


Abstract
The anti-inflammatory/antiallergic activity of a novel second-generation p38 mitogen-activated protein kinase inhibitor, SB 239063 [trans-1-(4-hydroxycyclohexyl)-4-(4-fluorophenyl)-5-(2-methoxypyridimidin-4-yl)imidazole], was investigated in vivo and in vitro. SB 239063 had an IC50 of 44 nM for inhibition of recombinant purified human p38α. In lipopolysaccharide-stimulated human peripheral blood monocytes, SB 239063 inhibited interleukin-1 and tumor necrosis factor-α production (IC50 values = 0.12 and 0.35 μM, respectively). A role for p38 kinase in cytokine-associated inflammation in the mouse was shown by p38 activation in the lung and inhibition of lipopolysaccharide-induced tumor necrosis factor-α production by SB 239063 (ED50 = 5.8 mg/kg p.o.). Antiallergic activity was demonstrated by essential abolition (∼93% inhibition) of inhaled ovalbumin (OA)-induced airway eosinophilia by SB 239063 (12 mg/kg p.o.), measured by bronchoalveolar lavage (BAL) in OA-sensitized mice. In addition, p38 kinase was found by Western analysis to be activated in guinea pig lung. Administration of SB 239063 (10 or 30 mg/kg p.o.) in conscious guinea pigs markedly reduced (∼50% inhibition) OA-induced pulmonary eosinophil influx, measured by BAL 24 h after antigen. SB 239063 (10 mg/kg b.i.d. p.o.) administered after leukotriene D4 inhalation, reduced by 60% the persistent airway eosinophilia seen at 4 days. Apoptosis of cultured eosinophils isolated from guinea pig BAL was increased by SB 239063 (1–10 μM) in the presence of interleukin-5. These results indicate that SB 239063 is a potent inhibitor of inflammatory cytokine production, inhibits eosinophil recruitment, in addition to enhancing apoptosis of these cells. Collectively, the results support the potential utility of p38 kinase inhibitors, such as SB 239063, for the treatment of asthma and other inflammatory disorders.
The mitogen-activated protein (MAP) p38 kinase is a ubiquitous and highly conserved, proline-directed serine-threonine protein kinase. It appears to play an important role in a variety of pathophysiological responses and has been suggested to be involved in many processes considered critical to the inflammatory response and tissue remodeling (for review, see Griswold and Young, 1996). Several of these events are hallmarks of pulmonary diseases such as chronic obstructive pulmonary disease and asthma (Barnes et al., 1998; Barnes, 1999)
Although there is a paucity of reports specifically addressing the role of p38 kinase in pulmonary disease, it is known that inflammatory cytokines play an important role in airways inflammation. Thus, cytokines such as tumor necrosis factor-α (TNF-α), interferon gamma (IFN-γ), interleukin (IL)-4, IL-5, and chemokines such as IL-8, regulated on activation normal T-cell expressed and secreted (RANTES), and eotaxin have all been shown to be capable of regulating or supporting chronic airway inflammation (Barnes et al., 1988, 1998).
There is also evidence for an involvement of p38 kinase in allergic mechanisms. For example, human monocyte IL-4-induced release of soluble cellular differentiation antigen 23 was dramatically inhibited by the p38 kinase inhibitor SB 203580 (Marshall et al., 1998), suggesting IgE synthesis also might be regulated by p38 kinase activity. In addition, aggregated IgA- or IgG-stimulated eosinophil degranulation has been shown to involve the MAP kinases and phosphatidylinositol 3-kinase (Bracke et al., 1998). As the eosinophil has been the focus of major recent antiasthma drug discovery efforts, the induction of apoptosis has been postulated to be a potential therapeutic objective in the resolution of the harmful activities of this granulocyte (Anderson, 1996). The recent demonstration of the enhancement of peripheral human eosinophil apoptosis by SB 203580 suggests a novel therapeutic approach and opportunity for p38 MAP kinase inhibitors (Kankaanranta et al., 1999).
Collectively, these data suggest there is considerable potential for a p38 kinase inhibitor in the treatment of inflammatory lung diseases. In this report, we describe the in vitro and in vivo pharmacology of a novel, potent, and selective inhibitor of p38 MAP kinase, SB 239063 [trans-1-(4-hydroxy-cyclohexyl)-4-(4-fluorophenyl)-5-(2-methoxypyridimidin-4-yl)imidazole] (Fig. 1), including effects in pulmonary disease models. The results support the contention that this class of compound may have promise in the treatment of chronic diseases of the airways.

Structure of SB 239063.
Materials and Methods
In Vitro Studies
Kinase Assays.
Yeast-expressed, activated, and purified p38α (3.55 nM) was added to the reaction mixture (30 μl) containing 25 mM HEPES, pH 7.5; 10 mM MgCl2; 0.2 mM sodium orthovanadate; 1 mM dithiothreitol; 0.1% BSA; 10% (w/v) glycerol; 0.17 mM/2.5 μCi of [γ-32P]ATP; 0.67 mM the endothelial growth factor receptor-derived T669 peptide as the substrate in the presence or absence of SB 239063. Incubation was for 25 min at 37°C in 96-well plates, the reactions were stopped by adding 10 μl of 0.3 M phosphoric acid, and phosphorylated peptide was isolated from the reaction mixture on phosphocellulose (p81) filters. Filters were washed three times with 75 mM phosphoric acid, followed by three times with H2O, and counted for bound32P. The inhibitor was preincubated with the reaction mixture on ice for 30 min before starting the reactions with [γ-32P]ATP.
The Km[ATP] for p38 was determined to be 0.166 mM. Other kinase assays (CSBP/p38 kinase, extracellular signal receptor-activated kinase, c-Jun NH2-terminal kinase 1, c-Raf, endothelial growth factor receptor tyrosine kinase, p56Lck, protein kinase C α, cdc2, cdk2, and cdk4) were performed as described previously (Lee et al., 1999).
Cytokine Production in Human Monocytes.
Highly enriched human peripheral blood monocytes were obtained from whole blood buffy coat and enriched by differential Ficoll and Percoll density centrifugation. Monocytes were cultured in RPMI 1640 (Life Technologies-BRL, Long Island, NY) and supplemented withl-glutamine and 1% human A-B serum and were stimulated with bacterial endotoxin [lipopolysaccharide (LPS); Escherichia coli, type W, 055:B5; Difco, Detroit, MI] in the presence or absence of different concentrations of SB 239063. Twenty-four-hour culture supernatants were assessed for cytokine content (IL-1, granulocyte/macrophage colony-stimulating factor, granulocyte colony-stimulating factor, TNF-α) with appropriate enzyme-linked immunosorbent assay (ELISA) kits (R & D systems, Minneapolis, MN) as described previously (Lee et al., 1999). Data were calculated from the standard curve, and IC50values were determined with regression analysis.
