


Abstract
2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) is a heterocyclic amine identified in the human diet and in cigarette smoke that produces prostate tumors in the rat. PhIP is bioactivated by cytochrome P-450 enzymes to N-hydroxylated metabolites that undergo further activation by conjugation enzymes, including theN-acetyltransferases, NAT1 and NAT2. To investigate the role of prostate-specific expression of human N-acetyltransferase 2 (NAT2) on PhIP-induced prostate cancer, we constructed a transgenic mouse model that targeted expression of human NAT2 to the prostate. Following construction, prostate, liver, lung, colon, small intestine, urinary bladder, and kidney cytosols were tested for human NAT1- and NAT2-specific N-acetyltransferase activities. Human NAT2-specific N-acetyltransferase activities were 15-fold higher in prostate of transgenic mice versus control mice, but were equivalent between transgenic mice and control mice in all other tissues tested. Human NAT1-specific N-acetyltransferase activities did not differ between transgenic and control mice in any tissue tested. Prostate cytosols from transgenic and control mice did not differ in their capacity to catalyze the N-acetylation of 2-aminofluorene, the O-acetylation ofN-hydroxy-2-aminofluorene andN-hydroxy-PhIP or theN,O-acetylation ofN-hydroxy-2-acetylaminofluorene. Transgenic and control mice administered PhIP did not differ in PhIP-DNA adduct levels in the prostate. This study is the first to report transgenic expression of human NAT2 in the mouse. The results do not support a critical role for bioactivation of heterocyclic amine carcinogens by humanN-acetyltransferase-2 in the prostate. However, the lack of an effect may relate to the level of overexpression achieved and the presence of endogenous mouse acetyltransferases and/or sulfotransferases.
Recent studies have shown that the dietary carcinogen, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) induces tumors in rat prostate (Shirai et al., 1997). Heterocyclic amine carcinogens such as PhIP may undergo bioactivation by cytochrome P-450 (CYP) 1A2 in the liver (Butler et al., 1989; Turesky et al., 1998) or by CYP1A1 and/or CYP1B1 in extrahepatic tissues (Crofts et al., 1998). TheN-hydroxy metabolite(s) formed can be further bioactivated by N-acetyltransferases (Hein et al., 1993, 1995; Turesky et al., 1991; Minchin et al., 1992; Lin et al., 1995) and sulfotransferases (Chou et al., 1995; Lin et al., 1995) to yield electrophilic arylnitrenium ions that bind covalently to DNA to form adducts that can initiate carcinogenesis.
Our laboratory has focused on the role of acetyltransferases in the bioactivation of heterocyclic amine carcinogens. Two majorN-acetyltransferase isozymes [EC 2.3.1.5], NAT1 and NAT2, are expressed in human and other mammalian species (Vatsis et al., 1995; Grant et al., 1997; Hein et al., 1997). Drugs such as sulfamethazine (SMZ) are selectively metabolized by human NAT2, whereas drugs such as p-aminobenzoic acid (PABA) are selectively metabolized by human NAT1 (Grant et al., 1991; Hein et al., 1993). Genetic polymorphisms exist for both human NAT1 andNAT2 (Vatsis et al., 1995; Grant et al., 1997; Hein et al., 1997). The NAT2 acetylation polymorphism is more completely characterized than NAT1. NAT2*4 is the most common allele associated with rapid acetylator phenotype (Vatsis et al., 1995) and recombinant human NAT2 4 acetyltransferase bioactivatesN-hydroxy-PhIP at higher rates than recombinant human NAT2 proteins encoded by NAT2 alleles from human slow acetylators (Hein et al., 1998). Furthermore, recombinant human NAT2 4 has a higher capacity to bioactivate N-hydroxy-PhIP than recombinant human NAT1 (Minchin et al., 1992; Hein et al., 1994).
