


Abstract
A toxicokinetic study was performed using rats to investigate the possible mechanism of 18 acute deaths in Japanese patients with cancer and herpes zoster by interactions of the new oral antiviral drug, sorivudine (SRV), with one of the oral 5-fluorouracil (5-FU) prodrugs within 40 days after approval of the use of SRV. Tegafur, an anticancer 5-FU prodrug suggested to be used by most of the patients who died, and SRV were orally administered to rats simultaneously once daily. All of these rats died within 10 days, whereas rats given SRV or tegafur alone under the same dosage conditions showed no appreciable change over 20 days compared with controls. In the rats given both drugs, bone marrow and intestinal membrane mucosa were greatly damaged at an early stage of the coadministration, and before death, the animals showed marked decreases in white blood cell and platelet counts, diarrhea with bloody flux, and severe anorexia, as was also manifested by the patients who subsequently died. In the rats given both drugs for 6 days, extremely enhanced 5-FU levels were observed from the first day of administration in plasma and in all tissues examined, including bone marrow and intestines. The extreme enhancement of the tissue 5-FU levels was attributable to the facile inactivation by (E)-5-(2-bromovinyl)uracil (BVU) of hepatic dihydropyrimidine dehydrogenase (DPD), a key enzyme regulating the systemic 5-FU level in the rat and human. BVU, a major metabolite formed from SRV by gut flora, was found at considerable levels in the liver of rats orally administered SRV alone or SRV and tegafur, and there was a marked decrease in hepatic DPD activity. In the presence of NADPH, DPD purified from rat liver cytosol was rapidly and irreversibly inactivated by [14C]BVU as a suicide inhibitor with concomitant incorporation of the radioactivity into the enzyme protein, although SRV showed no inhibitory effect on DPD under the same conditions. Human liver DPD was recently demonstrated by us to be inactivated with BVU in a manner very similar to rat DPD.
The Pharmaceutical Affairs Bureau, Japanese Ministry of Health and Welfare, reported that in 1993 fifteen deaths in Japanese patients occurred in association with the coadministration of the new antiviral drug for herpes zoster, SRV, with one of oral 5-FU prodrugs within 40 days after SRV was approved by the Japanese government and began to be used clinically (Pharmaceutical Affairs Bureau, 1994). In addition, the report noted that three additional patients died from the same drug interactions during phase II clinical trials of SRV. The report also mentioned that before death, all of these patients had severe symptoms of toxicity, such as diarrhea with bloody flux and marked decreases in white blood cell and platelet counts and that eight other Japanese patients receiving both drugs during this period had severe symptoms of gastrointestinal- and myelo-toxicity. All of these patients had received SRV for a period of several days although being administered continuous anticancer chemotherapy with one of the 5-FU prodrugs.
The clinical dose of SRV, 50 mg three times a day, is much lower than the minimal toxic doses for animals, e.g., >2000 mg/kg in rats (Nagasaka et al., 1990). Therefore, there were no reports of acute death or toxic symptoms as described above in patients who had herpes zoster and had received SRV alone or SRV and anticancer drugs other than 5-FU or its oral prodrugs, including FT. FT, the most widely used oral 5-FU prodrug in combination with uracil in Japan, is activated to 5-FU mainly by hepatic cytochrome P-450 after being absorbed from the intestinal membrane (Fujita et al., 1976). Actually, most of the patients who died from the drug interaction were likely to receive FT, although a few seemed to receive one of the other types of 5-FU prodrugs, such as carmofur (1-hexylcarbamoyl-5-fluorouracil) and doxifluridine (5′-deoxy-5-fluorouridine).
The anticancer drug, 5-FU, and its oral prodrugs have been well recognized to have severe toxic effects on the gastrointestinal tract and bone marrow, both of which have rapid cell proliferation, in patients on long-term treatment at clinical dosage levels. These toxic effects on the human and animals are characteristic of 5-FU as an inhibitor of DNA synthesis.
The 18 patient deaths would not have occurred if the following three facts had been carefully considered in the safety/risk assessment of drug interactions during development of the new antiviral drug SRV. First, BVU is a major metabolite of SRV in rats (Nishimoto et al., 1990) and humans (Ogiwara et al., 1990). Second, BVU markedly increased the plasma concentration of 5-FU and enhanced the toxicity of 5-FU with concomitant retardation of hepatic DPD activity when 5-FU and BVU were given only once successively i.p. to rats and mice (Desgranges et al., 1986). Third, an in vitro study using DPD partially purified from rat liver demonstrated that the enzyme was inactivated with BVU in the presence of NADPH and that its activity was not restored by dialysis (Desgrangeset al., 1986), although the inactivation mechanism is equivocal.
Hepatic DPD, a homodimeric 210-kDa flavoprotein with Fe-S clusters, has been recognized as a key enzyme regulating plasma and tissue concentrations of 5-FU administered to humans (Lu et al., 1992) as well as to rats (Shiotani and Weber, 1981; Fujimoto et al., 1991; Lu et al., 1993a). 5-FU is dihydrogenated at the 5,6-double bond by DPD and is rapidly decomposed to α-fluoro-β-alanine by subsequent enzymatic processes (Diasio and Harris, 1989). In the human, more than 85% of 5-FU administered i.v. is catabolized by DPD through this metabolic pathway (Diasio and Harris, 1989). The aforementioned facts imply that the 18 patient deaths might be due to an increase in the tissue 5-FU level by inactivation of hepatic DPD with BVU formed from the antiviral SRV that was coadministered.
Recently, we have undertaken a study on the possible mechanism of the patient deaths using rats and briefly reported in a communication that BVU rapidly inactivated DPD in the presence of NADPH by covalent binding of a reduced form as a reactive metabolite of BVU to the enzyme protein (Okuda et al., 1997). We also reported that oral coadministration of SRV and FT to rats elevated their blood and tissue levels of 5-FU, resulting in severe damage to bone marrow and intestinal membrane. More recently, we demonstrated that human DPD was also inactivated by BVU in the same manner as was rat hepatic DPD (Ogura et al., 1998).
