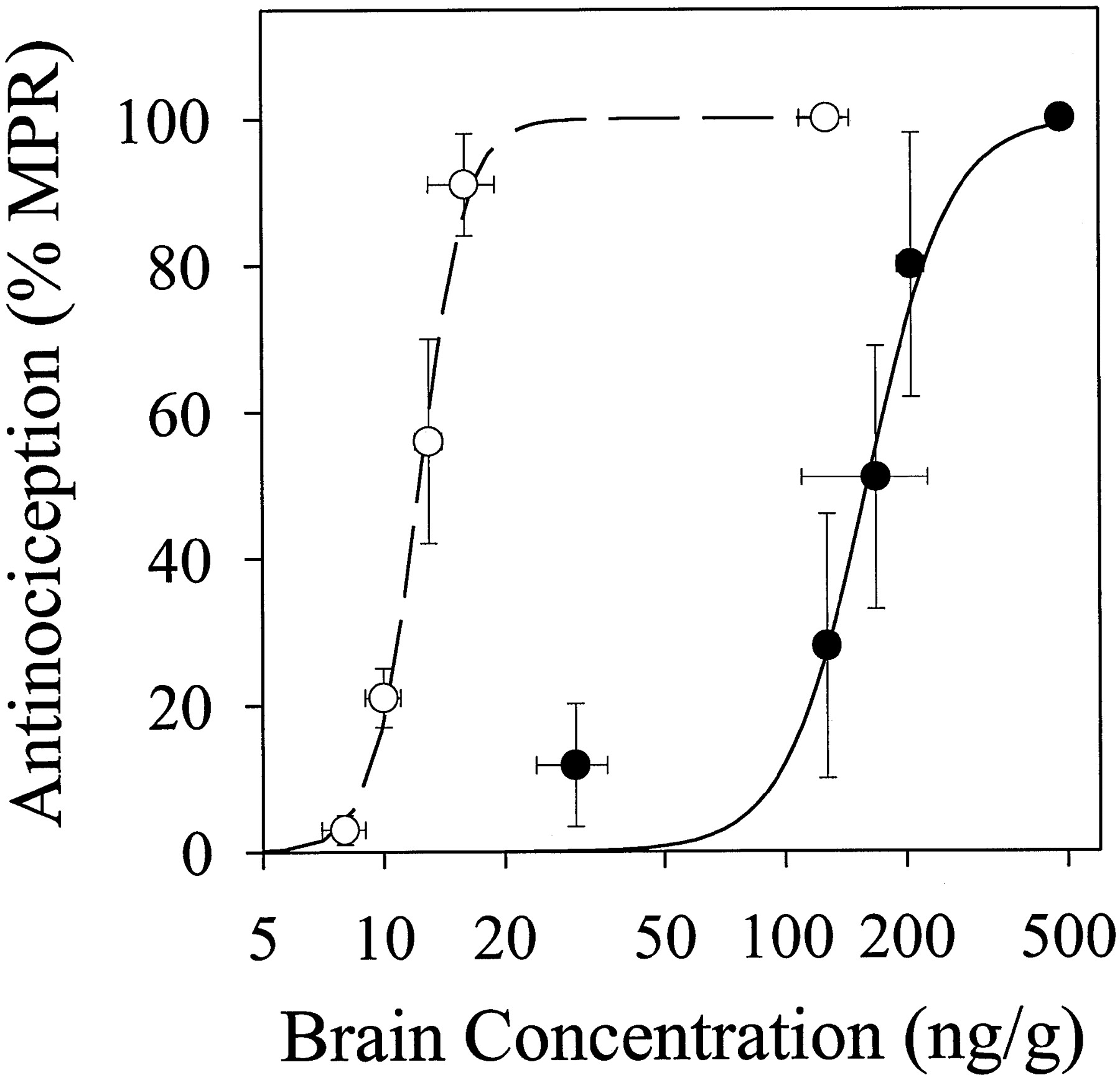
Antinociception vs. brain concentration from the dose-response study in mdr1a(−/−) (○) and FVB (•) mice. Symbols represent observed data (mean ± S.E.,n ≥ 4 per group); solid line is the fit of a sigmoidal Emax model to the data.