Individuals with rapid NAT2 acetylator phenotype who ingest heterocyclic amines in cooked meats exhibited a significantly higher incidence of colorectal cancer (Lang et al., 1994). Because heterocyclic amines produce prostate cancer in the rat (Shirai et al., 1997), it is of interest to investigate the role of human NAT2 in the bioactivation of heterocylic amine carcinogens within the prostate. The role and relative importance of hepatic and target organ acetyltransferases in the bioactivation of heterocyclic amines leading to prostate tumors is not well understood. A significant role for human NAT2 would suggest that the NAT2 acetylation polymorphism influences susceptibility to heterocyclic amine-related prostate cancer analogous to its effect on the susceptibility to colorectal cancer (Lang et al., 1994).
To investigate the role of human NAT2 in bioactivation within the prostate, we constructed a prostate-specific transgenic mouse model using a transgene expression system containing regulatory elements of the rat probasin (PB)-encoding gene (Rennie et al., 1993; Kasper et al., 1994). The PB gene encodes an androgen- and zinc-regulated protein that is specifically expressed in prostate epithelial cells (Kasper et al., 1994). The ability of the prostate-specific rat PB promoter to target heterologous genes specifically to the prostate in transgenic mice has been extensively characterized (Greenberg et al., 1994, 1995;Barrios et al., 1996). Hormonal induction of PB-transgenes yield maximal expression of targeted protein at sexual maturity (7 weeks of age). Following successful construction and characterization, we used the prostate-specific transgenic mouse model to investigate the effect of prostate-specific expression of rapid acetylator humanNAT2 (NAT2*4) on the bioactivation of PhIP and 2-aminofluorene (AF), an aromatic amine carcinogen.
Materials and Methods
Construction of PB-NAT2 Transgene.
Rat genomic DNA was isolated from a Wistar-Kyoto inbred rat (Charles River Laboratories, Wilmington, MA) heart by standard methods. Briefly, the heart was frozen in liquid nitrogen, pulverized with a mortar and pestal, digested in proteinase K, extracted with phenol and chloroform/isoamyl alcohol (24:1), and precipitated the genomic DNA with ethanol. The rat PB gene promotor was amplified by polymerase chain reaction (PCR) using primers (PB1: 5′-TCTGGATCCCTGTAGGTATCTGGACCTCACTGA-3′ and PB2: 5′-TCTGCGGAGCTCGCGGCCGCAAGCTTCCACAAGTGCATTTAGCCT-3′). PCR was performed with 100 ng DNA in a 100-μl reaction containing 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 200 mM each dNTP, 1 μg each of the appropriate primers, and 2.5 units ofTaq DNA polymerase (Perkin-Elmer, Norwalk, CT). The PCR product (−426/+28 base pair (bp) fragment of the rat PB promoter) was digested with SacI/BamHI and ligated into theSacI/BamHI sites of the eukaryotic expression vector pKS/RIP (Valera et al., 1994) to produce the intermediate construct pKS/PB. Human genomic DNA was isolated using the Instagene Genomic DNA kit (Bio-Rad, Hercules, CA) from whole blood obtained from a person possessing the NAT2*4 allele as determined by PCR restriction fragment length polymorphism genotyping (Doll et al., 1995). The NAT2*4 allele was amplified by PCR as described above using primers (NAT2A: 5′TTAGGAATTCATGGACATTGAAGCATATTTTGAAAGAAT-3′ and NAT-2B: 5′-TGTGAATTCAAGGGTTTATTTTGTTCCTTATTCTAAAT-3′). The 870-bp PCR product (NAT2*4) was inserted as an EcoRI fragment into the EcoRI site of pKS/PB (Fig.1). The combination PB promoter region and NAT2 gene was sequenced via a modified double-stranded dideoxy chain termination method (Sanger et al., 1977) using Sequenase (United States Biochemical, Cleveland, OH), to verify proper base insertion and orientation. A 2.9-kb NotI/XhoI fragment (Fig. 2) was excised by digestion with 10 U of NotI and XhoI in 100 mM NaCl, 50 mM Tris-HCl (pH 7.4), 10 mM MgCl2, and 1 mM dithiothreitol (DTT) for 1 h at 37°C. The 2.9-kb fragment (Fig. 2) was subjected to gel electrophoresis and purified using Prep-A-Gene (BioRad, Hercules, CA).