The report deals with 1) detailed results of our toxicokinetic study of 5-FU in the bone marrow and small intestine as well as in the plasma and liver of rats orally administered FT and SRV, 2) histological changes in these tissues of rats orally administered both drugs in addition to the hematological and toxicological findings in the animals and 3) the mechanism of the irreversible inactivation of purified rat liver DPD using [14C]BVU.
Materials and Methods
Materials.
SRV was prepared as reported previously (Sakataet al., 1980). [14C]BVU was prepared from 5-formyluracil with [2-14C]malonic acid (9.25 MBq, Du Pont NEN Research Products, Boston, MA) via 5-(2-carboxy-[2-14C]vinyl)uracil in the same manner as used for the synthesis of unlabeled BVU (Jones et al., 1979). [14C]BVU synthesized had a specific activity of 2.0 MBq/μmol and a radiochemical purity of more than 99% after purification by HPLC.
Animal treatment.
Female Wistar rats (5 groups of 90 animals each, 6 wk of age) were obtained from Japan SLC, Inc. (Hamamatsu, Japan) and housed with free access to food and water in a light- and dark-controlled room (lights on 6:00 a.m. to 6:00p.m. and lights off 6:00 p.m. to 6:00a.m.). The animals were orally administered FT alone (60 mg/kg, triple the daily dose in clinical use) (Taiho Pharmaceutical Co., Tokushima, Japan), SRV alone (30 mg/kg, 12 times the daily dose in clinical use), BVU alone (3.7 mg/kg) (Sigma Chemical Co., St. Louis, MO) or FT (60 mg/kg) and SRV (30 mg/kg) simultaneously at 9:00 to 10:00a.m. once daily for 6 days as a 1-ml suspension in 0.5% (w/v) sodium carboxymethylcellulose. The same volume of the vehicle was also used for the control animals. Blood and tissue samples were collected at 1, 2, 4, 8 and 24 hr after administration of the drugs on days 1, 2, 4 and 6. These samples were collected from three animals at each time point after they were anaesthetized with ether and killed.
Quantification of pyrimidine levels.
FT, 5-FU and uracil in the plasma, urine and tissue samples were determined as reported previously (Marunaka et al., 1980). SRV and BVU were analyzed by HPLC on an Inertsil ODS-2 column (150 × 4.6 mm, GL Sciences, Tokyo, Japan) eluted at a flow rate of 1 ml/min with 15% (v/v) acetonitrile-water containing 0.01% (w/v) trifluoroacetic acid after extraction with ethyl acetate from plasma (1 ml) and liver homogenates (0.3 g tissue/1 ml saline). SRV and BVU eluted at retention times of 7.4 and 9 min, respectively, from the HPLC column were determined with a detection limit of 0.1 μg/ml plasma or g liver as reported previously (Okuda et al., 1997).
Tissue preparation for optical microscopy.
Intestines and other tissues were obtained at the 24th hr on days 3 and 6 from the rats after the repeated oral administration of the vehicle, FT alone, SRV alone or FT and SRV at the same dose(s) as mentioned above. The tissues were fixed in a solution of 10% (w/v) formalin, dehydrated, and embedded in paraffin, and paraffin sections (5 μm in thickness) were stained with eosin and hematoxylin in the usual manner.
CFU-GM assay.
Bone marrow cells were collected at the 24th hr on days 1, 2, 4 and 6 from femurs of rats after repeated oral administration of the vehicle, FT alone, SRV alone or FT and SRV at the same dose(s) as mentioned above, seeded at 105 mononuclear cells/35-mm Petri dish, and cultured in the presence of a recombinant mouse granulocyte-macrophage colony-stimulating factor under the same conditions as reported previously (Okuda et al., 1997). After incubation for 7 days at 37°C in a humidified atmosphere of 5% (v/v) CO2, colonies containing 40 cells were counted as CFU-GM colonies with an inverted microscope.
Enzyme assay.
DPD was purified from young adult female Wistar rats as reported previously (Lu et al., 1993a). Activity of the purified enzyme toward 5-FU was assayed by a previously reported method (Desgranges et al., 1986) with modifications; the enzyme (5–50 ng of protein/5 μl) was incubated with 20 μM [14C]5-FU (2.1 MBq/μmol, Moravek Biochemicals Inc., Brea, CA) at 37°C for 5 min in the presence of 200 μM NADPH, 2.5 mM magnesium chloride and 10 mM 2-mercaptoethanol in a final volume of 50 μl of 30% (v/v) glycerol-35 mM K-phosphate buffer, pH 7.4. [14C]H2-5-FU formed as a metabolite was separated on a diethylaminoethyl cellulose TLC plate as reported previously (Traut and Loechel, 1984).
DPD activity in rat liver cytosol was assayed by a previously reported method (Ikenaka et al., 1979) with modifications; rat liver cytosol was incubated with 25 μM [14C]5-FU (29.6 kBq/mmol) in the presence of 500 μM NADPH, 1.25 mM magnesium chloride and 5 mM 2-mercaptoethanol in a final volume of 250 μl of 35 mM K-phosphate buffer, pH 7.4, at 37°C for 10 min. After the termination of the enzyme reaction by the addition of 60 μl of 2 M aqueous potassium hydroxide, the [14C]H2-5-FU formed was hydrolyzed to [14C]α-fluoro-β-ureidopropionate and/or [14C]α-fluoro-β-alanine as reported previously (Ikenaka et al., 1979). [14C]α-Fluoro-β-ureidopropionate and [14C]α-fluoro-β-alanine were separated from the substrate, [14C]5-FU, on a silica TLC plate, and their radioactivities were determined by liquid scintillation counting.
Reaction of purified DPD with BVU.
DPD (0.5 μg) purified from rat liver cytosol was preincubated with various concentrations of BVU in the presence and absence of 200 μM NADPH in a final volume of 50 μl of 30% (v/v) glycerol-35 mM K-phosphate buffer, pH 7.4, containing 2.5 mM magnesium chloride and 2.5 mM 2-mercaptoethanol at 37°C for 5 min. Aliquots (5 μl) of the reaction mixture were withdrawn and immediately assayed for residual DPD activity. A 10% decrease in the remaining activity was observed with the purified DPD preincubated with 50 μM BVU in the absence of NADPH when the preincubation mixture was diluted 10-fold and incubated for the enzyme assay with [14C]5-FU in the presence of NADPH (200 μM). The apparent decrease at various concentrations of BVU was subtracted from the data on the remaining activity to show that DPD was not influenced by BVU in the absence of NADPH during the preincubation.