Time course of response.
This experiment was designed to examine the relationship between the time course of antinociception and DPDPE blood concentrations after administration of doses that produced similar pharmacologic activity in mdr1a(−/−) and FVB mice. Peak effect was observed at 5 (83.0 ± 10.1% MPR) or 10 (100 ± 0% MPR) min after i.v. administration of DPDPE in wild-type (24 mg/kg) or knockout (0.9 mg/kg) mice, respectively. Antinociception remained >20% MPR for up to 40 [mdr1a(−/−)] or 60 [FVB] min; 20% MPR in each mouse strain was statistically higher than effect measured in control mice receiving saline (−13 ± 7% MPR, n = 5). Despite a 27-fold lower dose (0.9 vs. 24 mg/kg), mdr1a(−/−) mice had peak responses, as well as duration of action, similar to FVB mice. The concentration-time profiles for DPDPE in blood in mdr1a(−/−) and FVB mice are shown in figure 6. Similar to the 10-mg/kg dose, DPDPE concentrations in blood declined biexponentially in mdr1a(−/−) and FVB mice after administration of 0.9 or 24 mg/kg; both profiles were well described by a two-compartment model with first-order elimination from the central compartment. The pharmacokinetic parameters estimated by compartmental analysis are presented in table 2. The apparent volume of the central compartment was smaller in FVB mice (796 ± 55 ml/kg) than in mdr1a(−/−) mice (1493 ± 124 ml/kg), resulting in a larger elimination rate constant in the wild-type (0.266 ± 0.026 min−1) as compared to the knockout (0.149 ± 0.015 min−1) animals. No difference in apparent clearance was observed between the two strains (222 ± 29vs. 212 ± 25 ml/min/kg in mdr1a(−/−) and FVB mice, respectively).

Blood concentration-time profile of DPDPE after a 24-mg/kg i.v. bolus dose in FVB mice (•) or a 0.9-mg/kg i.v. bolus dose mdr1a(−/−) mice (○). Symbols represent observed data (mean ± S.E.; n = 5 per group); line is the fit of a two-compartment model to each data set (see table 2 for parameter estimates).
Pharmacokinetic parameters for DPDPE disposition based on compartmental analysis2-a
A plot of pharmacologic effect vs. DPDPE concentration in blood yielded a counterclockwise hysteresis loop (fig.7). Peak antinociceptive effect of DPDPE was not observed at the time of peak blood concentration, suggesting that blood was not in immediate equilibrium with the site of action for DPDPE. The magnitude (corrected for differences in concentration), as well as the shape, of the hysteresis loop was comparable betweenmdr1a(−/−) and FVB mice (fig. 7).

Relationship between antinociception and dose-normalized blood concentration of DPDPE after a 24- (FVB; •) or 0.9-[mdr1a(−/−); ○] mg/kg i.v. bolus dose. Arrows indicate the direction of time. Error bars have been omitted for clarity.
Development of an integrated PK-PD model for DPDPE.
The relationship between antinociception and brain tissue DPDPE concentration suggested that mdr1a(−/−) mice were more sensitive to the peptide than FVB mice. However, this conclusion is based on the assumption that whole brain concentrations of DPDPE are representative of concentrations at the receptor biophase. If P-gp is located at brain tissue sites other than capillary endothelia, the transporter may serve to recompartmentalize the peptide. To examine this possibility, two integrated PK-PD models (fig. 1) were fit to the various data sets obtained for each mouse strain as described in “Methods.”