Western Blot Analysis.
Western blot analyses for p38 and phospho p38 were conducted on mouse and guinea pig lung samples. With a Tissuemizer (Tekmar Company, Cinncinati, OH), lung homogenates were made by grinding 0.05 g of lung tissue in 1 ml of lysis buffer (200 mM Tris-HCl, pH 7.4; 1% Triton-X-100; 10% glycerol; 150 mM NaCl; 2 mM EDTA; 25 mM β-glycerophosphate; 20 mM NaF; 1 mM sodium orthovanadate; 2 mM pyrophosphate; 1 mM phenylmethylsulfonyl fluoride; 10 μg/ml Leupeptin). Homogenates were centrifuged at 25,000g for 15 min at 4°C, and the supernatant was collected and assayed for total protein concentration (Bio-Rad, Richmond, CA). For mouse lung, samples of 35 μg of total protein was loaded per lane, and for guinea pig lung, samples of 10 μg of protein were loaded. Proteins were resolved on a 10% Tris-HCl gel (Bio-Rad) and transferred onto a nitrocellulose membrane. The membranes were blocked for 1 h at room temperature with a membrane-blocking solution containing BSA and goat IgG (Zymed Laboratories, San Francisco, CA), and incubated overnight at 4°C in primary antibody, either anti-p38 or antiphospho p38 (1:1000, rabbit polyclonal IgG; New England Biolabs, Beverly, MA). The p38 antibody was raised against a synthetic peptide corresponding to residues 341 to 360 of human p38. The phospho-specific antibody detects p38 when phosphorylated at both Thr180 and Tyr182. The secondary antibody was an anti-rabbit IgG conjugated to horseradish peroxidase (1:2500; New England Biolabs), and the detection reagent was enhanced chemiluminescence (Amersham Life Sciences, Piscataway, NJ). Membranes were washed between incubations with 1× diphosphate-bufferred saline (DPBS) without calcium/magnesium containing 0.05% Tween 20. Total cell extracts from C6 glioma cells stimulated with or without anisomycin (New England Biolabs) were used as positive and negative controls, respectively.
Measurement of Guinea Pig Airway Eosinophil Apoptosis.
Cell suspensions of fresh lavage fluid preparations from guinea pigs that had been exposed 48 h earlier to an aerosol of leukotriene D4 (LTD4) (1 μg/ml for 1 min) were pelleted by centrifugation at 300g, and the cells were pooled by resuspension in 10 ml of saline in a 50-ml tube. Ten milliliters of Percoll working solution (9 ml of Percoll; Sigma P1644; 1 ml 10× DPBS; 5.52 ml of saline) was layered underneath the cell suspension and the tube was centrifuged at 400g for 30 min at room temperature. The top layers, including the mononuclear cell-containing interface, were aspirated and the pellet, containing mostly eosinophils, was resuspended in 9 ml of cold milli-Q water to lyse erythrocytes. One milliliter of 10× DPBS was immediately added and cells were pelleted at 300g. Cells were then washed with 10 ml of DPBS and cell number, viability, and purity were assessed by hemacytometer counts with trypan blue and differential staining of cytospin preparations. Yields were 1 to 2 × 107 total cells per two guinea pigs, and cells were >95% in terms of both viability and eosinophil purity. Cells were resuspended in medium (RPMI, 10% fetal bovine serum, and gentamycin) at 0.5 to 1.0 × 106/ml and 500 μl was added to wells of 24-well plates. Cells were treated by addition of 0.5 ml of medium with or without IL-5 (Pharmingen, San Diego, CA; final concentration 10 pM) and with SB 239063 or vehicle (0.1% ethanol). At various time points, cells were harvested and prepared for flow cytometric analysis with the ApoAlert Annexin V fluorescein isothiocyanate (FITC) Apoptosis Kit (Clontech, Palo Alto, CA) as per instructions. Briefly, each cell sample was washed with 200 μl of binding buffer, resuspended in 200 μl of binding buffer, and then 5 μl of Annexin V FITC and 10 μl of propidium iodide (PI) stocks were added. Samples were incubated for 10 min at room temperature in the dark with agitation. Two hundred microliters of the sample was diluted to 500 μl with binding buffer just before flow analysis. Samples were analyzed with a FACScan (Becton-Dickinson, San Jose, CA) and CellQuest cell analysis software (Becton-Dickinson). Forward angle light scatter and side scatter were used to gate on eosinophil population and exclude debris. Gate was large enough to ensure collection of all cells at all time points because changes in light scatter properties accompany the onset of apoptosis and necrosis. At each time point, an unlabeled control also was prepared to ensure that observed fluorescence was not due to any changes in autofluorescence. Color compensation was set to eliminate FITC spill into the FL2 detector and PI spill into the FL1 detector. Green (Annexin V FITC) versus Red (PI) fluorescence was assessed to monitor onset of apoptosis or necrosis. Analysis was accomplished by use of quadrant stats in CellQuest software. Quadrants were set so that cells in lower left (LL) quadrant were unlabeled, viable cells; cells in lower right (LR) were FITC positive only (apoptotic), and cells in upper right (UR) quadrant were both FITC and PI positive. Necrotic cells will be PI positive and also take up Annexin V FITC, but apoptotic cells, initially FITC positive/PI negative also will eventually have compromised membranes and incorporate PI. Preliminary studies at 29 h demonstrated that very few cells (<5%) were truly necrotic (“PI bright”), and this number varied by only 1 or 2% among treatment groups. Therefore, the LR quadrant numbers (definite apoptotic cells) were in approximately the same ratio between treatment groups as total FITC positive (LR + UR). Because the majority of PI-positive cells at all time points were not “true necrotic” (PI bright), but clearly FITC positive with increasing PI positivity, total FITC positive (LR + UR) was used for comparative quantitation and graphing of apoptosis among groups.
Cells isolated from guinea pig bronchoalveolar lavages (BALs) were cultured in the presence of IL-5 with and without various concentrations of SB 239063. Additionally, some cells were cultured without IL-5 so that maximal apoptosis levels could be observed. At designated time points, cultures were harvested, dual labeled with Annexin-V-FITC and PI and assessed by flow cytometry.
Histology.
The trachea and lungs of LTD4-challenged guinea pigs (according to in vivo methodology described in a following section) were collected intact for qualitative assessment of airway eosinophilia. The lungs were gently inflated by tracheal cannula with 10% buffered formalin until no pleural creases were visible, and the trachea was ligated followed by immersion in 10% buffered formalin. The lungs were sectioned longitudinally to include trachea, airways, and both right and left lungs. The tissue was paraffin embedded and sectioned at 5-μm thickness followed by Luna's stain for eosinophils (Luna, 1968); the microscopic image was taken at a magnification of 830×.