For isolation of the DPD protein incorporating the radioactivity of 4 μM [14C]BVU, the final volume of the incubation mixture was increased up to 500 μl without changing the concentrations of the constituents. The mixture was incubated at 37°C for 30 min, diluted with a large excess (25 times) of BVU and then rapidly chilled in an ice bath. The radioactivity incorporated into the enzyme protein was separated from unreacted [14C]BVU by HPLC and determined by liquid scintillation counting.
HPLC of DPD.
HPLC was carried out on a TSK GEL 2000 SWXL column (300 × 7.8 mm) (Tosoh Corporation, Tokyo, Japan) with a Gilson model 325 HPLC pump (Villiers-le-Bel, France). Rat liver DPD was eluted at a flow rate of 1 ml/min with 35 mM K-phosphate buffer, pH 7.4, at a retention time of 5.2 min. The DPD isolated by HPLC after incubation with 4 μM [14C]BVU for 30 min in the presence of NADPH was concentrated with a Centricon concentrator (Amicon, Inc., Beverly, MA) and rechromatographed under the same chromatographic conditions. The chromatogram was monitored by absorbance at 220 nm with a Gilson model 111B UV detector and by liquid scintillation counting of the column effluent collected every 30 sec.
Electrophoresis.
SDS-PAGE was carried out on a 10% (w/v) polyacrylamide gel slab by the previously reported method (Laemmli, 1970). Native PAGE was carried out on a 9% (w/v) polyacrylamide gel slab as reported previously (Lu et al., 1992). Proteins were stained with Coomassie Brilliant Blue R250.
Radioluminography.
A radiolabeled DPD fraction eluted from the HPLC column was subjected to SDS-PAGE after concentration of the effluent with a Centricon concentrator. After staining as mentioned above, the gel slab was dried in vacuo. Radioactivity migrating with DPD on the gel slab was visualized by radioluminography, using a Fuji Photo Co. model BAS 2000 (Tokyo, Japan) after exposing the dried gel slab to a Fuji imaging plate for 8 hr.
Statistical analysis.
The significance of differences was determined by Student’s t test.
Results
Marked increases in plasma and tissue levels of 5-FU in rats coadministered FT and SRV.
Toxicokinetics were investigated for the active metabolite 5-FU formed from its prodrug FT in rats orally coadministered FT (60 mg/kg) and SRV (30 mg/kg) simultaneously once daily for 6 days. In addition, plasma and urinary levels of uracil, an endogenous substrate for DPD, were also determined in the animals.
The daily doses of FT and SRV coadministered to the rats were determined through several preliminary experiments by adjusting the days at which severe toxicity appeared and death occurred in the rats to those reported for the patients: about 3 to 4 days after coadministration for toxicity and by 10 days for death. At these doses, a 6-day coadministration had to be selected for our toxicokinetic study because one third of the animals died on days 6 to 7 of treatment, and an increasing number of deaths occurred thereafter with prolongation of the duration of treatment.
FT was absorbed rapidly, with a Tmax of 1 to 2 hr, in rats orally administered FT alone or FT and SRV once daily and almost disappeared from plasma, with a t½ of 2.7 to 4.3 hr, within 24 hr throughout the days examined (fig.1A). The plasma FT level in the rats coadministered FT and SRV showed no appreciable difference from that in the animals administered FT alone. Plasma AUC0–24 andCmax of FT in the rats administered both drugs showed the almost same magnitudes as those in animals administered FT alone.

Time courses of FT, 5-FU and uracil levels in plasma of rats given FT alone or FT and SRV. Rats were orally administered FT alone (open circles) or FT and SRV simultaneously (closed circles) once daily for 6 days. The daily doses of FT and SRV were 60 and 30 mg/kg, respectively. A, B and C represent FT, 5-FU and uracil levels, respectively. Data are expressed as mean ± S.D. of three rats.
However, the plasma concentration of 5-FU formed from its prodrug FT was greatly increased to a highly toxic level when FT was orally coadministered with SRV, whereas rats administered FT alone showed a low plasma level of 5-FU (Cmax 1.4 ± 0.3 nmol/ml) similar to that observed in plasma of patients receiving FT clinically (fig. 1B). On day 1, the plasma AUC0–24 of 5-FU in the rats coadministered FT and SRV was 5.5 times more than that in the animals administered the same dose of FT alone, and the plasmaCmax was 5 times higher than that in the FT-treated animals. On days 4 to 6, the AUC0–24 was 7.7 to 8 times more than that in the rats administered FT alone, and the Cmax was 12.8 to 13.7 times higher than that in the FT-treated animals. In the plasma of the rats administered FT and SRV, 5-FU appeared with aTmax of 2 to 4 hr and was almost cleared, with at1/2 of 2.6 to 5 hr, within 24 hr therefrom throughout the days examined except day 1. On day 1, theTmax of 5-FU in the plasma was observed 8 hr after coadministration. In the plasma of rats administered FT alone, theCmax of 5-FU was reached at 2 to 4 hr postdose, and 5-FU was cleared with a t1/2 of 4.5 to 7.5 hr therefrom throughout the days examined.
The plasma level of uracil was also markedly increased by the oral coadministration of FT and SRV (fig. 1C). In the rats administered both drugs once daily, the plasma uracil level showed a markedly elevatedCmax, with a Tmax of 8 hr, whereas the animals given FT alone showed a low and constant plasma level of the pyrimidine throughout the days examined. Throughout the days examined, the average AUC0–24 of uracil in the rats administered both drugs was 692.5 ± 133.6 nmol × hr/ml which was 6.4 times larger than that in the animals administered FT alone (109.0 ± 9.4 nmol × hr/ml) or in the control animals (108.7 ± 11.2 nmol × hr/ml). The average Cmax of uracil in the rats administered both drugs was 37.5 ± 6.2 nmol/ml that was 6.2 times higher than that in the FT-treated animals (6.04 ± 1.1 nmol/ml) or in the controls (6.06 ± 1.2 nmol/ml).
