


Abstract
Interaction with the exsorptive transporter P-glycoprotein (P-gp) is a possible source of peculiarities in drug pharmacokinetics, including dose-dependent absorption, drug–drug interactions, intestinal secretion, and limited permeability of the blood-brain barrier. Among the established in vitro methods of the analysis of drug interactions with P-gp, none directly quantifies the affinity of ligands with P-gp. Instead, they measure the result of a membrane permeation and a receptor-binding process; this may lead to difficulties in the interpretation of results. An assay for quantification of drug affinity to the transporter is presented on the basis of the radioligand-binding assay principle. This has the advantage of directly quantifying the interaction between drugs and P-gp. Because of the reversible and competitive interaction of numerous substrates with P-gp, a radioligand-binding assay was developed by taking [3H]verapamil and [3H]vinblastine as radioligands and the human intestinal Caco-2 cells, overexpressed with P-gp by culturing in the presence of vinblastine or transfecting with multidrug resistance gene MDR-1 as receptor preparation. The assay was performed in 96-well plates and has the potential to be used as a high-throughput method. A clear induction of the expression of P-gp was demonstrated in the Caco-2 cells grown in the presence of vinblastine, as well as in the transfected cells, although to a lesser extent. Both radioligands were shown to bind to P-gp. Verapamil was the radioligand of choice for further investigations due to its lower nonspecific binding to the transporter preparation. Kinetics as well as specificity of the binding of verapamil to the P-gp preparation were demonstrated. A two-affinity model was found to adequately describe the data derived from saturation as well as from competition experiments, in accordance with previous findings on two exsorption sites for P-gp. The binding properties of [3H]verapamil and [3H]vinblastine to a P-gp preparation derived from induced Caco-2 cells are described. The concentration-dependent displacement of the radioligand by nonlabeled substrates for P-gp should be a suitable principle for the determination of drug affinity to the respective binding sites at the human intestinal multidrug transporter P-gp.
More than 20 years ago, the modulating effect of P-glycoprotein (P-gp) on drug permeability across cellular membranes was discovered (Juliano and Ling, 1976) and its role in the development of multidrug resistance in cancer chemotherapy identified. In spite of the known amino acid and gene sequence of P-gp (Chen et al., 1986), its functional properties are not yet fully understood, neither with respect to the ATP-driven mechanism of transport across the plasma membrane (Hsing et al., 1992) nor to the fact that a vast number of structurally unrelated compounds seem to be recognized by the transporter.
In addition to its expression in cancer cells (Chen et al., 1986), P-gp has been shown to be present under physiological conditions in different tissues, where it is supposed to contribute to the “compartmentation” in the body, e.g., at the gut wall or the blood-brain barrier (Schinkel et al., 1994; van Asperen et al., 1997). Two major fields of research have originated from these pathophysiological and physiological findings. The first field focuses on strategies of overcoming multidrug resistance in cancer treatment (Ayesh et al., 1996) by the use of drugs modulating the function of P-gp. The other more recent field is dedicated to investigating the influence of P-gp expression on the absorption in the gastrointestinal tract, distribution, and excretion of drugs (Schinkel et al., 1996; Hunter and Hirst, 1997; Spahn-Langguth et al., 1998).
Because of the growing interest in the determination of drug affinity to P-gp, various assay systems have been developed based on the influence of potential substrates on the ATPase activity of P-gp (Al-Shawi and Senior, 1993), the accumulation or efflux of fluorescent dyes transported by P-gp (Broxterman et al., 1997), or the modulation of toxicity of cytotoxic P-gp substrates (Scala et al., 1997). None of these assays allow interpretation of the results with respect to drug affinity to P-gp, although direct binding of substrates to P-gp has been shown with the use of photoaffinity labeling analogs of vinblastine and colchicine. This labeling was competitively inhibited by P-gp substrates (Beck and Qian, 1992; Bruggemann et al., 1992). Consequently, the observed binding characteristics seem to correspond well to the theory of competitive receptor–ligand interaction and may therefore be used for the development of a radioligand-binding assay (RBA) (Crevat-Pisano, 1986; Spahn et al., 1989).
In this investigation, the human colon carcinoma cell line Caco-2, which has previously been characterized with respect to its differentiation and expression of P-gp (Pinto et al., 1983; Anderle et al., 1998), was used. To increase its P-gp content, this cell line was adapted to grow in the presence of vinblastine. The vinblastine-induced cell line was characterized functionally by transport studies with polarized monolayers, saturation experiments, and fluorescence-activated cell sorting (FACS) analysis. Furthermore, Caco-2 cells transfected with MDR-1 were characterized with respect to the amplification of the expression of P-gp by transport and saturation experiments, as well as by Northern blotting. Important variables for the binding experiments such as cell permeabilization procedures have been optimized. Furthermore, binding characteristics of radioligands, as well as association and dissociation kinetics, were determined. Specificity of the radioligand binding was tested by competition experiments with ligands for other potential binding sites; moreover, the presence of cytochrome P-450 3A was investigated by using Western blotting.
Materials and Methods
Cell Culture Materials
Dulbecco’s modified Eagle’s medium, fetal calf serum,l-glutamine (200 mM), penicillin/streptomycin (10,000 U/ml, 10,000 μg/ml), trypsin/EDTA, minimum essential medium, nonessential amino acids, Hanks’ balanced salt solution (HBSS), and phosphate-buffered saline (PBS) were obtained from Life Technologies (Paisley, UK). Vinblastine sulfate and trypan blue solution (0.4%) were purchased from Sigma (Malmö, Sweden). Transwell cell culture inserts used for transport experiments (24 mm, 0.4-μm pore size, polycarbonate membrane) and all other cell culture materials were obtained from Costar (Cambridge, MA). The Caco-2 cells used were obtained from the American Type Culture Collection (Rockville, MD).