In Vivo Studies
Animals.
BALB/c mice and Hartley Guinea pigs, obtained from Charles River Breeding Laboratories (Raleigh, MA), were maintained in a barrier facility. All experimental procedures conform to Animal Care and Use Committee protocols filed at SmithKline Beecham Pharmaceuticals (King of Prussia, PA).
Mouse Studies.
LPS-Induced TNF-α Release.Age-matched, male BALB/c mice (n = 5 or 6/group, 22–25 g) from Charles River Breeding Laboratories were pretreated orally with compound or vehicle. Thirty minutes after pretreatment, the mice were given LPS (from Escherichia coli serotype 055-85; Sigma Chemical Co., St Louis, MO), 25 μg/mouse in 25 μl of PBS, pH 7.0 i.p. Two hours later, the mice were sacrificed by CO2 inhalation, and blood samples were collected by exsanguination into heparinized blood collection tubes and stored on ice. The blood samples were centrifuged, and the plasma collected for analysis by ELISA for TNF-α levels.
TNF-α levels were measured with a sandwich ELISA (Olivera et al., 1992) with a hamster monoclonal antimurine TNF-α (Genzyme, Cambridge, MA) as the capture antibody and a polyclonal rabbit antimurine TNF-α (Genzyme) as the second antibody. For detection, a peroxidase-conjugated goat anti-rabbit antibody (Pierce, Rockford, IL) was added, followed by a substrate for peroxidase (1 mg/ml orthophenylenediamine with 1% urea peroxide). TNF-α levels in the plasma samples from each animal were calculated from a standard curve generated with recombinant murine TNF-α (Genzyme).
Antigen-Induced Airway Eosinophilia. Male BALB/c mice (18–20 g) were sensitized by i.p. injections of 10 μg of ovalbumin (OA) in 200 μl of Rehsorptar aluminum hydroxide gel (Intergen, Purghase, NY) on days 0, 7, and 14. On day 21, mice were placed into a Plexiglas exposure chamber with an internal volume of 4 liters. An aerosol of OA (1% in normal saline), generated with an Opti-Mist nebulizer (Hospitak, Farmingdale, NY) was delivered into the box at a rate of 4 l/min for 30 min. SB 239063 (12 mg/kg) or vehicle (acidified 0.5% Tragacanth) was administered p.o. through a 22-gauge gavage needle 1 h before and 4 h after antigen challenge and b.i.d. thereafter for 3 days. BAL was performed 96 h after OA exposure. Mice were euthanized with an overdose of sodium pentobarbital (100 mg/kg i.p.), and the lungs were lavaged with 3.5 ml of Dulbecco's PBS with 100 μM EDTA (5 × 0.7 ml), which was aspirated after gentle chest massage. The BAL fluid was centifuged, and the pellet was resuspended in 1 ml of 0.9% NaCl. After a total cell count, slides were prepared, stained, and differentiated as eosinophils, neutrophils, and mononuclear cells by counting a minimum of 200 cells and expressing the results as percentage of total cells as well as actual numbers of each type. This measurement and expression technique has been previously validated as accurately reflecting endothelial and subendothelial airway eosinophilia documented by histological methodology (Underwood et al., 1996).
Guinea Pig Studies.
Sensitization Procedure. Male Hartley guinea pigs (200–250 g) were sensitized by i.m. injection of 0.35 ml of a 5% (w/v) OA/saline solution into each thigh (0.7 ml total) on days 1 and 4. Guinea pigs were available for use after day 25.
Antigen-Induced Bronchoconstriction and Airway Eosinophil Influx. Conscious animal body plethysmography. Male Hartley guinea pigs (550–750 g), actively sensitized to OA, were pretreated with chlorpheniramine (0.1 mg/kg s.c.) 15 min before antigen challenge and placed into a double-flow body plethysmograph (Penn-Century, Philadelphia, PA), consisting of a nasal (head) chamber and a thoracic (body) chamber, each equipped with a pneumotachograph. The plethysmograph was connected to a noninvasive respiratory analyzer (Buxco Electronics, Sharon, CN) via a Validyne differential pressure transducer (±2 cm) that calculated specific airway conductance. After a 10-min stabilization period, an aerosol of OA (1% in normal saline) was generated by an ultrasonic nebulizer (Pulmosonic; DeVilbiss Corporation, Somerset, PA) and delivered for 10 s at a rate of 250 ml/min via a nosecone built into the plethysmograph. Bronchoconstriction was calculated as average maximum decrease as well as area-under-the-curve analysis of the percentage decrease in specific airway conductance from baseline over the 10-min period after antigen inhalation. SB 239063 (3, 10, or 30 mg/kg) or vehicle (acidified 0.5% Tragacanth) was administered intragastrically via a size 8 French feeding tube 1 h before and 4 h after antigen challenge.
BAL. BALs were performed 24 h after OA exposure. Guinea pigs were euthanized by an overdose of sodium pentobarbital. The lungs were lavaged with 50 ml of Dulbecco's PBS (5 × 10 ml), which was aspirated after a gentle chest massage. The BAL fluid was centrifuged and the pellet was resuspended in 0.25% NaCl to lyse residual erythrocytes; after centrifugation, the pellet was resuspended again in 0.9% NaCl. After a total cell count, slides were prepared, stained, and differentiated as eosinophils, neutrophils, and mononuclear cells as outlined above.
Inhaled LTD4-Induced Persistent Airway Eosinophilia. Male Hartley guinea pigs were placed into a double-flow body plethysmograph, as outlined above for antigen challenge studies, and an aerosol of LTD4 (10 μg/ml) was administered to guinea pigs for 1 min. Inflammatory cell influx was measured with the aforementioned protocol in the antigen experiments. Because we have previously shown that a single exposure to LTD4 results in a persistent airway eosinophilia that peaks between 2 and 4 days and remains elevated for 2 to 4 weeks (Underwood et al., 1996), airway eosinophilia was measured on day 5 as outlined above. SB 239063 (10 mg/kg p.o.) or vehicle was administered 2 and 6 h after aerosol LTD4 exposure (10 μg/ml for 1 min) and b.i.d. thereafter for 3 days.
Statistical Analysis.
As appropriate, Student'st test, ANOVA, or Fisher's protected least significant difference (PLSD) were used to determine statistical significance, with *P < .05, **P < .01, or ***P < .001 considered to be statistically significant based on random probabilities of 5 in 100, 1 in 100, and 1 in 1000, respectively.
Drugs.
LPS, chlorpheniramine, and OA were obtained from Sigma Chemical Co. TNF-α was purchased from Genzyme. LTD4 and SB 239063 (Fig. 1) were synthesized by colleagues in the Department of Medicinal Chemistry, SmithKline Beecham Pharmaceuticals (King of Prussia, PA).
Results
p38 MAP Kinase Inhibition and Selectivity Profile.