Not only the blood levels of 5-FU and uracil, but also their urinary excretion were markedly increased by the oral coadministration of FT and SRV; the daily amounts of 5-FU and uracil excreted in the urine were 9.5 to 13.6 and 4.4 to 14.2 times higher, respectively, in the rats administered both drugs than in the animals administered the same dose of FT alone throughout the days examined. The urinary excretion of uracil in the rats given FT and SRV decreased almost linearly throughout the days examined. The average amounts of 5-FU and uracil excreted in the urine in the rats administered FT only were 184.0 ± 32.7 and 1691.5 ± 298.4 nmol/day, respectively. In the rats administered FT and SRV, 5-FU excreted in the urine for 6 days was 12.0 ± 7.1 μmol and estimated to be 7.1% of the total amount of FT administered during that period, whereas in the rats administered FT alone, it was 1.1 ± 0.2 μmol and to be only 0.6% of the total amount of the dosed FT. The daily amount of the prodrug FT excreted in the urine, however, was not affected by coadministration with SRV.
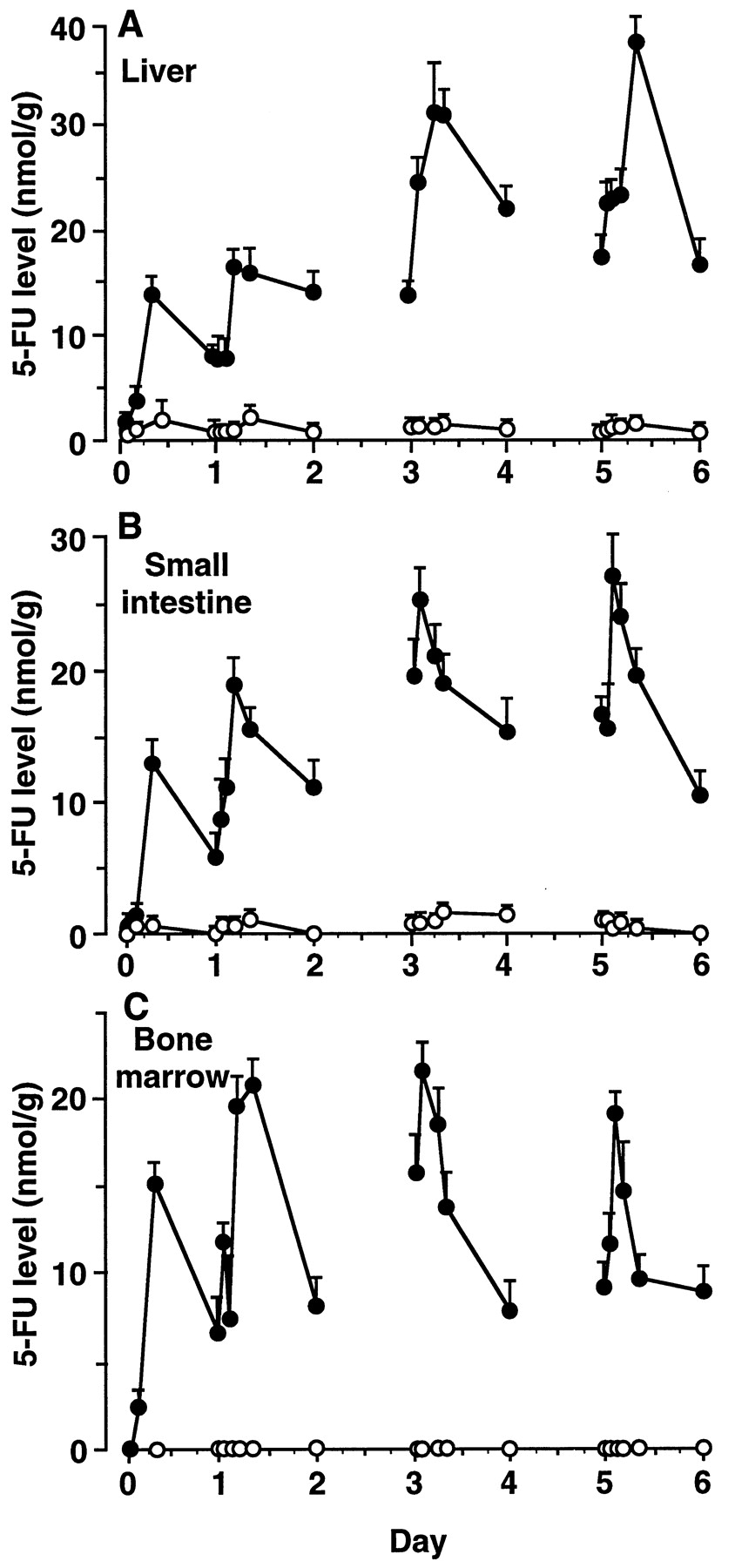
In rats orally given FT alone once daily, the 5-FU level was low in the liver (Cmax 1.4 ± 0.2 nmol/g tissue) and small intestine (Cmax 1.1 ± 0.3 nmol/g tissue) and was negligible in the bone marrow (fig. 2). However, in rats orally given FT and SRV once daily, the 5-FU level had greatly increased from day 1 in all tissues examined. On day 1,Cmax of 5-FU in the rats given both drugs were 12.3 and 16.7 times higher for the liver and small intestine, respectively, than in the rats given the same dose of FT alone. On day 6, the Cmax values were 26.4 and 26.2 times higher for the liver and small intestine, respectively, than in the rats given FT alone. Among tissues examined from rats administered FT and SRV, the 5-FU levels did not show a constant Tmax during the period of the administration; the Tmax values varied from 2 to 8 hr. However, in the liver and small intestine of the rats administered FT alone, Cmax of 5-FU was reached at 8 hr postdose throughout the days examined, except on day 6 when the Cmax of 5-FU was observed at 2 hr in the small intestine. Unlike in plasma, 5-FU was cleared slowly, with a t1/2 of 11 to 17 hr in all of the examined tissues of the rats coadministered both drugs, and these tissues contained 5-FU to a remarkable extent even 24 hr after coadministration.

Time courses of 5-FU levels in tissues of rats given FT alone or FT and SRV. Rats were orally administered FT alone (open circles) or FT and SRV simultaneously (closed circles) once daily for 6 days. The daily doses of FT and SRV were 60 and 30 mg/kg, respectively. A, B and C represent the 5-FU levels in liver, small intestine and bone marrow, respectively. Data are expressed as mean ± S.D. of three rats.