Compounds
[3H]Verapamil (25 μCi, 84 Ci/mmol) was purchased from New England Nuclear (Boston, MA); [3H]vinblastine (50 μCi, 16 Ci/mmol) was purchased from Amersham (Buckinghamshire, UK). Rhodamine 123 (R 123), calcein, and 2′,7′-bis(2-carboxyethyl)-5(6)-carboxyfluorescein (BCECF) were purchased from Molecular Probes (Leiden, the Netherlands). Metoprolol was obtained from Astra Hässle (Mölndal, Sweden). rac-Talinolol and R- and S-talinolol were gifts from Arzneimittelwerk Dresden (Radebeul, Germany). 2′-Deoxytubercidin (d-TUB) was obtained from TIB MolBiol (Berlin, Germany) and testosterone was obtained from Steraloids (Wilton, IA). 2-(N-Morpholino)ethanesulfonic acid (MES) was obtained from Fluka (Gothenburg, Sweden). Molecular biology single use plasticware was purchased from Sarstedt (Nürnbrecht, Germany). Enzymes for molecular biology purposes were obtained from Boehringer Mannheim (Mannheim, Germany). Hybond-N membranes and 32P-labeled cytidine triphosphate were purchased from Amersham. Scintillation fluid Optiphase “Highsafe” 3 was purchased from Wallac (Loughborough, UK). Monoclonal antibody MRK-16 was obtained from Kamiya Biomedical (Thousand Oaks, CA). Goat anti-mouse Ig-fluorescein isothiocyanate (catalog no. 349031) and mouse IgG2a Pure (catalog no. 349050) were purchased from Becton Dickinson (Glostrup, Denmark). Solvents used were of analytical grade and purchased from Skandinaviska Gentech (Kungsbacka, Sweden). All other compounds and reagents used were obtained from Sigma or BDH (Poole, UK).
Equipment
The MultiScreen 96-well plate assay system was obtained from Millipore (Eschborn, Germany). The MultiScreen 96-well plates with Durapore membrane of 0.22-μm pore size and the MultiScreen 96-well plate punch tips were purchased from Millipore (Malmö, Sweden). Liquid scintillation was determined by a WinSpectral 1414 counter from Wallac (Turku, Finland). For flow cytometric analysis a FACSCalibur from Becton Dickinson was used. The high-performance liquid chromatography (HPLC) system consisted of a Pharmacia LKB 2150 pump (Uppsala, Sweden), a Perkin/Elmer ISS-100 autoinjector (Allerød, Denmark), a Linear 206 PHD UV-absorbance detector from Linear (Reno, NV) or a Shimadzu RF 350 fluorescence monitor (Kyoto, Japan). An epithelial volt ohmmeter equipped with “chopstick” electrodes obtained from World Precision Instruments (Sarasota, FL) was used for monitoring transepithelial resistance of monolayers growing in Transwell inserts.
Optimization of the P-gp Containing Cell Preparation
Cell Culturing Conditions and Vinblastine-Induction of P-gp Expression in Caco-2 Cells.
For the induction of P-gp expression, Caco-2 cells were cultured as described previously (Anderle et al., 1998). Induction of P-gp was started at passage 74. Caco-2 cells were routinely subcultured once a week with trypsin-EDTA (0.25%, 0.3 mM) and seeded at a density of 4 × 105 cells per flask (75 cm2). Cells were grown at 37°C in a 5% CO2 atmosphere using Dulbecco’s modified Eagle’s medium with (induced cells) or without (reference and transfected cells) 10 nM vinblastine, 16.5% fetal calf serum, 1% nonessential amino acids, 100 U/ml penicillin, 100 μg/ml streptomycin, and 1% l-glutamine.
Transfection of Caco-2 Cells with MDR-1 for Elevation of P-gp Expression.
Plasmid construction and transfection. The human mdr1-cDNA (Linck et al., 1990) was obtained from the American Type Culture Collection and ligated into the mammalian expression vectorpMAMneo. The obtained construct was amplified inEscherichia coli, and plasmids were identified with restriction analysis and verified by sequencing on a 373 DNA Sequencer from Applied Biosystems, Perkin/Elmer (Foster City, CA) after a modified Sanger method.
For transfection, Caco-2 cells were grown to 80% confluence, harvested by regular trypsinization, and electroporated with a GenePulser apparatus (Bio-Rad, Hercules, CA) with a 250-V pulse at 960 μF. Cells were seeded in 100-mm dishes and after 24 h, G418 (GIBCO/BRL, Täby, Sweden) was added at a concentration of 950 μg/ml as selective agent. Medium was changed every other day and supplemented with fresh G418. Single colonies were picked from the dishes after 2 to 3 weeks and transferred to 24-well plates. Sublines were grown to higher cell numbers and characterized by Northern blotting and functionally by transport experiments with talinolol.
Northern blotting.
Total RNA was isolated from Caco-2 cells and the transfectants by the guanidinium-thiocyanate method (Trisolv, Biotecx Laboratories Inc., Houston, TX). The mdr1-RNA was detected with a 32P-labeled riboprobe; probe synthesis was carried out with the pGEM3Zf(−)Xba-MDR1 vector as a template after Dra restriction by using the Promega Riboprobe T7 Kit (SDS, Falkenberg, Sweden). Subclones obtained by transfection were cultivated in the same manner as noninduced Caco-2 reference cells.
Permeabilization of Cells for Radioligand-Binding Assay.
Induced cells were changed to vinblastine-free medium 12 to 16 h before binding studies. For permeabilization of membranes, freshly trypsinized Caco-2 cells were suspended in lysolecithin buffer (0.01% [w/v] in HBSS with 10 mM MES, pH 7.0), Triton X-100 buffer (0.1% [w/v] in HBSS with 10 mM MES, pH 7.0), or methanol buffer (70% [v/v] in HBSS with 10 mM MES, pH 7.0). For methanol and Triton X-100 a subsequent washing step with buffered HBSS was necessary to remove excess porating agent according to standard immunological cell poration protocols. Effectiveness of poration was checked by trypan blue exclusion tests and suitability of poration was evaluated considering total binding capacity for the radioligand (+/− inhibitors).
Characterization of the P-gp Expression of the Caco-2 Cell Lines.
Three different approaches were chosen for the characterization of the vinblastine-induced cells: estimation of maximal binding capacity of the radioligand [3H]verapamil, quantification of the increase in P-gp protein levels by FACS with the specific monoclonal antibody MRK-16, and performance of transport experiments where secretory transport of model substrates was determined across Caco-2 monolayers. For this functional characterization the P-gp substrates vinblastine, talinolol, and quinidine were chosen. MDR-1-transfected cells were characterized by maximal binding capacity for [3H]verapamil, transport experiments with talinolol, and Northern blotting.
Experimental Conditions for the Radioligand-Binding Assay.
The membranes of the MultiScreen 96-well plates were washed before incubation with 100 μl of HBSS buffered with 10 mM MES at pH 7.0 (no additives). Incubations with radioligand and competitors (for displacement experiments) were performed at 37°C under mild shaking (Incubator S.I. 60; Stuart Scientific, UK) in HBSS buffered with 10 mM MES at pH 7.0 with 50 μl of the porated Caco-2 P-gp preparation (1.25 × 106 cells per ml) in a total volume of 250 μl. Furthermore, 1 mM ATP was present, as well as an enzymatic system for the regeneration of ATP from ADP (ATP-system), containing magnesium chloride (10 mM), creatine phosphate (10 mM), and creatine kinase (100 μg/ml) (Horio et al., 1988). After 30 min the incubation was stopped by removing the liquid with gentle vacuum filtration and by washing the filters twice with 100 μl of ice-cold HBSS buffered with 10 mM MES, pH 7.0 (no additives). The filter membranes were incubated with 5 ml of scintillation fluid for 12 to 16 h at room temperature. Total radioactivity was determined by liquid scintillation counting.