SB 239063 is a potent and selective inhibitor of p38 MAP kinase. Thus, SB 239063 displayed specific and high-affinity binding to p38 MAP kinase, resulting in potent inhibition of its catalytic activity, with an ID50 of 44 nM (n = 4; Table1). Because p38 MAP kinase exists as four distinct isoforms (α, β, γ, and δ), SB 239063 exibits equipotent inhibitory activity against α- and β-isoform, and no activity (up to 100 μM) against the γ- and δ-kinase isoforms. A panel of protein kinases, including lipid kinases, tyrosine kinases, and extracellular signal receptor-activated kinase and c-Jun NH2-terminal kinase 1, which are closely related members of the MAP kinase family, were not inhibited by SB 239063 in concentrations up to 10 μM (Table 1).
Effect of SB 239063 on selected protein kinases
In Vitro Inhibition of Cytokine Production.
SB 239063 potently inhibited IL-1 and TNF-α production in LPS-stimulated human peripheral blood monocytes with IC50 values of 120 and 350 nM, respectively (Fig. 2). Importantly, inhibition of cytokine production by SB 239063 was also selective. Thus, GM-CSF production was inhibited by SB 239063, albeit less potently (IC50 = 1.6 μM) than for IL-1 and TNF-α, whereas G-CSF production was not affected.

Effects of SB 239063 on cytokine production from human monocytes. Human monocytes (as described in Materials and Methods) were incubated for 24 h with bacterial endotoxin in the presence and absence of SB 239063. Cytokine production was assayed by ELISA as described and means (without standard errors for clarity) are depicted; n = 4.
In Vivo Inhibition of TNF-α Production.
As seen in Table2, oral SB 239063 was a potent inhibitor of LPS-induced TNF-α production in the mouse peritoneal cavity with an ED50 of 5.8 mg/kg (2.8–10.3; 95% CL).
Effect of SB 239063 in LPS-induced TNF-α production in mice
Western Blot Analyses of Mouse and Guinea Pig Lungs.
As seen in Fig. 3A, lung homogenates from sensitized mice were electrophoresed (lane 3) along with anisomycin-stimulated C6 glioma cell lysates (lane 1) and a nonstimulated C6 cell lysate (lane 2). Western blotting of this gel and development with antibody detecting p38 (left) or phospho p38 (right) clearly showed the presence of both p38 and phospho p38 in mouse lung homogenates. Similar results are shown in Fig. 3B with guinea pig lung homogenates where p38 and phospho p38 were seen (lane 2 on the left and right, respectively). Lane 1 contains anisomycin-stimulated C6 glioma cell extracts as a positive control (Fig. 3B).
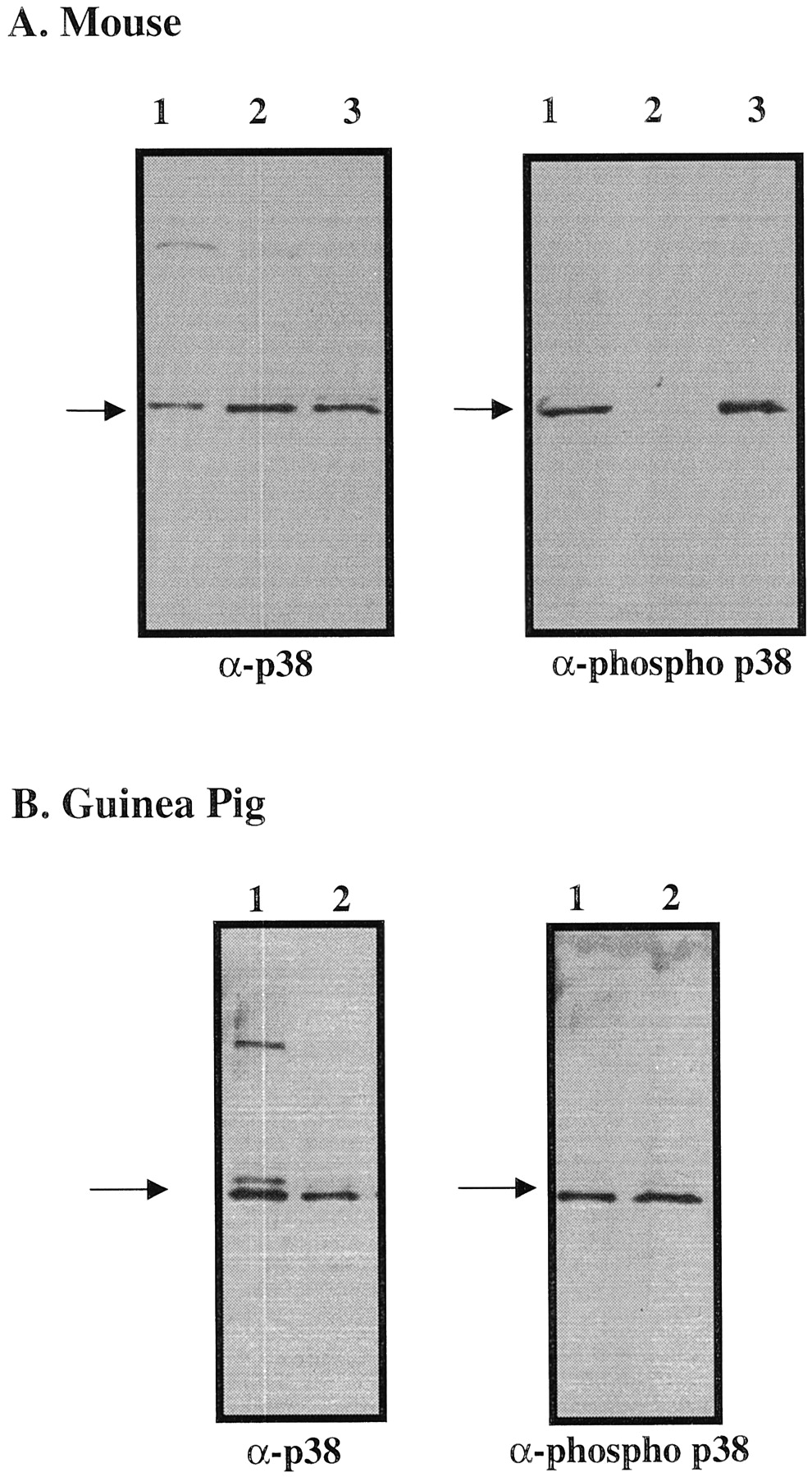
Western blots of mouse and guinea pig lung homogenates. A, Western blots of mouse lung homogenates stained with anti-p38 (left) and antiphospho p38 (right) indicate the presence of phospho p38 in mouse lung. Lane 1 was loaded with anisomycin-stimulated C6 cell extracts that serve as markers for both p38 and phospho p38. Lane 2 was loaded with nonstimulated C6 cell extracts (does not show band reacting with antiphospho p38). Lane 3 contains the mouse lung samples (35 μg of protein). B, Western blots of guinea pig lung homogenates stained with anti-p38 (left) and antiphospho p38 (right) indicate the presence of phospho p38 in guinea pig lung. Lane 1 was loaded with anisomycin-stimulated C6 cell extracts. Lane 2 contains the guinea pig lung samples (10 μg of protein).