Schematic diagram illustrating insertion of the rat PB promoter and human NAT2*4 into bluescript vector. For further details, see text.

Structure of the PB-NAT2 gene construct. Elements include the 5′-flanking region of the rat PB gene and a portion of the noncoding first exon (−426 to +28), regions from the rabbit β-globin gene that includes the last exon (open sections) and the humanNAT2*4 open reading frame (slashed section).
Production and Screening of PB-NAT2 Transgenic Mice.
Standard procedures were employed for construction of transgenic mice (Cameron, 1997). The 2.9-kb PB-NAT2 transgene (Fig. 2) was injected into one pronucleus of each one-cell mouse embryo of the inbred strain FVB (Harlan Sprague-Dawley, Indianapolis, IN). Microinjected embryos were implanted into pseudopregnant females and allowed to come to term. To identify transgenic founder mice, genomic DNA was isolated from 1-cm tail clips from 3- to 4-week-old mice. DNA was subjected to PCR as described above using primers specific for mouse NAT1 (M1: 5′-TGGTGTCTCCAGGTTAATCA-3′ and M2: 5′-GGTGGAGCCCACTAAACAGT-3′), to verify intact DNA, and primers specific for PB-NAT2 (1: 5′-TCAGTGAGGTCCAGATACCTACAG-3′ and 2: 5′-CTTCTGTCAAGCAGAAAATG-3′). Founder (mice generated from injected mouse embryos that contained the PB-NAT2 construct) mice were bred with FVB inbred mice and transgenic offspring were identified by PCR as described above. Transgenic positive (heterozygotes) and negative littermates were used for experiments at 7 weeks of age, when maximum transgene expression in the mouse prostate is observed (Greenberg et al., 1994; Barrios et al., 1996).
N-Acetyltransferase Assays.
SMZ, PABA, and AFN-acetyltransferase activities were measured using HPLC to separate N-acetyl-product from aromatic amine substrate using modifications of previous methods (Doll et al., 1997). Male mice (transgenic and control) were sacrificed at 7 weeks of age and organs were harvested and snap frozen. Tissue samples were homogenized (1 g/3 ml) in 20 mM sodium phosphate buffer (pH 7.4), containing 1 mM EDTA, 1 mM DTT, 100 μM phenylmethylsulfonylfluoride, and 10 mM leupeptin. Homogenates were centrifuged at 100,000g for 60 min to prepare cytosols.
SMZ and PABA N-acetyltransferase assays contained initial substrate concentrations of 300 μM for PABA or SMZ and 300 μM for acetyl coenzyme A. Reactions were carried out at 37°C and terminated by the addition of 15 μl perchloric acid, and neutralized with 11 μl of 1 M NaOH. The reaction tubes were centrifuged to precipitate protein and supernatant was injected onto a Lichrospher 100 RP-18 (5 μM) reversed phase column. Samples were eluted using a Beckman System Gold HPLC system equipped with a model 126 programmable solvent module pump and a model 166 programmable UV-visible detector, and the absorbance was measured at 260 nm (SMZ) and 280 nm (PABA). Briefly, the substrates and acetylated products were separated with a mobile phase of 20 mM sodium perchlorate (pH 2.5) (solvent A) and acetonitrile (solvent B). The elution was conducted with a linear gradient from 4% to 12% solvent B over 2 min at a flow rate of 2 ml/min to separate PABA and N-acetyl-PABA. A gradient from 9% to 29% solvent B over 5 min at a flow rate of 2 ml/min was used to separate SMZ andN-acetyl-SMZ. Under these conditions PABA andN-acetyl-PABA eluted at 2.8 and 7.2 min, respectively. SMZ and N-acetyl-SMZ were eluted at 5.2 and 8.4 min, respectively.
AF N-acetyltransferase assays were carried out as described above except that the AF and acetyl coenzyme A concentrations were 1 mM and the AF and N-acetyl-AF absorbances were measured at 290 nm. AF and N-acetyl-AF product were eluted from the column using a linear gradient from 25% to 75% solvent B over 5 min at a flow rate of 2 ml/min. Under these conditions, AF andN-acetyl-AF eluted at 4.8 and 6.6 min, respectively.