Saturation Experiments and Determination of Nonspecific Binding for Induced, MDR-Transfected, and Noninduced Caco-2 Cells.
The saturable binding of the radioligand [3H]verapamil to P-gp was characterized by analyzing samples of 12 different concentrations in the range from 0 to 15 μM (n = 2 each concentration). To determine total binding, solutions of radioligand obtained by spiking solutions of nonlabeled verapamil with nanomolar concentrations of radiolabeled drug were incubated with the P-gp preparation in the presence of the ATP system. Nonspecific binding was determined with the same experimental design, but in the presence of 1 mM R123 for saturation of specific binding (see competition experiment with R123 in Fig. 7).
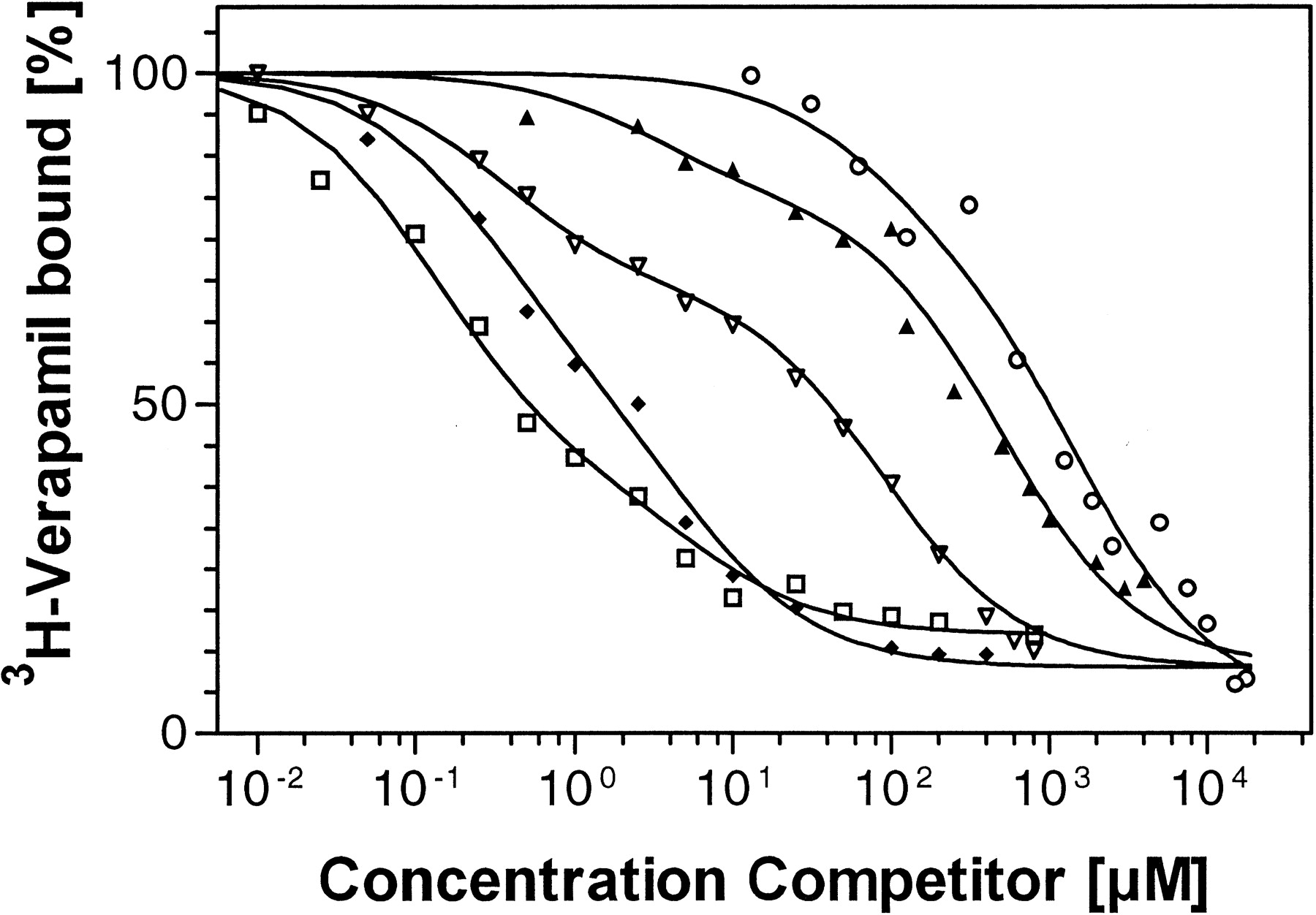
Results from competition experiments with [3H]verapamil as the radioligand. Radioligand bound is measured as percentage of radioligand bound in the absence of competitor for R123 (■), verapamil (♦), vinblastine (▿), quinidine (▴), or talinolol (○) and the respective fitted curves (—).
FACS.
Caco-2 cells were grown for 5 days to 90 to 95% confluence and trypsinized. The cells were washed once with PBS and then resuspended in PBS containing 5 mM d-glucose and 10% bovine serum albumin (PBS-BSA-G) to a concentration of 2 × 106 cells per 100 μl. The cells were incubated with 5 μl of MRK-16 anti-human P-gp monoclonal antibody for 30 min on ice. Cells incubated with 2.5 μl of IgG-2a isotype-control were prepared as control. After washing with PBS-BSA-G, cells were resuspended in 100 μl of PBS-BSA-G and mixed with 2 μl of goat anti-mouse Ig-fluorescein isothiocyanate and incubated on ice for another 30 min. After subsequent washing, labeled cells were resuspended in 500 μl of PBS-BSA-G and measured immediately (excitation wavelength 488 nm, emission 520 nm). The median fluorescence intensity of the cell populations was determined. For data evaluation, the fluorescence index was calculated by dividing the median fluorescence intensity of cells stained with MRK-16 by median fluorescence obtained using the isotype-matched control antibody (Den Boer et al., 1997).
Transport Experiments with Polarized Cell Monolayers.