Effect on Antigen-Induced Airway Eosinophilia in Mice.
Aerosol administration of OA to sensitized mice increased eosinophil number recovered by BAL, 96 h after OA challenge, from 0.02 ± 0.02 × 104 in control, unchallenged mice to 7.3 ± 1.1 × 104 in vehicle-treated animals that had received OA. Treatment with SB 239063 (12 mg/kg p.o., 1 h before and 4 h after OA challenge, then b.i.d. for 3 days) significantly inhibited (93% decrease) the resultant antigen-induced airway eosinophilia (0.25 × 104 eosinophils; P < .001, Fisher's PLSD; n = 4–6) (Fig.4). Antigen exposure also increased total leukocyte number recovered by BAL, 96 h after OA challenge, from 3.1 ± 0.3 × 105 in control, unchallenged mice to 8.6 ± 1.3 × 105. In animals treated with SB 239063, 4.6 ± 1.5 × 105 total leukocytes were recovered by BAL, representing a 47% reduction in leukocytes recovered compared with OA-exposed, vehicle treated animals (data not shown).

Effect of SB 239063 (12 mg/kg p.o., b.id. for 3 days) on inhaled OA-induced eosinophilia in mice. Data are expressed as total cell number of eosinophils (mean ± S.E.) recovered by BAL 96 h after challenge (n = 4–6). ∗∗∗, significantly different from vehicle control, P < .05 (Fisher's PLSD).
Effect on Antigen-Induced Airway Eosinophilia in Guinea Pigs.
Aerosol administration of OA to sensitized guinea pigs increased airway eosinophil number recovered by BAL from 0.50 ± 0.26 × 106 (∼4% of total leukocytes) in unchallenged animals to 3.22 × 106 (∼23%) in OA-challenged, vehicle-treated control animals, representing a 550% increase in airway eosinophilia 24 h after antigen challenge (Fig.5). SB 239063, administered orally 1 h before and 4 h postantigen inhalation, significantly inhibited, by ∼50%, the airway eosinophil influx at both 10 and 30 mg/kg; a lower dose (3 mg/kg) was without significant effect (Fig. 5). In contrast to the potent inhibitory activity of SB 239063 against OA-induced eosinophilia, there was no significant inhibition of the acute bronchoconstriction elicited by OA inhalation; thus, maximum decreases (occurring at 2–4-min post-OA exposure) or area-under-the-curve measurements (computed 0–10-min post-OA exposure) of specific airway resistance were not different (data not shown).
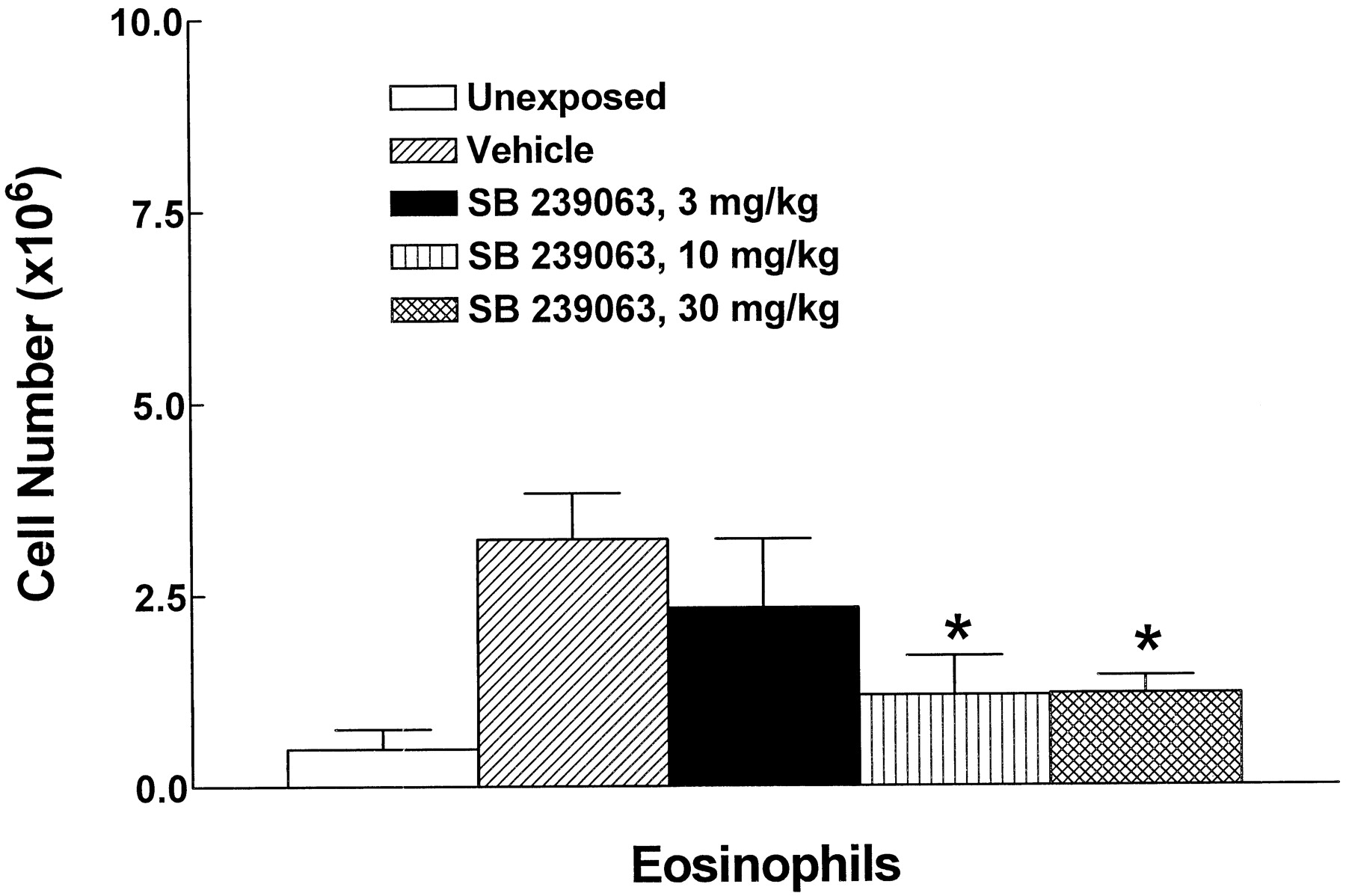
Effect of SB 239063 on inhaled OA-induced airway eosinophilia in guinea pigs. SB 239063 (3, 10, or 30 mg/kg p.o.) was administered 1 h before and 4 h after antigen challenge and airway eosinophilia was assessed by BAL 24 h after OA in guinea pigs. Data are expressed as total number of eosinophils (mean ± S.E.; n = 5–8. ∗, significantly different from vehicle control, P < .05 (ANOVA and Fisher's PLSD).