Parameters describing the systemic portion of each model (Vc, k12, k21, k10) were obtained from DPDPE disposition after a 10-mg/kg (fig. 2), 0.9-mg/kg (fig. 6) or 24-mg/kg (fig. 6) dose. Pharmacodynamic parameters (EC50 and γ) were obtained inmdr1a(−/−) mice (fig. 5), and were assumed to represent the true pharmacodynamics in both mouse strains (i.e., the brain tissue concentration of DPDPE was assumed to be homogeneous in the knockout animals). The systemic kinetic and pharmacodynamic parameters then were fixed in each integrated model. In the case ofmdr1a(−/−) mice, the model was fit to the concentration-time (brain tissue and blood) and effect-time data after a 10-mg/kg dose to obtain non-P-gp-mediated flux between blood and brain tissue (k13 and k31; fig.8). The model provided an adequate description of DPDPE disposition and antinociception. This simple model was not capable of describing DPDPE concentrations and effect in FVB mice with the pharmacodynamic parameters obtained frommdr1a(−/−) mice. It was necessary to differentiate the brain into three compartments, one in equilibrium with blood and containing P-gp-mediated efflux (presumably representing capillary endothelia), one representing the effect site and one representing brain tissue DPDPE at sites not in rapid equilibrium with the receptor biophase. This more complicated model provided a good description of the PK-PD data in FVB mice after a 10-mg/kg i.v. bolus dose (fig. 8). Final parameter estimates for the integrated PK-PD model are summarized in table 3.

Fit of the PK-PD models shown in figure 1 to the effect (diamonds), blood concentration (circles) and brain tissue concentration (triangles) vs. time data after a 10-mg/kg dose of DPDPE in mdr1a(−/−) (top) or FVB (bottom) mice. Symbols indicate mean ± S.E. (n = 4 per group); lines indicate fit of the model to the data. See table 3 for parameter estimates.
PK-PD parameters for DPDPE in mice3-a
To further test the performance of these two models, the blood concentration and effect data after a 0.9-mg/kg dose [mdr1a(−/−)] were simulated (fig.9). The model-predicted effectvs. time profile was in good agreement with the observed data. In the case of the 24-mg/kg dose in FVB mice, blood concentrations were in the range previously shown to be associated with saturable brain uptake in rats (Thomas et al., 1997). To overcome this difficulty, a saturable uptake process was added to the model, although all other parameters were held constant. Again, the model provided an excellent description of the observed data (fig. 9). Moreover, the kinetic parameters associated with brain uptake of DPDPE were in reasonably good agreement with the parameters obtained from previous experiments in rats (table 4).

Time course of DPDPE blood concentrations (circles) and antinociceptive effect (diamonds) in mdr1a(−/−) (0.9 mg/kg; A) and FVB (24 mg/kg; B) mice. Symbols indicate mean ± S.E. (n = 5 per group); lines indicate model predictions based on the parameter estimates presented in table 3[mdr1a(−/−) and FVB] and table 4 (FVB only).
Comparison of DPDPE brain uptake kinetics in mice and rats
Discussion
Previous experiments (Chen and Pollack, 1997a) indicated that the duration of action of DPDPE would be short based solely on systemic pharmacokinetics. DPDPE was eliminated rapidly (half-life of ∼14 min) due to extensive biliary excretion of intact peptide; ∼80% of the dose was recovered in bile (Chen and Pollack, 1997a). Rapid DPDPE elimination could explain, in part, the minimal availability of DPDPE at the site of action. Further studies were focused on DPDPE distribution between brain tissue and blood. These experiments suggested that active efflux from brain might be responsible for the low brain tissue partitioning (Chen and Pollack, 1997b). Based on these results, we hypothesized that P-gp may be responsible for the saturable biliary excretion in the rat and the active efflux of DPDPE from brain in the mouse.