PhIP N-acetyltransferase assays were measured using a modification of a more sensitive radiolabel method described previously (Trinidad et al., 1990). Briefly, undiluted transgenic and control mouse prostate cytosols were incubated at 37°C with 2 mM PhIP and 2 mM [3H]acetyl-coenzyme A.
O-Acetyltransferase Assays.
The activation ofN-hydroxy-PhIP and N-hydroxy-AF (viaO-acetylation) to species that are capable of binding DNA were evaluated in prostate cytosols from transgenic and control mice as previously described (Hein et al., 1995). Briefly, prostate tissue cytosols were incubated at 37°C for 20 min in a reaction that included [ring-3H]N-hydroxy-PhIP (100 μM) or [ring-3H]N-hydroxy-AF (100 μM), 20 mM sodium phosphate buffer (pH 7.4), 2 mM acetyl coenzyme A, 1 mM DTT, 1 mM EDTA, and 1 mg/ml calf thymus DNA. In controls, distilled water was substituted for acetyl coenzyme A.
N,O-Acetyltransferase assays.
The activation (via N,O-acetylation) ofN-hydroxy-N-acetyl-AF to species capable of binding DNA were evaluated in prostate cytosols from transgenic and control mice as previously described (Hein et al., 1995). The reactions were carried out with 100 μM [3H]-N-hydroxy-N-acetyl-AF as described above except that water was substituted for acetyl coenzyme A and controls used heat-denatured enzymes.
Measurement of PhIP DNA-Adduct Levels In Vivo.
PhIP (50 mg/kg) was dissolved in 100% dimethyl sulfoxide and administered i.p. to transgenic and control mice (7 weeks of age). Controls were administered vehicle. Mice were sacrificed 6 h after injection and the prostate tissues were collected, snap frozen in liquid nitrogen, and DNA isolated by proteinase K digestion followed by phenol/chloroform extraction. DNA was treated with RNase A and T1 and quantified by spectrophotometric analysis at 260 nm. DNA isolated from prostate glands (10 μg) or reference DNA (1.48 pmol of PhIP-DNA adducts/mg DNA) was hydrolyzed to 3′-monophosphates by incubating with micrococcal nuclease (3.3 μg; Sigma, St. Louis, MO) and spleen phosphodiesterase (3.3 μg; Sigma) in 10 mM sodium succinate, 5 mM calcium chloride, pH 6.0, at 37°C for 5 h. DNA adducts were enriched by n-butanol extraction method (Gupta, 1985). PhIP-DNA adducts were 32P-labeled in the presence of T4 polynucleotide kinase and [γ-32P]ATP (50 μCi/sample, specific activity 7000 Ci/mmol; ICN, Costa Mesa, CA). PhIP-DNA adducts were resolved by polyethyleneimine-cellulose thin-layer chromatography using slight modifications of previously published methods (Peluso et al., 1991). Briefly, DNA was separated using thin-layer chromatography on PEI Cellulose F (Alltech, Deerfield, IL) in D1 (top to bottom; 1 M sodium phosphate, pH 5.7), followed by D3 (bottom to top; 4 M lithium formate/8.5 M urea, pH 3.5), followed by D4 (left to right; 0.8 M LiCl, 0.5 M Tris-HCl, and 7 M urea, pH 8.0). PhIP-DNA adducts were quantified by Instant Imager electronic autoradiography (Packard Instruments, Chicago, IL) and compared with the intensity of the PhIP-DNA adduct standard;N-(deoxyguanosin-8-yl)-PhIP (Lin et al., 1992).
Data Analysis.
Differences in enzyme activity and PhIP-DNA adduct levels measured in vivo between transgenic and control mice were analyzed for significance by Student’s ttest.