Effect on Inhaled LTD4-Induced Persistent Airway Eosinophilia in Guinea Pigs.
Inhalation of LTD4 (10 μg/ml for 15 min) by guinea pigs resulted in a persistent airway inflammation as demonstrated by a doubling of total leukocyte recovery and a 6-fold increase in eosinophil number by BAL 96 h after LTD4challenge (Fig. 6). SB 239063 (10 mg/kg p.o.), administered 2 and 6 h after LTD4inhalation and b.i.d. for 4 more days, substantially reduced (by 50%) the recovery of eosinophils by BAL of the airways at 96 h post-LTD4 challenge (ANOVA, Fisher's PLSD;n = 4–6; P < .05; Fig. 6).

Effect of SB 239063 on inhaled LTD4-induced persistent airway eosinophilia in guinea pigs. Data are expressed as total number of eosinophils (mean ± S.E.) recovered by BAL 96 h after LTD4 challenge (n = 4–6). ∗, significantly different from vehicle control, P < .05 (Fisher's PLSD).
Effect on Apoptosis of Cultured Guinea Pig Airway Eosinophils.
Using a technique of flow cytometric analysis of apoptosis, a dose-related increase in apoptosis of eosinophils was observed at multiple time points with SB 239063 (0.1–10 μM) (**P < .01 or ***P < .001; Fig. 7). The time points depicted in Fig. 7 are 29- and 47-h postintroduction of guinea pig eosinophils into culture in the presence of IL-5. As observed in preliminary studies, very few cells (<5%) were truly necrotic (“PI bright”), and necrotic cell number varied less than 2% among treatment groups. A statistically significant increase in apoptotic cells was observed (P < .05 or less) with either 1 or 10 μM SB 239063 compared with untreated control, at every time point from 21 h onwards.
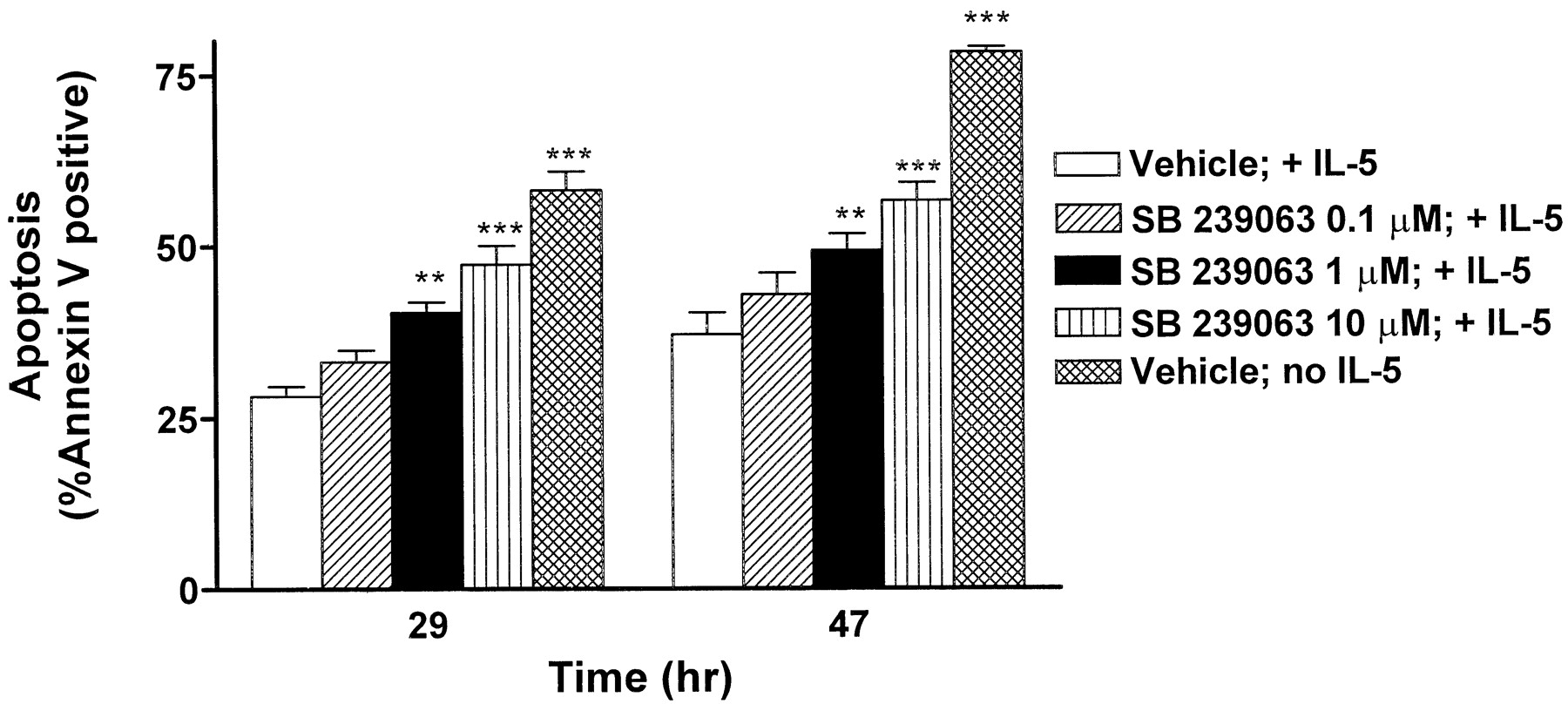
Effect of SB239063 on the onset of apoptosis in cultured eosinophils isolated from guinea pig BALs. Eosinophils were isolated from lavages as described in Materials and Methods and cultured in the presence of 10 pM IL-5 with and without various SB 239063 concentrations. At various time points, cultures were harvested and dual-labeled with PI and Annexin-V-FITC. Each column represents the mean percentage of Annexin-V-positive cells from three separate experiments at the 29 and 47 h time points. Asterisks indicate differences from vehicle control + IL-5 (**P < .01; ***P < .001).
Histology.