Cell culture and study design. Ninety to 95% confluent monolayers of vinblastine-induced, transfected, or noninduced Caco-2 cells were trypsinized and seeded into Transwell cell culture inserts (300,000 cells/well) and cultured for 14 to 17 days. Formation of tight monolayers was monitored by measuring transepithelial electrical resistance. Transport experiments were performed with vinblastine concentrations of 0.1, 1, 10, and 100 μM, a rac-talinolol concentration of 500 μM, and quinidine concentrations of 0.25, 2.5, 25, and 250 μM. To start the experiment, the compound being tested was added to the apical (a) or basolateral (b) compartment, and its appearance was monitored in the opposite compartment. Samples of vinblastine were taken every 30 min for 2.5 h from the acceptor compartments, those of talinolol were taken every 15 min for 1 h, and those of quinidine were taken every 10 min for 1 h. Sampling intervals and total experimental periods for the compounds were chosen depending on individual permeabilities and limit of quantification of the respective assay. To quantitate the fluxes across the monolayer, [3H]vinblastine was analyzed by liquid scintillation counting; talinolol and quinidine were analyzed using HPLC methods.
HPLC Methods.
Precision and accuracy of the HPLC methods used were determined for three different concentrations in the high, medium, and low concentration range of the respective calibration range, as well as for the lower limit of quantification by repeated analysis of spiked samples. Precision was evaluated by relative standard deviation and accuracy by the mean deviation of individual results from nominal value. Calibration curves were prepared daily; quality control samples were analyzed in every run. Acceptance criteria were applied according to international recommendations (Shah et al., 1992).
Talinolol was determined as described previously (Wetterich et al., 1996), by an enantioselective method with a chiral stationary phase (LiChrospher 100 ChiraSpher NT, 250 × 4 mm i.d.; Merck, Darmstadt, Germany) with ethanol/triethylamine (1000:0.5, [v/v]) and UV absorbance detection at 245 nm after liquid-liquid extraction into dichloromethane/isopropanol (95:5 [v/v]) and reconstitution with methanol. Pindolol (Sandoz, Basel, Switzerland) was used as the internal standard.
A reversed phase HPLC method (Carignan, 1995) was used for the determination of quinidine in samples obtained from transport experiments. Separation was achieved by a LiChrospher RP-18 HPLC analytical column, 150 × 4.6 mm i.d., using methanol/acetonitrile/0.01 M sulfuric acid (350:100:450 [v/v/v]), containing 10 mM 1-octanesulfonic acid as the mobile phase and fluorescence detection at an excitation wavelength of 350 nm and an emission wavelength of 450 nm.
Talinolol was determined with a lower limit of quantification of 2.5 ng/ml per enantiomer (precision for R- andS-talinolol, 11.9 and 6.7%; accuracy, 15.4 and 8.4%;n = 5). Precision of the method was determined to be 11.3 and 12.7% (20 ng/ml), 3.7 and 3.5% (125 ng/ml), and 3.8 and 3.8% (500 ng/ml). Accuracy for R- andS-talinolol was 1.2 and 10.8% (20 ng/ml), 5.0 and 6.5% (125 ng/ml), and 10.9 and 7.3% (500 ng/ml) (n = 6 for each concentration). The analytical method for the determination of quinidine had a lower limit of quantification of 1 ng/ml (precision, 9.0%; accuracy, −4.9%; n = 6). The precision and accuracy of the assay for quinidine were determined to be 1.9 and 6.8% (10 ng/ml), 7.0 and −0.1% (100 ng/ml), and 5.6 and 0.1% (1000 ng/ml).
Kinetic Experiments: Optimization of Incubation Time for [3H]verapamil as Radioligand
For [3H]verapamil the association and dissociation kinetics of the binding to the P-gp preparation were determined from 0.5 to 40 min (n = 4 for each time point). In studying association, all ingredients for incubation except the permeabilized cells were incubated and P-gp preparation was added at the appropriate time before terminating incubation by vacuum filtration (0.5, 5, 10, 15, 20, 25, 30, 35, and 40 min). Verapamil was used at a concentration of 10 μM (containing 2.4 nM [3H]verapamil); nonspecific binding was determined in the presence of 1 mM R123. Specific binding was obtained by subtracting nonspecific binding from total binding. For the determination of dissociation kinetics, verapamil (concentration as for association kinetics) was preincubated with P-gp preparation for 60 min to complete association; afterwards, dissociation was started by adding 500 μM nonlabeled verapamil at 0.5, 5, 10, 15, 20, 25, 30, 35, and 40 min before filtration.
Selection of an Appropriate Radioligand: [3H]Verapamil versus [3H]Vinblastine
Saturation experiments with the vinblastine-induced Caco-2 cells were performed for both radioligands, [3H]verapamil and [3H]vinblastine, in the concentration range from 0 to 15 μM. These experiments included the determination of nonspecific binding for both radioligands, according to the protocol described above in Characterization of P-gp Expression of the Caco-2 Cell Lines.
Specificity of Radioligand Binding to P-gp
Other Transporters.
To exclude the presence of relevant amounts of binding sites for verapamil other than P-gp but with overlapping substrate recognition, competition experiments with calcein (1 mM), d-TUB (5 mM), and BCECF (0.5 mM) were performed. Concentrations were chosen on the basis of available data for competitive displacement from the respective binding sites (Nelson et al., 1995; Draper et al., 1997). Where necessary, dimethyl sulfoxide (Sigma) or ethanol (Kemethyl, Haninge, Sweden) was added to solutions up to a concentration of 1% in the incubation medium. Control experiments for those samples were performed with identical concentrations of dimethyl sulfoxide or ethanol.
Detection of CYP3A in the Vinblastine-Induced Caco-2 Cells.
Because a coregulation of P-gp and CYP3A expression has been hypothesized (Wacher et al., 1995; Schuetz et al., 1996), a potential increase in CYP3A levels was evaluated via both displacement experiments with testosterone at a concentration of 2 mM (Waxman et al., 1991) and Western blotting. These two experiments were used because the selected radioligand 3H-verapamil may also bind to CYP3A in the cell (Kroemer et al., 1993), yielding higher saturable, but not P-gp-related, binding to the cell preparation. For the Western blots, solubilized human liver microsomal samples or Caco-2 cells were separated by SDS-polyacrylamide gel electrophoresis with a 7.5% separation gel (Laemmli, 1970). Proteins separated by SDS-polyacrylamide gel electrophoresis were transferred to a nitrocellulose membrane. The membrane was washed and unspecific sites were blocked using 5% nonfat milk in Tris-buffered saline. Specific sites were detected by incubating the membrane with CYP3A4 antibodies raised in goat (Dai-ichi Pure Chemicals, LTD, Tokyo, Japan) followed by anti-goat Ig-biotinylated antibody and streptavidin-horseradish peroxidase conjugate. Visualization of specific binding was performed using an immunodetection system (enhanced chemiluminescence) purchased from Amersham Life Science.