The markedly increased Cmax and prolonged time for elimination of 5-FU resulted in its extremely large AUC0–24 in the tissues of rats coadministered FT and SRV,e.g., the AUC0–24 were 340 (liver), 327 (small intestine) and 353 (bone marrow) nmol × hr/g tissue on day 2 (table 1). In contrast, AUC0–24 of 5-FU in rats orally administered FT alone once daily for 6 days were 22 to 30 (liver) and 8 to 33 (small intestine) nmol × hr/g tissue throughout the days examined, and at an undetectable level (less than 0.3 nmol × hr/g tissue) in the bone marrow.
AUC of 5-FU in plasma and tissues of rats given FT alone or FT and SRV
Marked decrease in hepatic DPD activity in rats coadministered FT and SRV.
There was no appreciable difference in hepatic DPD activity (1.0–1.2 nmol 5-FU reduced/mg protein/min) between control rats and rats administered FT alone once daily. However, the DPD activity in rats orally administered FT and SRV once daily for 6 days was markedly decreased to 34.2, 21.1 and 8.9% that of the controls 24 hr after coadministration on days 1, 4 and 6, respectively.
SRV and its metabolite BVU were detected at significant levels in the plasma and liver of rats orally given SRV alone as well as FT and SRV. For SRV, plasma and hepatic Cmax occurred 2 hr after dosage and averaged 23.3 nmol/ml (range: 18.3–30.6 nmol/ml) and 34.3 nmol/g tissue (range: 21.5–59.5 nmol/g tissue), respectively, throughout the days examined. For BVU, plasma and hepatic Cmax were reached at 8 hr after dosage of SRV and averaged 12.6 nmol/ml (range: 10.8–14.7 nmol/ml) and 25.5 nmol/g tissue (range: 22.3–31.4 nmol/g tissue), respectively. SRV and BVU were cleared from the plasma and liver with t1/2 of 1.5 to 5 and 9 to 14 hr, respectively, throughout the days examined.
In rats orally administered BVU at a dose of 3.7 mg/kg once daily for 6 days, Cmax of BVU in the plasma and liver were 13.2 nmol/ml and 27.0 nmol/g tissue, respectively, which were approximately equal to those from SRV orally administered once daily at a dose of 30 mg/kg. In the animals administered BVU at the aforementioned dose, hepatic DPD activity was markedly decreased throughout the days examined,e.g., the DPD activity was 14% of the controls on day 6.
Marked increase in toxicity by increased tissue levels of 5-FU in rats coadministered FT and SRV.
Body weight and dietary intake of rats orally administered FT and SRV once daily for 6 days decreased after day 2 (fig. 3). From the third day, body weight rapidly deceased, accompanied by a marked decrease in dietary intake. On days 5 to 6, the dietary intake was negligible, and on days 6 to 7, one third of the rats administered FT and SRV died; none of the animals remained alive on day 10. However, the animals given the same dose of FT or SRV alone once daily for 20 days showed no appreciable change in vital signs compared with the control animals.

Decreases in body weight and dietary intake in rats given FT and SRV. Rats were orally administered FT alone (open circles) or FT and SRV simultaneously (closed circles) once daily for 6 days. The daily doses of FT and SRV were 60 and 30 mg/kg, respectively. Closed squares represent control rats. A and B represent body weight and dietary intake, respectively. Data are expressed as mean ± S.D. of three rats.
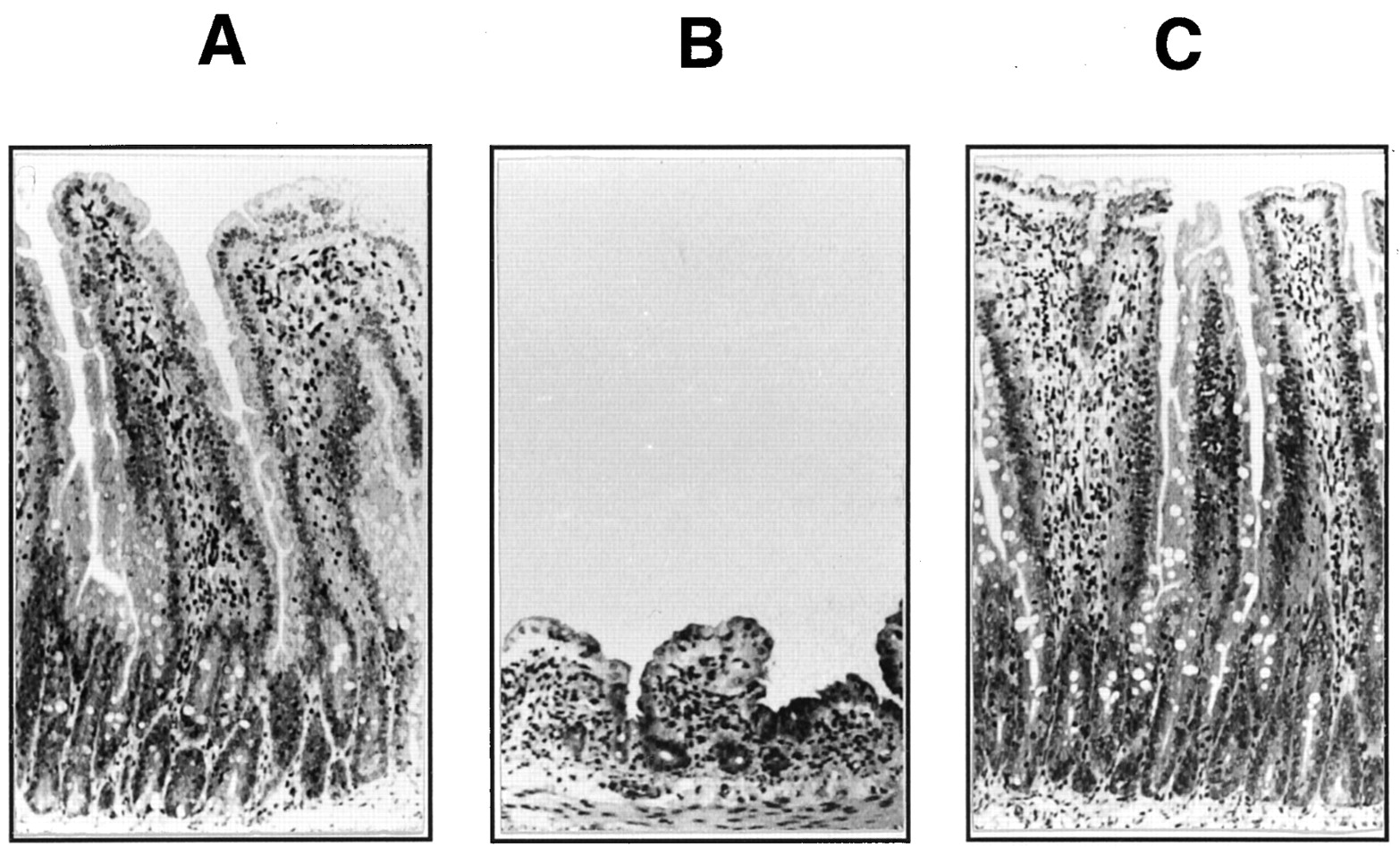
On days 3 to 4, most of the rats orally administered FT and SRV once daily had diarrhea with bloody flux and remarkable atrophy of the mucosal membrane in intestines, such as in the jejunum, ileum and colon. On day 6, the rats given FT and SRV showed serious pathologic histological changes in the intestines; i.e., for the jejunum, marked atrophy of mucous membrane and significant edema in the proper mucous membrane (fig. 4); for the ileum, significant atrophy and blood stasis in mucous membrane and edema in the proper mucous membrane; and for the colon, significant atrophy of mucous membrane. Rats given the same dose of FT or SRV alone did not show any pathological changes in intestines compared with the control rats (fig. 4).