P-gp is an energy-dependent efflux pump that reduces intracellular accumulation of chemotherapeutic agents in drug-resistant cells (Gottesman and Pastan, 1989). P-gp displays a specific pattern of expression in normal tissues, including brain capillary endothelium and bile canaliculi (Bradley et al., 1990). In vitrostudies have shown that morphine, a mu opioid agonist, is a P-gp substrate. Morphine accumulation in Chinese hamster ovary cells that overexpress P-gp was more than 3-fold lower than in cells that did not overexpress P-gp (Callaghan and Riordan, 1993). Morphine accumulation in bovine brain microvessel endothelial cells also was enhanced significantly by the P-gp inhibitors GW918 and verapamil (Letrent et al., 1997). Thus, opioids as a class may be substrates for P-gp.
Several studies have demonstrated that P-gp interacts with peptides (Sharma et al., 1992; Saeki et al., 1993; Aungst and Saitoh, 1996). Cyclosporine effectively reverses the multidrug resistant phenotype (Goldberg et al., 1988) by binding to P-gp (Foxwell et al., 1989). P-gp appears to be a functional barrier to the intestinal absorption of the cyclic peptide DMP728 in rats (Aungst and Saitoh, 1996); mucosal-to-serosal flux was 4-fold lower than serosal-to-mucosal flux, suggesting net export from blood to the intestinal lumen. Considering the apparent role of P-gp in the disposition and action of opioids and peptides, it is not unlikely that opioid peptides are substrates for this transporter.
Mice with homozygously disrupted mdr1a gene represent a unique in vivo model to examine the role of P-gp in the pharmacology of drugs that act in the CNS. However, only one relevant study published to date. Loperamide, a mu opioid agonist with virtually no CNS effects due to limited brain penetration, displayed profound CNS activity in mdr1a(−/−) mice, presumably due to a 6-fold higher brain tissue concentration as compared to control mice (Schinkel et al., 1996).
Our study was designed to test the hypothesis that P-gp-mediated efflux limits the antinociceptive effect of DPDPE. Consistent with this hypothesis, a 10-mg/kg i.v. dose of DPDPE elicited 100% MPR inmdr1a(−/−) mice vs. only 20% MPR in FVB mice. The duration of antinociception also was substantially longer inmdr1a(−/−) mice; >85% MPR remained for 40 min inmdr1a(−/−) mice, although measurable effect lasted only 15 min in FVB mice.
A striking difference (50- to 80-fold) was observed in the doses of DPDPE required to elicit comparable antinociception inmdr1a(−/−) and FVB mice (fig. 4). The shift in the dose-response relationship could be due to a variety of differences between the two mouse strains (e.g., decreased systemic clearance, decreased brain efflux). Although blood concentrations of DPDPE were similar in the two mouse strains, an ∼3-fold higher brain concentration was observed in mdr1a(−/−) compared to FVB mice. The significantly higher brain:blood concentration ratio inmdr1a(−/−) as compared to FVB mice (0.265 ± 0.025vs. 0.045 ± 0.008, respectively; P < .0001) is consistent with the hypothesis that brain-to-blood efflux of DPDPE is mediated by P-gp, which is virtually absent in brain capillary endothelial cells of mdr1a(−/−) mice (Schinkel et al., 1995). However, pharmacological effect per unit brain tissue concentration also was much higher for mdr1a(−/−) compared to FVB mice (fig. 5), resulting in a substantial difference in EC50 (12 vs. 160 ng/g). This unanticipated observation suggested that mechanisms other than whole-organ accumulation must be responsible, in part, for the observed differences in antinociception.
The dose-normalized mass of DPDPE excreted in the gallbladder was dose-independent in mdr1a(−/−) and FVB mice, suggesting that biliary excretion was not saturable in the dose range (0.4–10 mg/kg for mdr1a(−/−) and 10–80 mg/kg for FVB mice) investigated. A significantly higher (4-fold) dose-normalized mass of DPDPE in the gallbladder was observed in mdr1a(−/−) mice as compared to FVB mice. Both mdr1a and mdr1bexist in bile canaliculi, and mdr1b is overexpressed inmdr1a(−/−) mice (Schinkel et al., 1995). If biliary excretion of DPDPE was mediated only by the protein product of the mdr1a gene, then the dose-normalized amount of DPDPE in the gallbladder should have been lower in mdr1a(−/−) than in FVB mice. Because recovery of DPDPE in the gallbladder was higher inmdr1a(−/−) than FVB mice, mdr1b might be involved in the biliary excretion of DPDPE.