Results
To investigate whether the PB promoter fragment could regulate the expression of human NAT2*4, transgenic mice were constructed by microinjection of a 2.9-kb transgene carrying the −426/+28 PB promoter fragment upstream of the 870-bp intronless NAT2*4(Fig. 2). As shown in Fig. 3, founder transgenic mice were identified by PCR amplification of the PB-NAT2 transgene using genomic DNA isolated from tail clips. Intact DNA was verified from tail clips by amplification of mouse NAT1. ThePB-NAT2 construct was used as a control to verify proper amplification of the PB-NAT2 fragment from mouse genomic DNA. Two founder mice were identified (Fig. 3, lane 7 [DWH2.1] and lane 11 [DWH2.2]) that possessed the PB-NAT2 transgene. Both founder transgenic mice transmitted the transgene to approximately 50% of their offspring.

Identification of transgenic mice by PCR. Lane 1,PB-NAT2 construct control. Lanes 2–6, PCR amplification of mouse NAT1 (to verify intact DNA) from possible founder mice. Lanes 7–11, PCR amplification of PB-NAT2using DNA from lanes 2–6. Founder mice were identified in lane 7 (DWH2.1) and lane 11 (DWH2.2).
To determine whether PB-NAT2 transgene expression yielded functional NAT2 enzyme activity restricted to the prostate, male transgenic and control mice were sacrificed at 7 weeks of age. In comparisons between transgenic and control mice, significant increases in SMZ (human NAT2-specific) N-acetyltransferase activity were observed in the prostate but not in any other tissue. As shown in Fig.4A, transgenic line DWH2.1 demonstrated a 15-fold (p < .001) increase in SMZN-acetyltransferase activity compared with control mice. In contrast, no significant increase in SMZ N-acetyltransferase activity was observed in any other tissue tested (Fig. 4A). Transgenic line DWH2.2 did not express an increase in SMZ N-acetyltransferase activity in any tissue (data not shown).

Expression of SMZ and PABAN-acetyltransferase activity in tissue cytosols of transgenic and control mice. Open bars represent transgenic mice and solid bars represent control mice. A, mean ± S.E.M. for SMZ (human NAT2-specific) N-acetyltransferase activities in prostate, liver, lung, colon, small intestine, urinary bladder, and kidney. *Transgenic mice showed 15-fold higher (p< .001) SMZ N-acetyltransferase activities than control mice in the prostate, but activities did not differ significantly (p > .05) in the other tissue cytosols of control and transgenic mice. B, mean ± S.E.M. for PABA (human NAT1-specific) N-acetyltransferase activities in prostate, liver, lung, colon, small intestine, urinary bladder, and kidney. PABAN-acetyltransferase activities did not differ significantly (p > .05) between transgenic and control mice in any tissue, including the prostate. n = 2 for each tissue, except prostate where n = 4.
Transgenic mice did not demonstrate significantly higher levels of PABA (human NAT1-specific) N-acetyltransferase activity in the prostate compared with control mice (Fig. 4B). Moreover, no significant differences were observed between transgenic and control mice for PABA N-acetyltransferase activity in other tissues analyzed (Fig. 4B). These data suggest that the elevated SMZ N-acetyltransferase activities observed in the prostate of the transgenic mice were due to specific overexpression of human NAT2 in the prostate.
As shown in Table 1, transgenic and control prostate cytosols did not differ significantly in their ability to catalyze the O-acetylation of N-hydroxy-PhIP or N-hydroxy-AF. Moreover, there were no significant differences observed in the N-acetylation of AF and theN,O-acetylation ofN-hydroxy-N-acetyl-AF between transgenic and control prostate cytosols. PhIP N-acetyltransferase activities were below the limit of detection in both transgenic and control prostate cytosols.
Acetyltransferase activities in prostate cytosols of transgenic and control mice
PhIP-DNA adducts were detected in the prostate of both transgenic and control mice treated with PhIP, but not in those treated with vehicle (Fig. 5). The primary DNA adduct found in prostate was identified as N-(deoxyguanosin-8-yl)-PhIP adduct by chromatographic comparisons with the synthetic standard. The PhIP-DNA adduct levels did not differ significantly between transgenic and control prostate DNA (Fig. 6).

32P-Postlabeling autoradiograms of PhIP-DNA adducts. A, autoradiogram of DNA isolated from prostate tissue of a transgenic mouse treated with vehicle. B, autoradiogram of DNA isolated from a transgenic mouse treated with 50 mg/kg b.wt. PhIP. Spot 1 represents N-(deoxyguanosin-8-yl)-PhIP; identity of spot 2 is unknown. For further details, see text.