Atrophy of jejunal mucosa in rats given FT and SRV. A, B and C represent jejunal mucosa in rats given a vehicle, FT and SRV simultaneously, and FT alone, respectively, once daily for 6 days. Photographs (×33) were taken on day 6 after coadministration. The daily doses of FT and SRV were 60 and 30 mg/kg, respectively.
No appreciable change was observed in the liver of the rats administered FT and SRV once daily for 6 days compared with the control rats, whereas marked atrophy was found in the spleen and thymus in these animals. On day 6, tissue weights (mean ± SD) of the spleen and thymus were 104 ± 45.1 and 27.3 ± 14.6 mg, respectively, in rats administered FT and SRV, 323 ± 16.5 and 340 ± 29.5 mg, respectively, in animals given FT alone, and 319 ± 11.4 and 354 ± 16.6 mg, respectively, in the control animals; the ratio of tissue to body weight was decreased to 54% for the spleen and 13% for the thymus of those of controls. There was no appreciable difference in these tissue weights between the rats administered FT or SRV alone and control animals.
The coadministration of FT and SRV to rats for 6 days severely affected the blood components with vital importance; i.e., white blood cell and platelet counts in the rats decreased to 18 and 26% of those in control rats, respectively (table2). However, the coadministration had little effect on red blood cells, which have a relatively long life span, i.e., mean corpuscular volume, mean corpuscular hemoglobin, and mean corpuscular hemoglobin concentrations were almost equal to those in the controls or in the FT-treated rats.
Hematological data in rats administered FT alone or FT and SRV
As early as day 2, bone marrow cells of the animals were demonstrated to be severely suppressed by the coadministration of FT and SRV (fig.5). On day 2, the cells collected from these rats showed only the slight formation of CFU-GM in the presence of a granulocyte-macrophage colony stimulating factor, and on day 6, no detectable colony formation was observed. Bone marrow cells in the rats given the same dose of FT or SRV alone, however, showed no appreciable change for 6 days.

Decrease in colony-forming activity of CFU-GM isolated from bone marrow of rats given FT and SRV. Cells were collected at the 24th hr on days 1, 2, 4 and 6 from bone marrow of rats orally administered FT alone (open bars), SRV alone (hatched bars) or FT and SRV simultaneously (closed bars) once daily for 6 days. The daily doses of FT and SRV were 60 and 30 mg/kg, respectively. Data are expressed as percent of CFU-GM colonies from control rats. Number of colonies from control rats on days 1, 2, 4, and 6 were 114.3, 118.6, 126.4 and 82.7/105 mononuclear cells/dish, respectively. Significantly different from mean values in FT-treated rats, **P < .01. ND, Not detectable.
Inactivation of DPD by covalent binding of BVU in the presence of NADPH.
DPD, isolated from rat liver cytosol and purified to homogeneity, had an activity to catalyze the hydrogenation of [14C]5-FU to [14C]H2-5-FU at a rate of 816 nmol/mg protein/min in the presence of NADPH. Native PAGE of the purified enzyme protein showed a single band with an apparent molecular mass of 210 kDa, whereas the enzyme migrated as two bands with molecular masses of 105 and 97.7 kDa on SDS-PAGE.
The DPD activity was strongly inhibited by preincubation with BVU in the presence of NADPH for 5 min before incubation with the substrate [14C]5-FU for determining the enzyme activity (fig.6). Under the conditions used, IC50 of BVU for inactivation of DPD was 4 μM. Omitting the cofactor NADPH from the preincubation medium, BVU showed no inhibitory effect on DPD. However, in the presence of NADPH, SRV showed no inhibitory effect on the 5-FU-reducing activity of the DPD even at 50 μM under the same preincubation conditions (fig. 6).

Inactivation of purified rat liver DPD by preincubation with BVU. Purified rat liver DPD was preincubated with various concentrations of BVU in the presence (open circles) and absence (closed circles) of NADPH (200 μM) as described in “Materials and Methods.” The DPD was also preincubated with SRV in the presence of NADPH (open squares).
Radioactivity of [14C]BVU used as a substrate was detected in the DPD (280 dpm/μg protein) isolated from the preincubation mixture by HPLC on a gel filtration column, after 30-min incubation in the presence of NADPH under the same conditions as used for the above preincubation. No detectable amount of radioactivity was found in the DPD protein in the absence of NADPH.