Displacement of Radioligand by P-gp Substrates: Competition Experiments
For the determination of the affinity of nonlabeled test compounds to P-gp, displacement of the radioligand at a fixed concentration of 100 nM verapamil (containing 2.4 nM concentration of labeled verapamil) or 160 nM vinblastine (containing 60 nM concentration of labeled vinblastine) by increasing concentrations (n = 16) of the respective compound was determined in duplicate. Concentrations of radioligands were chosen as 10 to 20% of the high affinity Kd, in compliance with recommendations for radioligand-binding assays to obtain maximum sensitivity and low ratio of nonspecific to specific radioligand binding (Enna, 1985).
Data Analysis
Data Derived by Radioligand Binding Assay.
One-, two- and three- affinity models were evaluated empirically and the improvements of the fit by the more complicated models were tested by calculating the F-test ratio. Based on these criteria, for saturation as well as for competition data, a two-affinity model was chosen to describe the data derived from the respective experiments.
Saturation Experiments.
Specific binding was calculated by subtracting nonspecific binding from total binding. A descriptive two-affinity Hill equation was fitted to these data for specific binding (Wells, 1992).
Competition Experiments.
The following descriptive two-affinity model was fit to the data obtained from competition experiments without any data transformation (Kenakin, 1993):
Transport Experiments Using Caco-2 Monolayers.
Net secretory flux rate was determined as the difference of the flux rate in basolateral to apical (b→a) and apical to basolateral (a→b) direction (Karlsson et al., 1993).
Results
Optimization of P-gp Containing Cell Preparation
Culturing of the Vinblastine-Induced and Transfected Cell Lines.
The vinblastine-induced and the MDR-1 transfected Caco-2 cell line showed normal behavior in growth and differentiation forming typical domes of epithelial cell monolayers, except for a slight reduction in proliferation rate for the cells growing in the presence of vinblastine. The formation of tight and polarized monolayers were monitored by microscopy and direction-specific transport of P-gp substrates. The transepithelial resistance of the monolayers formed from the vinblastine-induced cells was similar to that of the noninduced and nontransfected cells, whereas the resistance of the MDR-1 transfected cells was higher than that of the noninduced Caco-2 cells (vinblastine-induced [n = 18], 2555 ± 529; transfected [n = 6], 3886 ± 257; reference cells [n = 18], 2322 ± 420 Ω × cm2, mean ± S.D.).
Permeabilization of Cells.
All three methods for the poration of Caco-2 cells resulted in more than 99% poration efficacy as determined by a trypan blue exclusion test. Additionally, the method using lysolecithin resulted in the highest total binding capacity for the radioligand, whereas permeabilization using methanol or Triton X-100 resulted in a loss of 15 ± 6 and 18 ± 4% (n = 3 each) of binding capacity, respectively. This loss is most probably due to solubilization and/or protein denaturation and loss during the washing step that was needed when methanol or Triton X-100 was used. In addition, the lysolecithin method was highly reproducible; no washing steps were necessary to remove excess porating agent.
Characterization of P-gp Expression in Caco-2 Cells.
The maximum binding capacity as determined by saturation experiments with verapamil increased by a factor of 2.5 in the vinblastine-induced cells (Table 1). Similarly, for the vinblastine-induced Caco-2 cells, FACS analysis by direct detection of P-gp with monoclonal antibody MRK-16 (Fig.1) resulted in an overall 2.1-fold shift of the fluorescence index (9.0 for MRK-16-treated vinblastine-induced Caco-2 cells versus 4.2 for MRK-16-treated noninduced Caco-2 cells). In addition, the functional characterization of the induced cell line by transport experiments showed increases in secretory flux by factors of 1.7, 2.4, 1.6, and 1.7 for vinblastine, quinidine, andR- and S-talinolol (Table2). For the MDR-1 transfected Caco-2 cells, the increase in mRNA for P-gp was investigated by Northern blotting (Fig. 2). Total RNA from untransfected Caco-2 cells contained the mRNA coding (approximately 4500 bases long) for the P-gp, which was found to a greater extent in cell clones transfected with the vector bearing the MDR-1 gene.
Characterization of noninduced Caco-2, MDR-1-transfected Caco-2, as well as vinblastine-induced Caco-2 cells with respect to expression of P-gp
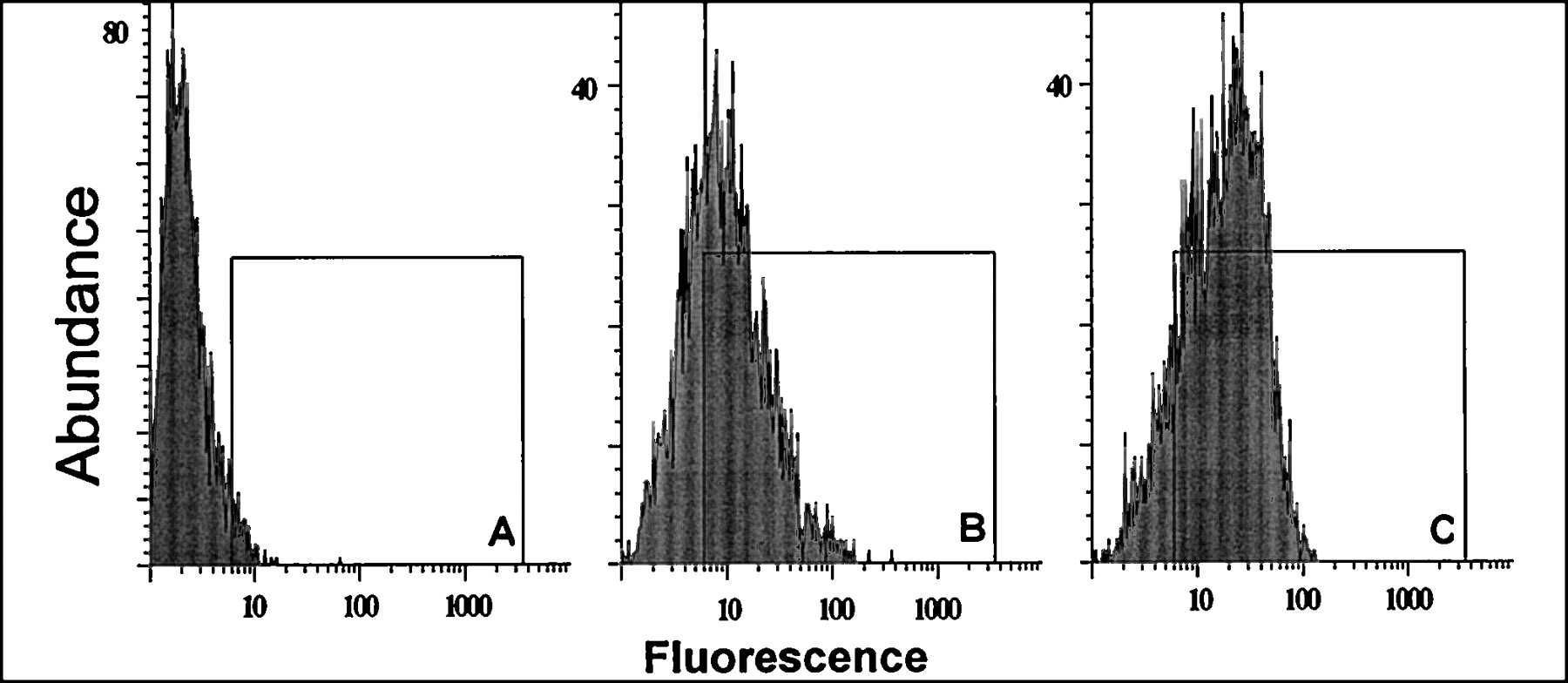
FACS analysis of P-gp expression using monoclonal antibody MRK-16 (B,C) or isotype-control antibody (A) for non-induced (B) and vinblastine-induced (A,C) Caco-2 cells (isotype-control for non-induced cells not shown). Induced cells show distinct right-shift in median of distribution.