A counterclockwise hysteresis loop characterizing the relationship between antinociception and blood concentration was observed for bothmdr1a(−/−) and FVB mice (fig. 7), suggesting that the site of action in both strains was pharmacologically distinct from the central compartment; peak blood concentrations were not associated with maximal antinociception. It is important to note that the degree of hysteresis was similar between the two mouse strains. No statistical difference (P > .05) was observed in the area bounded by the hysteresis loop between FVB and mdr1a(−/−) mice. Because blood-brain translocation kinetics of DPDPE differed betweenmdr1a(−/−) and FVB mice, it is unlikely that this hysteresis behavior represents time-dependent equilibration between brain tissue and blood. Rather, the kinetic-dynamic dissociation likely resulted from a temporal delay between presentation of DPDPE to the site of action and the onset of measurable antinociception. In this case, the similarity in hysteresis would provide evidence that thedelta opioid receptor system per se was not altered inmdr1a(−/−) mice.
If the intrinsic responsivity of the opioid system is not affected by the genetic alteration in P-gp expression, then the difference in the effect vs. brain tissue concentration relationship between knockout and wild-type mice must have a pharmacokinetic basis. Although the mechanism underlying a pharmacokinetic perturbation of DPDPE-associated antinociception remains to be elucidated, two potential explanations may be proposed. If DPDPE binds to P-gp, then P-gp in brain tissue would decrease the unbound (pharmacologically active) concentration of DPDPE. Whole brain tissue concentrations of DPDPE would not be reflective of unbound concentrations in the organ. Alternatively, P-gp could redistribute DPDPE away from the receptor biophase, either on a regional (recompartmentalization to brain regions devoid of the delta opioid receptor) or local (essentially serving as a barrier to peptide presentation to the receptor) basis. The development of an integrated PK-PD model (fig. 1) for DPDPE was pursued to address this issue. It was possible to describe the time course of antinociception in both mdr1a(−/−) and FVB mice with a single set of pharmacodynamic parameters (fig. 9), as would be expected in the opioid system was not perturbed in the knockout animals, only through introduction of a secondary brain tissue compartment in relatively slow equilibrium with the effect site. Although this modeling exercise does not provide direct evidence for a regional pharmacokinetic difference between the two mouse strains, it does suggest that a pharmacokinetic explanation is viable. Mechanistic studies are underway to address the hypothesis that P-gp in brain tissue, as opposed to capillary endothelium, redistributes DPDPE from the receptor biophase.
In summary, our results implicate P-gp as a factor that limits the accessibility of DPDPE to brain tissue. In addition, these data suggest that P-gp might be located in brain sites other than capillary endothelium, consistent with recent reports of P-gp in glial cells (Dietzmann et al., 1994) and astrocyte foot processes on the ablumenal side (Golden and Pardridge, 1997). Future studies of the interaction between opiates, including opioid peptides, and P-gp will provide insight into the role of this transport protein in opioid pharmacology.
Acknowledgments
DPDPE was generously provided by National Institute on Drug Abuse.
Footnotes
-
Send reprint requests to: Dr. Gary M. Pollack, Division of Drug Delivery and Disposition, School of Pharmacy, Beard Hall CB#7360, The University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-7360.
- Abbreviations:
- DPDPE
- [d-penicillamine2,5]enkephalin
- P-gp
- P-glycoprotein
- MPR
- maximum possible response
- mdr1a(−/−)
- mdr1a gene-deficient
- ANOVA
- analysis of variance
- PK-PD
- pharmacokinetic-pharmacodynamic
- Vss
- steady-state volume of distribution
- MRT
- mean residence time
- Cl
- systemic clearance
- CNS
- central nervous system
- Received February 9, 1998.
- Accepted June 15, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}