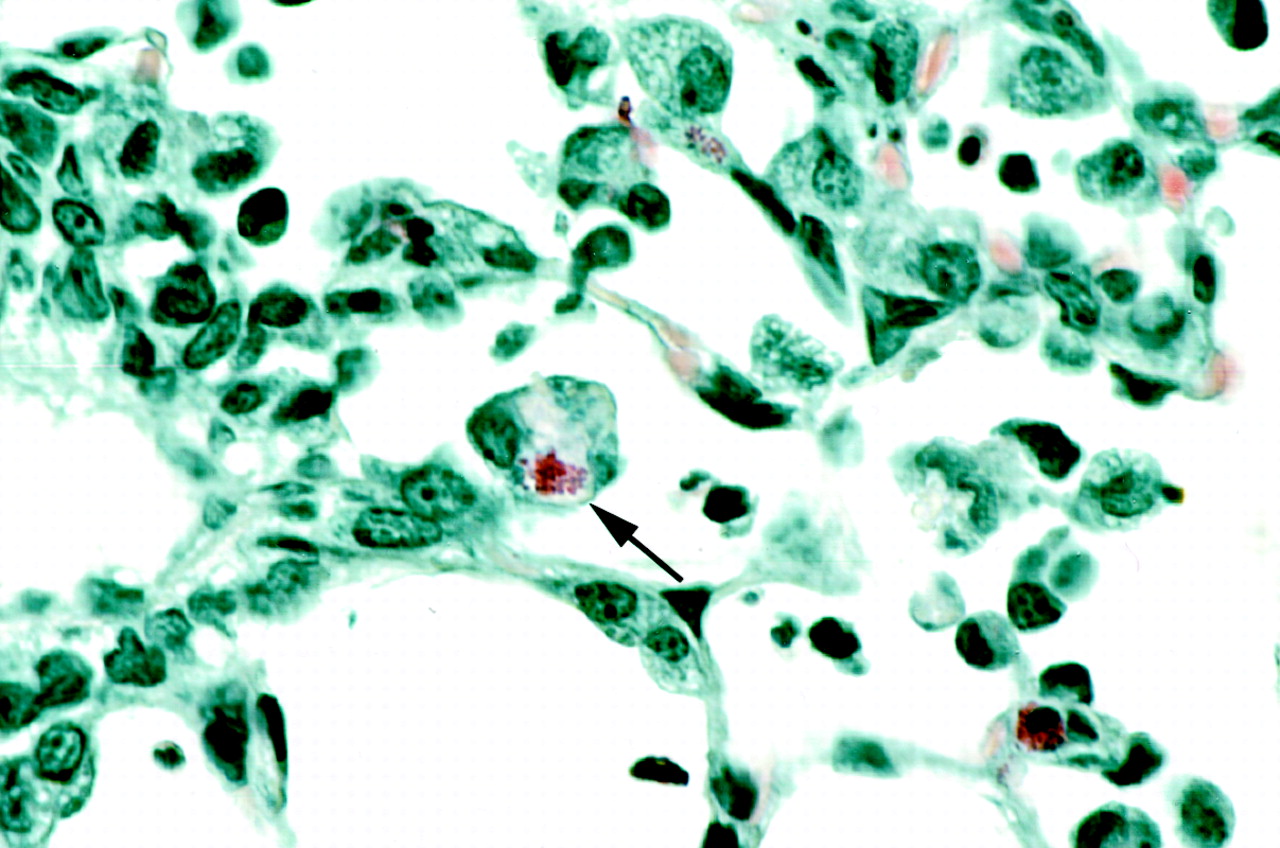
The microscopic scan of the airways of LTD4-challenged guinea pigs revealed several unique observations of phagocytosis of eosinophils by alveolar macrophages (Fig. 8), not recognized in a previous study (Underwood et al., 1996). As shown in Fig. 8, the granules of the macrophage-engulfed eosinophils remained intact. Although histological quantitation of airway eosinophilia was not the focus of the present studies, generally increased numbers of eosinophils were observed in the epithelium and subepithelial connective tissue of bronchi and bronchioles in LTD4-exposed guinea pigs compared with those of representative unexposed animals.

Phagocytosis of eosinophils by alveolar macrophages in the guinea pig airway. Guinea pig trachea and lungs were fixed, sectioned at 5-μm thickness, and stained with Luna's stain for eosinophils. The photomicrograph shown was taken at 830× magnification. The arrow indicates an engulfed eosinophil with intact granules.
Discussion
The results of the present study clearly demonstrate the efficacy of the novel, potent, and selective p38 MAP kinase inhibitor SB 239063 in reducing proinflammmatory cytokine production, leading to diminished leukocytes trafficking toward and enhanced clearance from a pulmonary inflammatory site. The major findings of this study include 1) the demonstration of potent inhibitory activity against p38 MAP kinase (α- and β- but not γ- and δ-isoforms) without direct inhibition of other kinases involved in the inflammatory process, e.g., c-Jun NH2-terminal kinase; 2) inhibition of proinflammatory cytokine production in intact cells; 3) in vivo inhibition of LPS-induced TNF-α production in mice; 4) inhibition of antigen-induced airway eosinophilia in both mice and guinea pigs; 5) enhanced clearance of inhaled LTD4-induced persistent airway eosinophilia; 6) augmented apoptosis of eosinophils cultured from guinea pig airways; and 7) demonstration of activated p38 kinase in mouse and guinea pig lung.
SB 239063 demonstrates comparable potency, but improved selectivity, toward the p38 MAP kinase than the earlier generation p38 kinase inhibitor SB 203580. Unlike SB 203580, SB 239063 was inactive against c-Raf; SB 203580 had an IC50 of 360 nM (Lee et al., 1999). Although multiple cell types, both inflammatory and structural, have been postulated to be involved in the pathology of asthma, clearly the eosinophil has received the most attention recently. In airways disease, especially asthma, the accumulation and activation of eosinophils appear to contribute to the development and maintenance of airway inflammation by releasing proinflammatory cytokines, lipid mediators, cytotoxic cationic proteins, and oxygen radicals (Giembycz and Lindsay, 1999). In addition, although more than 50 different mediators have been implicated in asthma, the eicosanoids (e.g., leukotriene B4, LTD4), cytokines (TNF-α, IL-5, etc.), and chemokines (RANTES, eotaxin, and IL-8) have received recent focus because of the availability of appropriate detection antibodies and the demonstrated efficacy of selective antagonists and inhibitors (Barnes et al., 1998). The ability of the p38 MAP kinase inhibitor SB 239063 to inhibit antigen-induced accumulation of eosinophils in the airways of both mice and guinea pigs was demonstrated in the present study, suggesting an inhibition of chemotaxis. Over the past decade, eosinophil trafficking in the guinea pig lung has been comprehensively studied in our laboratory. Although other mediators are probably involved, much of the activity has been associated with the formation and action of lipoxygenase products, especially the cysteinyl leukotrienes, and cytokines such as IL-5 (Underwood et al., 1996). Similar studies in mice have provided strong evidence of the involvement of Th2-dependent cytokines IL-4 and IL-5 in allergen-mediated lung eosinophilia (for review, see Selig and Chapman, 1999). Although these cytokines were not measured in the present study, the potent inhibitory activity of a close structurally related p38 kinase inhibitor HEP689 (SB 235699) against IL-4 production in a murine allergic skin model has been demonstrated (Aaes et al., 1999). In other studies from our own laboratory with a murine chronic contact sensitivity model featuring a Th2-dependent up-regulation of IL-4 production that correlates with an inflammatory infiltrate that includes eosinophils (Webb et al., 1998), we demonstrated a marked reduction (52%) in tissue levels of IL-4 and inflammation by topical treatment with SB 239063 (unpublished observations). In addition, chemokines such as RANTES and eotaxin have been recognized as potential contributors to the pathophysiology of asthma (Barnes et al., 1998). Additional support for a role for p38 MAP kinase comes from a recent study by Hashimoto et al. (1999) who demonstrated that TNF-α stimulates RANTES production in human pulmonary vascular endothelial cells. This effect is inhibited by the p38 MAP kinase inhibitor SB 203580. Because RANTES plays an important role through its chemotactic activity for eosinophils, this study supports the contention that the p38 pathway is important in the production of allergic inflammation of the airways. Further evidence for a role for p38 kinase in allergic disorders was the demonstrated inhibition of IgE synthesis via inhibition of cellular differentiation antigen 23 expression by the p38 MAP kinase inhibitor SB 203580 (Marshall et al. 1998).