Characterization of noninduced Caco-2 as well as vinblastine-induced and MDR-1-transfected Caco-2 cells with respect to expression of P-gp

Northern blotting of MDR-1 mRNA (approximately 4.5 kb) in total RNA isolated from MDR-1 transfected Caco-2 cells as well as from untreated Caco-2 reference cells, for demonstration of increase in P-gp mRNA in the transfected cell line.

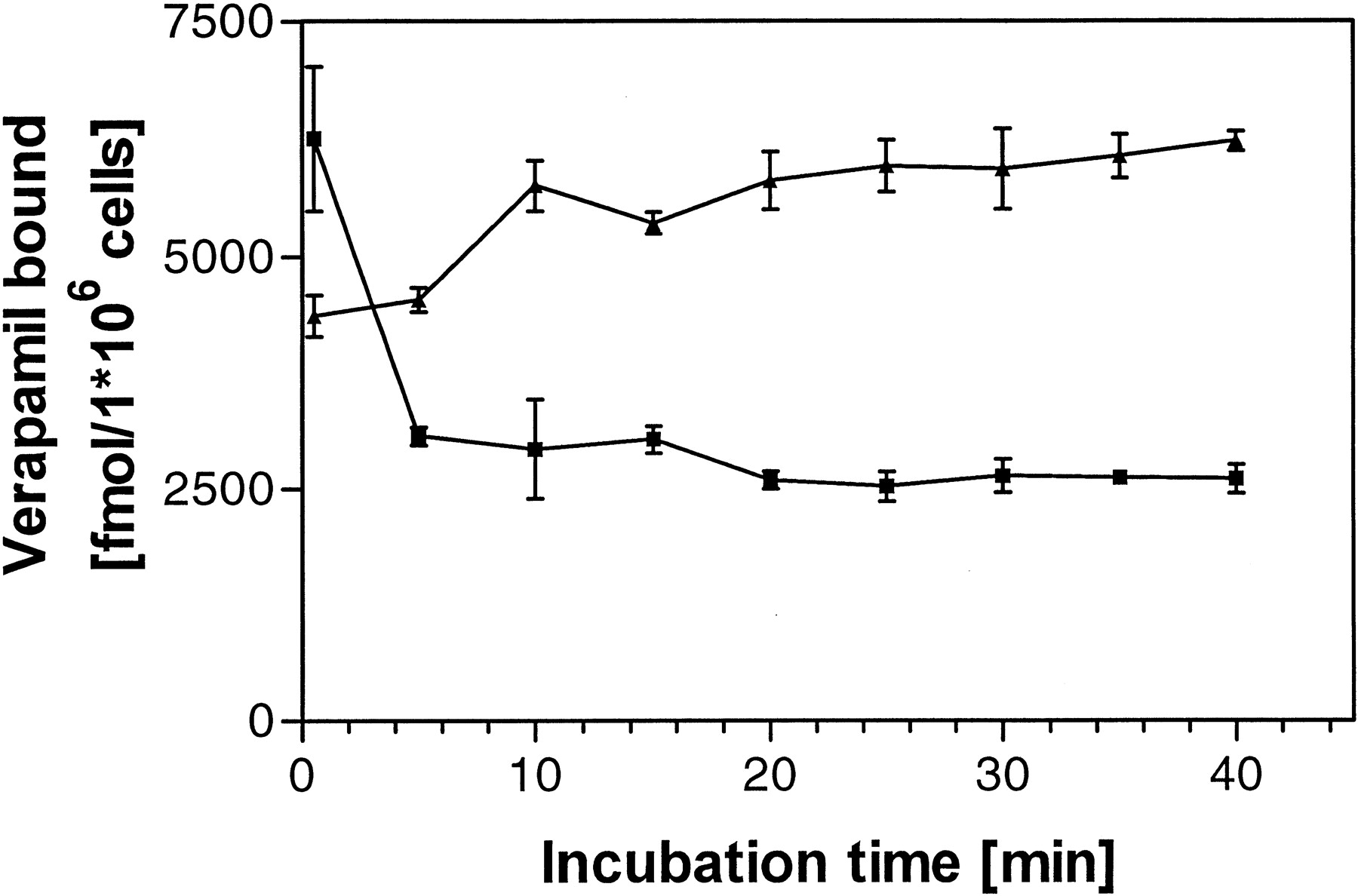
Association (▴) and dissociation (▪) kinetics of the radioligand verapamil to porated vinblastine-induced Caco-2 cells as preparation of P-gp (mean ± S.D., n = 4 each).
In summary, all methods applied (binding capacity of radioligand, FACS analysis by monoclonal antibody MRK-16, and quantification of polarized flux of P-gp substrates across cell monolayers) showed a pronouncedly higher expression of P-gp in vinblastine-induced Caco-2 cells than in the noninduced reference cells and even higher levels of P-gp than in the MDR-1-transfected Caco-2 cells. Consequently, vinblastine-induced cells were chosen as a source for P-gp preparation for further assay optimization.
Kinetics of Radioligand Binding
The kinetic experiments performed revealed a rapid onset of association as well as of dissociation of verapamil with the P-gp preparation; equilibria were reached within 10 to 15 min (Fig.3). Therefore, the incubation time of 30 min was chosen for the saturation and competition experiments.
