


Abstract
Multidrug resistance in tumor cells is often associated with reduced drug accumulation resulting from increased expression of the 190-kDa multidrug resistance protein 1 (MRP1) or the 170-kDa P-glycoprotein. However, unlike P-glycoprotein, MRP1 is a primary active transporter of many conjugated organic anions, including the cysteinyl leukotriene LTC4. Moreover, agents such as verapamil that reverse P-glycoprotein-mediated resistance are often poorly, or not at all, effective in MRP1-overexpressing cells. In the present study, we investigated the effects of verapamil on MRP1-mediated transport processes. We found that verapamil inhibited LTC4 transport into inside-out membrane vesicles prepared from MRP1-transfected cells in a competitive manner, but only in the presence of reduced glutathione (GSH) or its nonreducing S-methyl derivative. In the presence of 1 mM GSH, the apparentKi for verapamil was 1.2 μM, and in the presence of 100 μM verapamil, the apparentKi for GSH was 77 μM. Verapamil itself was not transported by MRP1 in either intact cells or membrane vesicles. However, verapamil strongly stimulated MRP1-mediated GSH uptake by membrane vesicles in a concentration-dependent and osmotically sensitive manner that was inhibitable by MRP1-specific monoclonal antibodies. In the presence of 100 μM verapamil, the apparentKm and Vmax for GSH uptake were 83 μM and 55 pmol mg−1min−1, respectively. It is proposed that the variable ability of verapamil to modulate MRP1-mediated resistance in different cell lines may be more closely linked to its effect on the GSH status of the cells than on its ability to inhibit the MRP1 transporter itself.
Resistance to multiple chemotherapeutic agents is a major problem that impedes or prevents the successful treatment of many infectious and malignant diseases. Multidrug resistance is often accompanied by reduced drug accumulation, and in mammalian tumor cells, increased expression of either the 170-kDa P-glycoprotein or the 190-kDa multidrug resistance protein 1 (MRP1) is frequently observed (Lautier et al., 1996; Hipfner et al., 1999a). Several clinical studies indicate that the presence of these drug resistance proteins can be of prognostic significance. Of particular interest is a recent report where detection of either MRP1 or P-glycoprotein in retinoblastoma samples was found to correlate with failure of patients to respond to chemotherapy, whereas the absence of both proteins correlated with long-term survival of patients with this disease (Chan et al., 1997).
Both MRP1 and P-glycoprotein are members of the ATP-binding cassette (ABC) superfamily of transport proteins but share <20% amino acid identity (Cole et al., 1992). P-glycoprotein, like many ABC proteins, contains two membrane-spanning domains and two nucleotide-binding domains arranged in a tandemly duplicated configuration and with both the NH2 and COOH termini of the protein located in the cytoplasm (Gottesman and Pastan, 1993). In contrast, MRP1 and several other closely related ABC proteins contain an additional third NH2 proximal membrane-spanning domain with an extracytosolic NH2 terminus (Cole et al., 1992;Hipfner et al., 1999a). Despite the lack of similarity between the primary and secondary structures of MRP1 and P-glycoprotein, tumor cells expressing either of these proteins display resistance to many commonly used natural product oncolytic drugs. However, there are significant differences in the substrate transport characteristics and certain other pharmacological properties of the two proteins (Hipfner et al., 1999a). For example, studies with membrane vesicles or purified reconstituted protein have demonstrated that P-glycoprotein binds and actively transports the drugs to which it confers resistance (Shapiro and Ling, 1995; Sharom, 1997). In contrast, under similar conditions, direct transport of drugs by MRP1 is not observed although there is considerable evidence that certain xenobiotics may be transported by MRP1 in association with reduced glutathione (GSH) (Loe et al., 1996,1997, 1998; Renes et al., 1999). MRP1 is also a primary active transporter of a variety of structurally diverse conjugated organic anions (Hipfner et al., 1999a). Potential physiological substrates of MRP1 include both GSH and glucuronide-conjugated endobiotics [e.g., the cysteinyl leukotriene LTC4, 17β-estradiol 17-(β-d-glucuronide), and GSH-conjugated prostaglandin A2] as well as the oxidized form of GSH, glutathione disulfide (GSSG). Conjugates of various xenobiotics also have been shown to be actively transported by MRP1 (Loe et al., 1997;Keppler et al., 1997; Suzuki and Sugiyama, 1998; Hipfner et al., 1999a). In contrast, these anionic molecules are poorly, if at all, transported by P-glycoprotein.