In the conscious guinea pig model, SB 239063 had no inhibitory activity against the antigen-induced acute bronchoconstriction. Acute bronchospasm in this model is produced almost exclusively by mast-cell mediators, predominantly histamine, with modest contributions from eicosanoids such as prostaglandin D2 and cysteinyl leukotrienes in later phases (Selig and Chapman, 1999). The present animals had been pretreated with only enough antihistamine to prevent apnea and collapse due to anaphylaxis; we have demonstrated that higher doses of antihistamine provide additional attenuation of bronchospasm (Underwood et al., 1998b). Therefore, acute treatment with SB 239063 appears to provide no substantial direct stabilization of mast cell release of preformed histamine or H1 histamine receptor antagonism or inhibition of cysteinyl leukotriene release at the doses used in these experiments. Because modest release of cysteinyl leukotrienes may occur with mast cell degranulation, this direct chemotactic activity of this eicosanoid may account for the small residual eosinophilia remaining in SB 239063-treated animals.
Only recently has the persistence and maintenance of lung eosinophilia been evaluated. Although allergen-induced eosinophilia may be fleeting (i.e., 12–48 h) with a single provocation, and multiple provocations may help maintain some degree of low-level chronicity, the recent development of a chronic model in guinea pigs exposed to inhaled LTD4 in which airway accumulation peaks after 96 h and remains plateaued for at least 2 weeks has allowed us to test the efficacy of drugs on the persistence of eosinophils in the airways (Underwood et al., 1996, 1998a). In the present experiments, SB 239063 substantially reduced the persistent airway eosinophilia, suggesting a more complex activity additional to simple inhibition of chemotaxis into the airways. The demonstration of enhanced apoptosis that may signal for phagic capture by alveolar macrophages (Stern et al., 1992), as shown histologically in the present study, provides an additional unique mechanism by which p38 kinase inhibitors may provide a therapeutic benefit in chronic airways inflammation. It has been consistently demonstrated that human cultured eosinophils purified from peripheral blood undergo rapid apoptosis when placed into primary culture (Stern et al., 1992; Walsh, 1997; Kankaanranta et al., 1999); this phenomenon is diminished with IL-5 supplementation (Yamaguchi et al., 1991). Although some variability occurred with respect to spontaneous apoptosis among eosinophil populations from subsets of patients, enhanced in vitro apoptosis was clearly shown in the presence of the earlier generation p38 MAP kinase inhibitors, SB 203580 and SB 202190 Kankaanranta et al., 1999). The present study with the more selective inhibitor SB 239063 in a persistent population of guinea pig eosinophils isolated from the lung is consistent with that finding. However, SB 239063 was capable of enhancing apoptosis in guinea pig eosinophils isolated from lung in the presence of IL-5, thereby overcoming the survival-enhancing activity of low concentrations of this cytokine, an observation different from the findings by Kankaanranta et al. (1999). Whether this difference is due to the source of eosinophils (human peripheral versus guinea pig lung), the greater selectivity of SB 239063 compared with SB 203580 relative to other kinase pathways such as c-Raf, or to other technique differences is not known. Nonetheless, both studies have demonstrated that p38 activation plays an important role in eosinophil survival.
Of major importance is the present demonstration of activated p38 MAP kinase in the lung where cell-cell interactions may play an equally important role in not only the survival of inflammatory cells but also in the activation state of these cells. The consistent histologic finding of eosinophils, infiltrated in response to aerosol exposure to LTD4, in lung tissue engulfed by macrophages, with demonstrable granules intact, is evidence of the potential to enhance a naturally occurring neutralization of the pathogenesis of this granulocyte.
The combined activities of p38 kinase inhibition, that is, attenuation of proinflammatory cytokine formation, inhibition of chemotaxis of eosinophils into the airways, and the enhancement of apoptosis to signal phagic engulfment by alveolar macrophages, may provide a multipronged potential therapeutic approach toward chronic airways disease management. Thus, potent and selective p38 kinase inhibitors, such as SB 239063, may have therapeutic utility in lung diseases such as chronic obstructive pulmonary disease and asthma.
Acknowledgments
We thank Brendan O'Connell and Thomas Covatta for technical assistance in tissue analysis and photography, respectively, for this manuscript.
Footnotes
-
Send reprint requests to: David C. Underwood, Ph.D., SmithKline Beecham Pharmaceuticals, Department of Pulmonary Pharmacology, UW2532, 709 Swedeland Rd., King of Prussia, PA 19406-0939. E-mail: David_C_Underwood{at}sbphrd.com
- Abbreviations:
- MAP
- mitogen-activated protein
- TNF-α
- tumor necrosis factor-α
- IFN-γ
- interferon-γ
- IL
- interleukin
- RANTES
- regulated on activation normal T-cell expressed and secreted
- SB 239063
- trans-1-(4-hydroxycyclohexyl)-4-(4-fluorophenyl)-5-(2-methoxypyridimidin-4-yl)imidazole
- LPS
- lipopolysaccharide
- ELISA
- enzyme linked immunosorbant assay
- DPBS
- diphosphate-bufferred saline
- LTD4
- leukotriene D4
- FITC
- fluorescein isothiocyanate
- PI
- propidium iodide
- BAL
- bronchoalveolar lavage
- OA
- ovalbumin
- PLSD
- protected least significant difference
- Received October 6, 1999.
- Accepted December 21, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}