Characteristics of Radioligand Binding
Both radioligands investigated, [3H]verapamil and [3H]vinblastine, were comparable with respect to maximum binding capacity to the P-gp preparation (Fig.4; Table3); however, under identical incubation conditions verapamil showed a considerably lower nonspecific binding [90 ± 12 versus 136 ± 6 (fmol/106cells)/μM]. Saturation experiments revealed a biphasic dependence of saturable binding of the radioligands from total ligand concentrations. The resulting data could be described empirically by a two-affinity Hill equation. At the radioligand concentrations chosen for competition experiments, the nonspecific binding of [3H]verapamil amounted to 6% of the total binding of the radioligand. For [3H]vinblastine 20% of total binding represented nonspecific binding.

Saturable binding of verapamil (A) and vinblastine (B) to P-gp derived from vinblastine-induced Caco-2 cells. Specific (▪, mean, n = 2 each concentration) and non-specific binding (┈) to P-gp, as well as fit obtained by two-affinity model (—).
Binding characteristics of [3H]verapamil and [3H]vinblastine to preparations of P-gp,Kd values for the two-affinity model, Hill coefficients of the respective affinity (n), maximum binding capacity of specific binding (Bmax), and slope of nonspecific binding (mean ± S.D., n = 4 for [3H]verapamil and individual results, n = 2 for [3H]vinblastine)
Specificity of Radioligand Binding
Competition experiments with specific ligands for other receptors likely to be expressed in Caco-2 cells revealed no significant displacement of the radioligand [3H]verapamil (Fig. 5). Calcein and BCECF were used for exclusion of the presence of multidrug resistance-related protein, 2′-deoxytubercidin for the organic cation transporter (OCT), and testosterone for CYP3A. Furthermore, Western blotting of the vinblastine-induced cells with antibody against human CYP3A revealed the absence of considerable amounts of this cytochrome in the cell line (Fig. 6). The compounds 5-fluorouracil, floxuridine (up to 8 mM each), and acetaminophen (up to 4 mM) as weak bases, baclofen (up to 4 mM) as an amino acid, and phenobarbital and indomethacin (up to 4 mM) as acidic compounds served as negative controls, showing no significant displacement of3H-verapamil in the RBA. Overall, these experiments demonstrated good specificity of the method. No displacement of the radioligand verapamil by progesterone was detected in the investigated concentration range.

Competition experiments for testing specificity of radioligand binding to the P-gp preparation of permeabilized, vinblastine-induced Caco-2 cells. Radioligand bound in presence of test compound as percentage of radioligand bound in absence for calcein,d-Tub, BCECF, progesterone, and testosterone, as well as R123 as prototype of a P-gp substrate.

Western blot analysis of Caco-2 cells and human liver microsomes using anti CYP3A4 polyclonal antibodies. Untreated solubilized Caco-2 cells were added to wells 1 to 3 (50,000, 100,000, and 200,000 cells) and vinblastine-treated solubilized Caco-2 cells were added to wells 5 to 7 (50,000, 100,000, or 200,000 cells). In well 4, four μg of solubilized human liver microsomal proteins was added. In lane 4, anti-CYP3A4 recognized a protein band at 52 kDa which is the molecular mass for CYP3A4. In lanes 1 to 3 and 5 to 7 no specific binding can be seen in the area around 50 kDa. However, several proteins were stained in the area between 72 and 120 kDa. The numbers in the figure refer to the molecular mass of standards (in Da).
Displacement of Radioligand by P-gp Substrates: Competition Experiments
Competition experiments for the radioligand [3H]verapamil with known P-gp ligands, e.g., verapamil, quinidine, and talinolol, showed distinct differences in their affinities to P-gp (Fig.7). A two-affinity model in accordance with two distinct binding sites accurately described the observed data for competition and saturation experiments. Calculated Ki values are depicted in Table 4, together with the fractions of high-affinity binding sites, reflecting the relative contribution of the high-affinity binding to total binding.
Results for competition experiments of selected drugs with [3H]verapamil as the radioligand
Competition experiments with the second radioligand [3H]vinblastine were performed for some compounds to investigate correlation of results obtained with the two radioligands. Results are depicted in Table5. A strong correlation was observed between the IC50-values of several P-gp substrates using vinblastine or verapamil as radioligand.
Competition experiments with [3H]verapamil and [3H]vinblastine as radioligand (mean ± S.D.,n = 3)
Discussion
There is a growing interest in the quantification of drug affinity to P-gp present in various tissues under physiological and pathophysiological conditions. Benefits of rapid affinity assay systems are to be expected not only in the search for multidrug resistance modifiers in cancer research but also for relationships between drug affinity to a particular binding site at the P-gp and certain pharmacokinetic characteristics of that compound, including intestinal membrane permeability, blood-brain barrier permeation, and intestinal secretion. The influence of such a transport process is currently a well accepted phenomenon in (cellular) pharmacokinetics not only for cytostatic drugs, but also for a number of very frequently used drugs (Karlsson et al., 1993, Fricker et al., 1996). From a drug development perspective, knowledge of the affinity of xenobiotics to P-gp is desirable at an early stage in drug development. In this article, the development of an RBA based on the competitive binding and displacement of ligands for P-gp (Tamai and Safa, 1990) is described.
Previously described assays usually provide surrogate parameters, such as cytotoxicity assays (Al-Shawi and Senior, 1993) or functional efflux studies using fluorescent P-gp substrates (Scala et al., 1997). These parameters are a mixture of cell membrane permeability and affinity (Al-Shawi and Senior, 1994).
The preparation of P-gp was obtained by gentle but effective perforation (Ymer and Jans, 1996) of cell membranes of P-gp-expressing cells, conserving the physiological lipid environment as much as possible to preserve binding properties. The poration was undertaken under the assumption that an intracellular binding site for P-gp achieves identical concentrations of radioligand and competitor at the binding sites. The question of intracellular or intramembranous binding sites of P-gp is still, however, under discussion. There is theoretical and practical support for both theories (Twentyman et al., 1994; Stein, 1997). One of the striking arguments for the intramembranous theory is the rapid onset of outward-directed transport, i.e., the delayed occurrence of intracellular concentrations of P-gp substrates in influx experiments compared to P-gp-deficient cells. But this phenomenon may also be explained by a biphasic binding characteristic of P-gp for substrates as found with our assay, resulting in an onset of pumping activity at low concentrations of substrate (high-affinity binding) and a shallow increase in pumping activity to reach its maximum at higher concentrations compared with a simple one binding site model. By this simple mechanism the “working range” of the transporter may be conveniently enhanced.