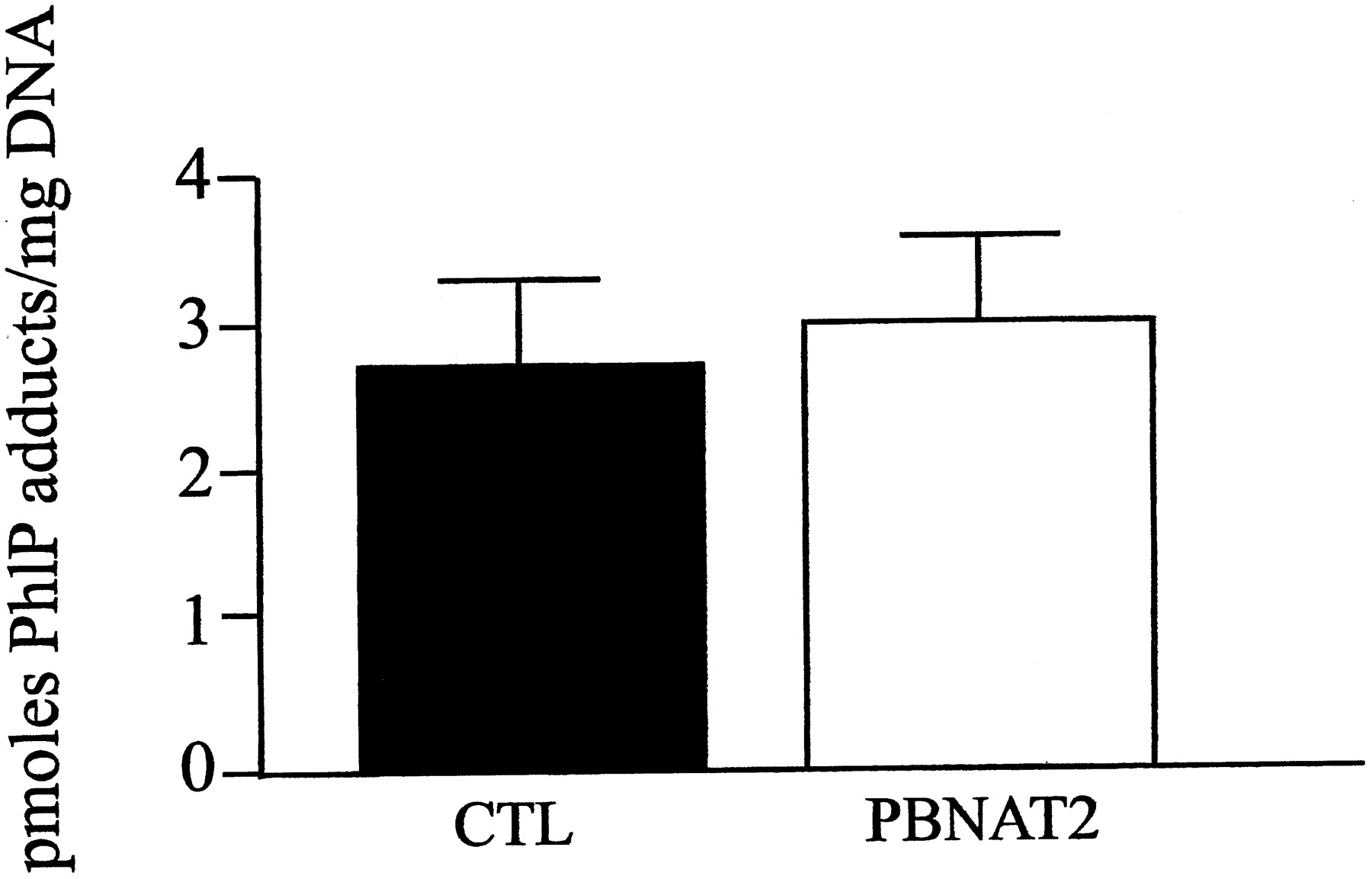
PhIP-DNA adduct levels in transgenic versus control mouse prostate. PhIP (50 mg/kg) was administered to transgenic and control mice. Levels of PhIP-induced DNA adducts were determined as described in the text. Significant differences (p> .05) in prostate DNA-adducts between transgenic (PBNAT2) and control (CTL) animals were not observed. All data represent mean ± S.E.M. in pmol PhIP adducts/mg DNA (n = 4).