Substantial efforts have been directed toward the identification of agents capable of reversing resistance mediated by MRP1 and P-glycoprotein because of the potential clinical importance of these proteins. The use of antisense oligonucleotides to inhibit synthesis of these proteins has been one strategy with great promise because of its potentially high degree of specificity (Stewart et al., 1996). However, the more commonly taken approach has been to screen small molecules for their chemosensitizing activity. A large number of structurally diverse chemicals have been reported to inhibit the drug efflux activity of P-glycoprotein (Sharom, 1997), but the most extensively studied is the calcium channel blocker verapamil, a diphenylalkylamine derivative commonly used to treat mild hypertension and certain other cardiovascular disorders (Tsuruo et al., 1981). Verapamil has long been known to be capable of inhibiting the binding of photoactivatable drug analogs to P-glycoprotein, restoring drug accumulation and enhancing drug sensitivity of cultured cells that overexpress this protein (Tsuruo et al., 1981; Sharom, 1997). Verapamil also has been investigated for its drug resistance reversing activity in several clinical trials but optimal testing of its efficacy has been hampered by the dose-limiting cardiovascular toxicity associated with its administration (Dalton et al., 1989). In contrast to P-glycoprotein-mediated resistance, verapamil has been reported in most instances to be only weakly, or not at all, effective at restoring drug sensitivity in cells overexpressing MRP1, or the compound has been found to have a comparable effect on parental drug-sensitive control cells (Cole et al., 1989, 1994; De Jong et al., 1990; Cole, 1992;Barrand et al., 1993). However, in some MRP1-expressing model systems, verapamil has been observed to have a moderate chemosensitizing effect (Binaschi et al., 1995; Davey et al., 1995;Doyle et al., 1995). The basis for this apparently variable effect of verapamil on MRP1-associated resistance is unknown.
To clarify the effects of verapamil on MRP1-mediated drug resistance, we have investigated the ability of this drug to inhibit direct transport of the well characterized MRP1 substrate LTC4. We have determined that verapamil inhibits LTC4 transport into inside-out MRP1-enriched membrane vesicles in a competitive manner, but only in the presence of GSH or its nonreducing S-methyl derivative. We also have established that verapamil itself is not transported by MRP1 in either intact cells or membrane vesicles in the presence or absence of GSH. In contrast, we found that verapamil is a potent stimulator of MRP1-mediated GSH uptake by inside-out membrane vesicles. Our observations suggest that the ability of verapamil to enhance the drug sensitivity of MRP1-overexpressing cells may be more closely linked to its effect on cellular GSH levels than on its ability to inhibit the MRP1 transporter itself.
Experimental Procedures
Materials.
[N-methyl-3H]verapamil (60.8 Ci mmol−1), [glycine-2-3H]GSH (40–44.8 Ci mmol−1), and [3H]LTC4 (165 Ci mmol−1) were obtained from DuPont NEN (Markham, Ontario, Canada). Nucleotides, GSH, verapamil,S-methylglutathione, GSSG, 2-mercaptoethanol, and dithiothreitol (DTT) were purchased from Sigma Chemical Co. (St. Louis, MO). LTC4 was purchased from CalBiochem (La Jolla, CA). The MRP1-specific murine monoclonal antibodies (mAbs) QCRL-1, QCRL-2, QCRL-3, and QCRL-4 have been described previously and were purified before use (Hipfner et al., 1999b).
Cell Culture.
T14 cells are a multidrug resistant population of HeLa cells obtained by stable transfection with the episomally replicating MRP1 cDNA expression vector pCEBV7-MRP1 (Cole et al., 1994). C6 cells are a control population of HeLa cells transfected with the pCEBV7 vector alone. Cells were maintained in RPMI 1640 medium with 10% bovine calf serum and 100 μg ml−1hygromycin.
Preparation of Membrane Vesicles.
Plasma membrane vesicles were prepared as described with modifications (Loe et al., 1996). Cells were homogenized in buffer containing 50 mM Tris-HCl, 250 mM sucrose, 0.25 mM CaCl2 (pH 7.5), and protease inhibitors. Cell pellets were frozen at −70°C for at least 1 h, thawed, and then disrupted by N2 cavitation. EDTA was added to 1 mM and after centrifugation at 500g for 15 min, the supernatant was layered over 35% (w/w) sucrose in 10 mM Tris-HCl, 1 mM EDTA, and centrifuged at 100,000g for 2 h. The interface was collected and washed twice by centrifugation. The membrane pellet was resuspended in transport buffer (50 mM Tris-HCl and 250 mM sucrose, pH 7.5) and passed 20 times through a 27-gauge needle for vesicle formation.