The rationale for selecting Caco-2 cells as source for P-gp was the inducibility of the acceptor protein content. This increased by approximately 150% due to the addition of vinblastine, allowing optimization of the ratio of specific to nonspecific binding. The induction by vinblastine was shown to be more efficient than using MDR-1 transfected Caco-2 cells. This was shown by binding capacity determinations for verapamil, functional experiments, and FACS analysis. The mechanism of induction of P-gp is not fully understood yet. The role of the inducer on the coregulation of a second detoxification system, CYP3A, needs to be clarified. Potentially the effect on this CYP 450 is dependent on the inducer (Schuetz et al., 1996) which would be a strong hint against the theory of a strict coinduction of both proteins, exhibiting a strong overlap in substrate recognition (Horio et al., 1988). For the vinblastine-induced cell line established here, the presence of significant amounts of CYP3A were excluded by competition experiments with the well known CYP3A substrate testosterone, as well as by immunoblotting of the protein.
The IC50 values determined by competition experiments with [3H]verapamil or [3H]vinblastine, respectively, showed a strong correlation for both radioligands. The correlation of the results obtained with these physicochemical and pharmacological distinct radioligands gives further evidence of the specificity of the assay presented here.
The binding properties of verapamil and vinblastine, two known ligands of P-gp, revealed similar maximum binding capacities to the preparation, but for verapamil the extent of nonspecific binding turned out to be approximately 35% lower than that of vinblastine. Thus, verapamil was chosen as radioligand for further optimization of assay parameters.
The specificity of the saturable binding of verapamil to the P-gp preparation was tested with respect to other binding sites for lipophilic, weak bases that have been described to be present in intestinal cells, the multidrug resistance-related protein and the OCT. The fluorescent dyes calcein and BCECF have been shown to be transported by the multidrug resistance-related protein, but not by P-gp (Twentyman et al., 1994; Draper et al., 1997), whereasd-Tub has been identified as a specific substrate for the distinction between the OCT and P-gp (Nelson et al., 1995). The expression of relevant amounts of these proteins were excluded by lack of significant displacement of verapamil with these specific ligands.
The concentration dependence of displacement of the radioligand by unlabeled compounds allowed rapid quantitative determination of the interaction with P-gp. In saturation as well as in competition experiments, curved shapes indicated a distinct deviation from a one-affinity model for the underlying process, evaluated by computing the F-test ratio for the models under consideration. The data obtained were adequately described by a two-affinity model, for saturation as well as for competition; a three-affinity model showed no significant improvement in the fits obtained (F-test ratio). The binding of vinblastine and vincristine to P-gp expressing tumor cells has been described previously by a two-affinity model (Tamai and Safa, 1990) using accumulation studies with [3H]vinblastine and [3H]vincristine. Two affinities were found for vincristine with parameters for half-maximum saturation of 0.14 ± 0.05 and 25 ± 7 μM, which is roughly comparable to the Kd values found in competition experiments using the method described in this communication (0.43 ± 0.05 and 148 ± 20 μM). Furthermore, the competition data for the compound SR33557 presented by Martin et al. (1997) were fit more adequately by a two- than by a one-affinity model, evaluated by the F-test ratio (data not shown).
The number of binding sites at P-gp is still under debate (Bruggemann et al., 1992). The formation of P-gp by two highly homologous six-transmembranous domains. Results of photoaffinity labeling experiments (Stein, 1997) indicate that intact functional P-gp forms at least two substrate recognition areas with different affinities for some substrates. This model is further supported by studies employing fluorescence or photolabeling assay systems using different P-gp preparations and overexpressing cell lines (Dey et al., 1997; Shapiro and Ling, 1997). There are also indications that, in addition to the known binding sites for verapamil and vinblastine, at least one more binding site exists for compounds that are bound to but not transported by P-gp, e.g., the allosteric modulator progesterone (Garrigos et al., 1997; Seelig, 1998). The allosteric nature of the interaction of progesterone with P-gp in the vinblastine-induced Caco-2 cells used in this study is consistent with the lack of displacement of the radioligand verapamil detected by competition experiments with progesterone (tested in concentrations up to 200 μM; data not shown). Further work including transport experiments is necessary to investigate the role of progesterone and other allosteric modulators of P-gp. However, irrespective of the number and type of binding sites at P-gp, it is evident that all drugs that interact with the verapamil binding site on P-gp will also be transported by the multidrug transporter, whereas drugs that interact with the progesterone binding site may reverse multidrug resistance but may not be transported by P-gp. This has important consequences for the application of a screening assay. Although in screening programs for multidrug resistance modifiers, the “loss of hits” due to the use of verapamil as radioligand in an RBA would be obvious and undesirable, this fact should not be problematic when screening for compounds that are transported by P-gp. Because the aim of most recent pharmacokinetic screening programs is devoted to the latter, radioligands other than verapamil may not be necessary for that purpose. If, however, the binding of different substrates to P-gp should be fully characterized with respect to their binding properties and sites, binding studies with different radioligands appear to be necessary. In the future, data on the relevance of the binding data for the passage across biological membranes will be provided.
In summary, by using the affinity data to the human intestinal P-gp obtained here, it should be feasible to 1) rapidly screen new compounds for their secretion potential by interaction with human P-gp, 2) evaluate quantitatively the impact of P-gp-mediated exsorption on the overall membrane permeability of drugs, and 3) use the quantitative binding characteristics of several P-gp ligands for extended structure-binding studies by means of molecular modeling.
Acknowledgments
We thank Sven Nylander for valuable support for the FACS analysis and Sibylle Neuhoff for her help with the Western blots.
Footnotes
-
Send reprint requests to: Prof. Dr. Peter Langguth, School of Pharmacy, Biopharmaceutics and Pharm. Tech., Staudingerweg 5, D-55099 Mainz, Germany.
-
1 This study was supported by the Fonds der Chemischen Industrie, Frankfurt and Dr. Robert Pfleger Stiftung, Bamberg (to H.S.L.).
- Abbreviations:
- P-gp
- P-glycoprotein
- RBA
- radioligand-binding assay
- FACS
- fluorescence-activated cell sorting
- R123
- rhodamine 123
- BCECF
- 2′,7′-bis(2-carboxyethyl)-5(6)-carboxyfluorescein:d-TUB, 2′-deoxytubercidine
- MES
- 2-(N-morpholino)ethanesulfonic acid: PBS, phosphate-buffered saline
- HBSS
- Hanks’ balanced salt solution
- HPLC
- high-performance liquid chromatography
- PBS-BSA-G
- bovine serum albumin
- OCT
- organic cation transporter
- Received February 20, 1998.
- Accepted July 20, 1998.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}