Discussion
N-acetyltransferases bioactivate heterocyclic amines via O-acetylation to ultimate carcinogens and individuals segregate into rapid, intermediate, or slow acetylator phenotypes (Grant et al., 1997; Hein et al., 1997). Thus, rates of heterocyclic amine O-acetylation may be increased in rapid acetylators (Hein et al., 1995). Molecular models that increase carcinogen biotransformation, as it relates to genetic polymorphisms in activating enzymes such as the N-acetyltransferases, may aid in the understanding of complex pathways that either propagate elimination or enhance genotoxicity of carcinogens. The prostate is a target organ for the carcinogenic effects of PhIP in rats (Shirai et al., 1997). However, the underlying mechanisms that initiate heterocyclic amine-induced prostate tumor formation are unclear.
A working hypothesis includes PhIP bioactivation in the liver to formN-hydroxy-PhIP, and transport to extrahepatic organs where further bioactivation, via O-acetylation orO-sulfation, forms electrophilic species that bind DNA and initiates neoplasia. To study the role of extrahepaticO-acetylation in the bioactivation ofN-hydroxy-PhIP within the prostate, we constructed a transgenic mouse model in which human NAT2 was specifically overexpressed in the prostate.
Our results are consistent with a previous study that found that mice express relatively high levels of PABA N-acetyltransferase activity and very low levels of SMZ N-acetyltransferase activity (Glowinski and Weber, 1982). Another study found that recombinant mouse NAT1 and NAT2 proteins were unable to catalyze SMZN-acetylation at detectable levels (Martell et al., 1992). Because SMZ is a very selective substrate for human NAT2 (Grant et al., 1991; Hein et al., 1993), we used SMZ N-acetyltransferase activity to assess the functional expression of the humanNAT2*4 transgene. The specific expression of this transgene was clearly shown by the 15-fold increase observed in prostate cytosols of transgenic mice, whereas significant increases were not observed in other tissues. These results provide further evidence of the specificity of the PB promoter in targeting prostate-specific transgene expression.
Previous studies found that transgene copy number does not correlate with expression of two different transgenes using the same PB promoter employed in our study (Greenberg et al., 1994, 1995). Consequently, copy number was not determined. However, the Mendelian inheritance observed in our study suggests a single insertion site for the transgene.
Previous studies have shown relatively high levels of PABAN-acetyltransferase activity in rat (Hein et al., 1991) and hamster (Hein et al., 1992) prostate that was significantly higher in rapid versus slow acetylator animals. Other studies in rodents have shown that genetic differences in N-acetyltransferase activity can affect DNA adduct levels. For example, higher levels of aromatic amine DNA adduct levels were observed in urinary bladder of rapid acetylator versus slow acetylator congenic mice (Levy and Weber, 1989) and congenic hamsters (Feng et al., 1996) following administration of AF. Assuming that bioactivation of PhIP toN-hydroxy-PhIP occurs in the liver with further bioactivation in the prostate, we tested the hypothesis that overexpression of rapid acetylator human N-acetyltransferase in mouse prostate would also result in higher levels of DNA adducts both in vitro and in vivo.