The radioactive DPD obtained by the incubation with [14C]BVU was subjected to SDS-PAGE after being isolated by HPLC (fig. 7). The DPD protein isolated from the reaction mixture appeared at 105 and 97.7 kDa as major and minor dye-stained bands, respectively, on SDS-PAGE (fig. 7A). The radioactivity of the DPD was also visualized as two bands by radioluminography of the same dye-stained gel slab (fig. 7B). In the absence of NADPH, DPD was not radiolabeled with [14C]BVU.

SDS-PAGE and radioluminography of rat liver DPD incubated with [14C]BVU in the presence and absence of NADPH. Purified rat liver DPD was incubated with 4 μM [14C]BVU for 30 min in the presence (lane +) and absence (lane −) of NADPH as described in “Materials and Methods.” After incubation, the hepatic DPD was isolated from the mixture by HPLC on a gel filtration column as described in “Materials and Methods.” A and B represent SDS-PAGE and radioluminography, respectively, of the radiolabeled DPD. Lane M represents molecular mass markers. Radioactivity incorporated into the DPD was visualized by radioluminography of the SDS-PAGE gel plate stained with Coomassie Brilliant Blue.
Discussion
Our study using rats strongly suggests that the 18 patient deaths caused by the interactions of the 5-FU prodrugs with the new antiviral drug SRV were due to extremely high concentrations of 5-FU in various tissues, especially in bone marrow and intestines, as a result of the facile inactivation of hepatic DPD by the covalent binding of an active metabolite formed in liver from BVU which is generated from SRV (fig.8).

Proposed mechanism for lethal toxicity exerted by oral coadministration of SRV and FT to rats and humans. BVU, (E)-5-(2-bromovinyl)uracil, formed from SRV by gut flora is absorbed through the intestinal membrane, reduced in liver by DPD (dihydropyrimidine dehydrogenase) to H2-BVU (dihydro-BVU) as a reactive intermediate with potential allyl bromide type of structure, and instantly inactivates hepatic DPD through covalent binding. The markedly decreased level of DPD markedly increases tissue 5-FU levels, leading to death in rats and humans.
Female Wistar rats were required to be used in our study to reinvestigate an incomplete study reported by previous workers who were engaged in the SRV development. After the 18 patient deaths, they (Yoshifune et al., 1994) reported, without showing any data on the 5-FU levels in plasma and tissues, that neither diarrhea with bloody flux nor deaths occurred in female Wistar rats by repeated oral coadministration of FT and SRV under the same dosage conditions, including the same volume of the same vehicle for suspending both drugs, as used in our study. However, our study, including several preliminary experiments for determination of the dosage conditions, demonstrated that all the animals given both drugs died following the severe diarrhea in 10 days so far as treated under the same dosage conditions as reported by them. They, however, also demonstrated the marked decreases in platelet and white blood cell counts in the rats given both drugs to the almost same extent as demonstrated in our study.
In the rat, BVU was reported to be generated from SRV by gut flora but not to be formed in tissues and was demonstrated to showCmax at 8 hr in plasma after oral administration (Nishimotoet al., 1990). Similar evidence also has been provided for the microbial formation and plasma level of BVU from SRV in the human (Ogiwara et al., 1990). Bacteroides species, such as Bacteroides eggerthill and Bacteroides vulgatus, which abundantly exist in human intestines, were identified as the major bacteria which generate BVU from SRV (Nakayamaet al., 1997).
Recently, Yan et al. (1997) reported the strong suppression of DPD activity in humans administered SRV; i.e., patients having herpes zoster and administered SRV at a dosage of 40 mg/day for 10 consecutive days did not show any DPD activity in their peripheral blood mononuclear cells during the period of administration. During the period of SRV administration, BVU appeared in blood at a remarkably high level and was eliminated from the circulation within 7 days after the last dose of SRV.
DPD is most abundant in the liver and occurs at very low concentrations in other tissues, including the colon in the human (Ho et al., 1986) and bone marrow in the rat (Ikenaka et al., 1979). The primary structure of DPD has been elucidated by molecular cloning of cDNA encoding the porcine (Yokota et al., 1994), human (Yokota et al., 1994), bovine (Albin et al., 1996) and rat (Kimura et al., 1996) enzyme subunits, which share very strong identity, more than 89%, in amino acid sequence. The DPDs have been demonstrated to have a potential pyrimidine-binding domain including the sole cysteinyl residue, which is highly conserved in amino acid sequence across mammalian species (Yokota et al., 1994).
Previously, we reported, using purified rat liver DPD and radiolabeled BVU, that rapid inactivation of DPD by BVU occurred in the presence of NADPH with concomitant incorporation of the radioactivity of BVU into the enzyme protein (Okuda et al., 1997). The radioactivity was incorporated into the DPD protein in a manner reciprocal to the enzyme inactivation and was inseparable from the enzyme protein by HPLC on a gel filtration column (Okuda et al., 1997). These results indicated that the enzyme inactivation is caused by covalent binding to DPD of a reduced form of BVU.
DPD purified from rat liver in our study showed a single band on native PAGE but migrated as two sharp bands, an intact subunit as a major band at 105 kDa and a degradation product as a minor one at 97.7 kDa, on SDS-PAGE, as had already been shown with purified specimens of human (Lu et al., 1992) and porcine (Lu et al., 1993a) DPDs; the purified enzymes were also demonstrated to be degraded, in part, to peptides with a little smaller molecular mass under the denaturing conditions used for SDS-PAGE. Radioactivity incorporated into DPD protein after the incubation with [14C]BVU in the presence of NADPH was also detected in the two bands on SDS-PAGE, after isolation on a gel filtration column. In our study, the radioactivity was found to be inseparable from the enzyme protein even by SDS-PAGE, after gel chromatography.
The reactive metabolite formed from BVU by DPD was suggested to act as a suicide inhibitor on the enzyme, because various thiol compounds (20 mM), such as cysteine, glutathione and dithiothreitol, added to the incubation mixture had no retarding effect on enzyme inactivation and on the radiolabeling of DPD by [14C]BVU (data not shown). It is reasonable to suppose that the reactive metabolite, dihydro-BVU (H2-BVU), would be 5-(2-bromoethyliden)uracil, a reactive allyl bromide type of electrophile, rather than the direct hydrogenation product, 5-(2-bromovinyl)-5,6-dihydrouracil, possibly with considerable stability (fig. 8). The reactive H2-BVU is formed at the potential pyrimidine-binding domain of DPD, and it might react instantly with the sulfhydryl group of the sole cysteinyl residue in the domain.