The results of this study did not support this hypothesis. Although we successfully overexpressed human NAT2*4 in mouse prostate, we did not observe higher levels of PhIP DNA adducts in transgenic versus control mice. Similarly, higher levels of N-,O-, or N,O-acetyltransferase activity were not observed in vitro. The significant increase in SMZN-acetyltransferase activity in the transgenic mouse prostate is most likely due to the specificity of human NAT2 toN-acetylate SMZ (Grant et al., 1991; Hein et al., 1993), and the very low ability of endogenous mouseN-acetyltransferases to N-acetylate SMZ (Glowinski and Weber, 1982; Martell et al. 1992). Furthermore, we did not observe a significant increase in O-acetylation in the prostates of transgenic mice. One explanation is thatN-hydroxy amine derivatives, such as N-hydroxy-AF and N-hydroxy-PhIP, are not specific substrates for human NAT2 and these compounds may be activated by endogenous mouse acetyltransferases or sulfotransferases. The relative importance of acetylation versus sulfation in this bioactivation pathway varies with species (Lin et al., 1995). Compared with mice and rats, humans have high O-acetyltransferase activity, but the lowestO-sulfotransferase activity for the bioactivation ofN-hydroxy-PhIP (Lin et al., 1995). Mice, in contrast, have the lowest O-acetyltransferase activity, but the highestO-sulfotransferase activity toward the bioactivation ofN-hydroxy-PhIP (Lin et al., 1995). Previous studies have suggested that O-sulfotransferase predominates overO-acetyltransferase in the bioactivation ofN-hydroxy heterocyclic amine carcinogens in the mouse (Buonarati et al., 1990; Lin et al., 1995).
The results of this study suggest that bioactivation viaO-acetylation within the mouse prostate is of limited importance in the generation of PhIP-DNA adducts, and presumably tumors. These results are consistent with a recent report by Agundez et al. (1998) that found very low levels of SMZN-acetyltransferase activity in human prostate that was independent of the NAT2 genotype. However, our results need to be interpreted with caution. As described above, mice express very high levels of sulfotransferase activity, and it is possible that the high levels of sulfotransferase overwhelmed the increase in acetyltransferase activity achieved in the transgenic mice. In addition, mice have endogenous N-acetyltransferase activities that also contribute to the bioactivation (Fretland et al., 1997; Hein et al., 1997). These endogenousN-acetyltransferases may also have overwhelmed the modest increase (15-fold) in human NAT2 4 that we achieved in this transgenic model.
In summary, to our knowledge, this is the first study to report construction of a transgenic mouse that overexpresses humanN-acetyltransferase. The overexpression was successfully targeted to the prostate. Differences were not observed between transgenic and control mice in bioactivation of heterocyclic amine carcinogens within the prostate. Although these results do not support a role for bioactivation of heterocyclic amine carcinogens by human NAT2 within the prostate, the lack of an effect may relate to endogenous mouse N-acetyltransferases or sulfotransferases in the prostate and the modest level of human NAT2 over-expression observed.
Acknowledgments
We thank Dr. Fred Kadlubar, National Center for Toxicological Research, Jefferson, Arkansas, for his generous donation ofN-hydroxy arylamine and arylamide substrates and the PhIP-DNA adduct standard. We also thank Dr. Fatima Bosch, University of Barcelona, Bellaterra, Spain, for kind donation of the pKS/RIP expression vector.
Footnotes
-
Send reprint requests to: David W. Hein, Ph.D., Department of Pharmacology and Toxicology, University of Louisville School of Medicine, Louisville, KY 40292. E-mail:d.hein{at}louisville.edu
-
↵1 This work was partially supported by United States Public Health Service Grant CA34627 from the National Cancer Institute. A preliminary report of this work was presented at the 1998 annual meeting of the Society of Toxicology (Leff et al., 1998; Toxicol Sci42:318).
-
↵2 This work constitutes partial fulfillment by Matthew Leff for the Ph.D. in Pharmacology and Toxicology at the University of Louisville.
-
↵3 Present address: Toxicology and Pathology Services, Inc., 10424 Middle Mount Vernon Rd., Mt. Vernon, IN 47620.
- Abbreviations:
- PhIP
- 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine
- NAT2
- N-acetyltransferase 2
- NAT1
- N-acetyltransferase 1
- SMZ
- sulfamethazine
- PABA
- p-aminobenzoic acid
- PB
- probasin
- PCR
- polymerase chain reaction
- bp
- base pair
- AF
- 2-aminofluorene
- DTT
- dithiothreitol
- Received December 29, 1998.
- Accepted March 23, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}