The above hypothesis on the suicide inhibition of DPD may be supported by the fact that 5-iodouracil (Porter et al., 1991) and 5-ethynyluracil (Porter et al., 1992) inactivate bovine liver DPD in the presence of NADPH by covalent binding of their reactive metabolites to the sulfhydryl group of the cysteinyl residue located in the pyrimidine-binding domain of the enzyme.
Very recently, we demonstrated, using recombinant human liver DPD expressed in Escherichia coli and purified to homogeneity, that BVU covalently binds to and inactivates the DPD in the presence of NADPH (Ogura et al., 1998). Radioactivity of [14C]BVU was incorporated in the presence of NADPH into the human DPD with concomitant loss of enzyme activity. In the absence of NADPH, the human DPD was not inactivated nor radiolabeled. SRV showed no inhibitory effect on DPD in the presence of NADPH as demonstrated with rat DPD in the present study.
5-FU is as good a substrate for DPD as are the endogenous pyrimidines, uracil and thymine (Lu et al., 1993a). Tissue 5-FU levels in patients administered anticancer chemotherapy with 5-FU or its prodrugs are strongly suggested to depend on hepatic DPD activity (Diasio and Harris, 1989). Actually, patients with very low genetically determined hepatic DPD activity have been reported to die during anticancer chemotherapy by 5-FU infusion (Lu et al., 1993b; Milano and Etienne, 1994). Therefore, it has been noted that 5-FU-based anticancer chemotherapy should not be administered to patients who are DPD-deficient or have very low DPD activity in liver or in peripheral blood mononuclear cells, for otherwise they will suffer from severe toxic symptoms or die from markedly elevated tissue 5-FU levels (Luet al., 1993b; Chazal et al., 1996).
The clinically observed typical toxic symptoms of 5-FU and its prodrugs, marked decreases in white blood cells and platelets, were reported for all of the patients who had received both SRV and one of the oral 5-FU prodrugs and who subsequently died (Pharmaceutical Affairs Bureau, 1994). The severe hematotoxicity mentioned above was also found in the rats administered FT and SRV, but not in the animals treated with the same dose of FT or SRV alone. On the basis of the present CFU-GM assay for bone marrow in the rats given both drugs, proliferation of bone marrow cells was suppressed at an early stage of the coadministration.
The average Cmax of hepatic BVU generated from SRV in rats was much higher than for the facile and almost complete inactivation of the purified rat liver DPD in the presence of NADPH. However, liver cytosol from the rats orally administered SRV and FT still had DPD activity at a level of about 9% that of the controls. Moreover, in the liver of rats orally given BVU for 6 days, the DPD activity remained at a level of 14% of the controls on day 6. There may be two reasons for the remaining DPD activity in the liver of the rats given FT and SRV or BVU alone. One is the markedly elevated levels of the pyrimidines, 5-FU and uracil as demonstrated in our study, which act as competitive inhibitors for the inactivation of DPD by BVU. In connection with this, the inhibition of the DPD activity by BVU in rat liver cytosol containing NADPH has been demonstrated to be significantly retarded in the presence of 5-FU or uracil (Desgranges et al., 1986;Tatsumi et al., 1987). Another reason is the circadian rhythm of hepatic DPD activity in the rat. DPD activity in rat liver was demonstrated to vary over a 24-hr period in association with a light-dark cycle (Harris et al., 1988, 1989). Although the physiological mechanism for the circadian rhythm is unclear, facile synthesis and decomposition of the DPD protein could occur.
Thus, our study provides the first evidence for lethal interaction of respective metabolites from two drugs. One of the two drugs gives an active metabolite with high toxicity, the elevated tissue levels of which readily result in death, and the other gives a metabolite that irreversibly inactivates the enzyme catabolizing the active metabolite. In other words, in case of the interaction of SRV with one of 5-FU prodrugs, the patients who died are strongly suggested to become extremely poor metabolizers for the active metabolite, 5-FU, with high toxicity as a result of irreversible inactivation of the key enzyme, DPD, by BVU from SRV. Except for the metabolic pathway depending on DPD, there is no major alternative pathway for the 5-FU catabolism in the human as well as in the rat. Studies on the synthesis of the reactive metabolite from BVU and on the mode of its covalent binding to DPD are now in progress in our laboratory.
Acknowledgments
The authors appreciate Sekio Nagayama, Kazumasa Ikeda, Shuji Yamaguchi, Takahito Nishiyama, Yoshimasa Nakamura and Yasuro Kawaguchi of Tokushima Research Center, Taiho Pharmaceutical Co., Ltd. for their great contributions to our toxicokinetic experiments.
Footnotes
-
Send reprint requests to: Dr. Tadashi Watabe, Department of Drug Metabolism and Molecular Toxicology, School of Pharmacy, Tokyo University of Pharmacy and Life Science, 1432-1 Horinouchi, Hachioji-shi, Tokyo 192-03, Japan.
- Abbreviations:
- AUC0–24
- area under the curve 0 to 24 hr after administration
- BVU
- (E)-5-(2-bromovinyl)uracil
- CFU-GM
- colony-forming unit granulocyte-macrophage
- Cmax
- maximum concentration
- DPD
- dihydropyrimidine dehydrogenase
- FT
- tegafur [1-(2-tetrahydrofuryl)-5-fluorouracil]
- 5-FU
- 5-fluorouracil
- H2-BVU
- dihydro-BVU
- H2-5-FU
- 5,6-dihydro-5-FU
- HPLC
- high-pressure liquid chromatography
- IC50
- 50% inhibitory concentration
- NADPH
- nicotinamide adenine dinucleotide phosphate, reduced form
- SDS-PAGE
- sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- SRV
- sorivudine [1-β-d-arabinofuranosyl-(E)-5-(2-bromovinyl)uracil]
- TLC
- thin-layer chromatography
- Tmax
- time to maximum concentration
- Received November 14, 1997.
- Accepted June 3, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}