Membrane Vesicle Transport Studies.
ATP-dependent uptake of [3H]LTC4 was measured by a rapid filtration technique as described previously (Loe et al., 1996). Transport assays were performed at 23°C with 50 nM substrate (50 nCi per reaction) in a 50-μl reaction containing 2 to 4 μg of vesicle protein. Uptake was stopped after 30 s by rapid dilution in ice-cold buffer, and then the reaction was filtered through glass fiber (Type A/E) filters (Gelman Sciences, Dorval, Québec, Canada) that had been presoaked in transport buffer. All data were corrected for the amount of [3H]LTC4 that remained bound to the filter, which was usually 5 to 10% of the total radioactivity, as well as for the amount of vesicle-associated [3H]LTC4 in the presence of AMP alone. For kinetic analysis of LTC4transport in the presence of verapamil and/or GSH, LTC4 was included at concentrations ranging from 12.5 nM to 1 μM and ATP-dependent [3H]LTC4 uptake was determined as described above.
Transport of [3H]verapamil into membrane vesicles also was measured by rapid filtration with membrane vesicles (20–25 μg of protein in a 50-μl reaction volume) that were incubated at 37°C in the presence of 200 nM [3H]verapamil (0.3 μCi per reaction), 10 mM DTT, 10 mM MgC12, and 4 mM ATP or AMP. Where indicated, GSH was added to 5 mM. Uptake was stopped by filtration through glass fiber filters as before. All data were corrected for the amount of [3H]verapamil that remained bound to the filter, which was usually 10 to 15% of the total radioactivity.
Transport of [3H]GSH into membrane vesicles also was measured by rapid filtration as described above with membrane vesicles (20–25 μg of protein per 50-μl reaction volume) incubated at 37°C with 20 nM [3H]GSH (40 nCi per reaction), 10 mM DTT, 10 mM MgCl2, and 4 mM ATP or AMP. To minimize GSH catabolism by γ-glutamyltranspeptidase during transport, membranes were preincubated in 0.5 mM acivicin for at least 1 h before measuring [3H]GSH uptake. Verapamil was dissolved in dimethyl sulfoxide and added to 100 μM unless otherwise indicated. The final concentration of dimethyl sulfoxide did not exceed 1%, and in control experiments, had no effect on [3H]GSH uptake. MRP1-specific mAbs were added to 10 μg ml−1 where indicated. All data were corrected for the amount of [3H]GSH that remained bound to the filter, which was usually <5% of the total radioactivity. Data also were corrected for the amount of vesicle-associated [3H]GSH in the presence of AMP or ATP alone, but in the absence of 100 μM verapamil, where indicated. For kinetic analysis of GSH transport in the presence of verapamil (100 μM), GSH was included at concentrations ranging from 16 nM to 675 μM and ATP-dependent [3H]GSH uptake was determined as described above.
[3H]Verapamil Accumulation in Intact Transfected HeLa Cells.
Accumulation of 3H-labeled verapamil in intact cells was measured as previously described (Cole et al., 1994). Briefly, HeLa C6 or T14 cells (1.25 × 106 cells ml−1) were incubated at 37°C in the presence of 1 μM [3H]verapamil (0.3 μCi ml−1) in RPMI 1640 medium supplemented with 5 mM HEPES (pH 7.0), 10 mM glucose, and 5% fetal bovine serum. Aliquots of suspended cells were removed at selected times and accumulation of [3H]verapamil was stopped by rapid dilution into ice-cold PBS. The earliest time point measured was ∼10 s. Cells were centrifuged and washed twice with 1 ml of ice-cold PBS. After recentrifugation, cell pellets were solubilized in 1% SDS and cell-associated radioactivity was determined by liquid scintillation counting. Results were expressed as picomoles of verapamil per 106 cells.
Results
Effect of Verapamil and GSH on [3H]LTC4Uptake by MRP1-enriched Membrane Vesicles.
Uptake of LTC4 into MRP1-enriched membrane vesicles was examined, and as shown in Fig. 1 was unaffected by verapamil alone. We and others have observed previously that although chemotherapeutic agents by themselves are relatively poor inhibitors of MRP1-mediated LTC4 transport, the inhibitory potency of some of them could be enhanced by GSH (Loe et al., 1996, 1998). For this reason, we investigated whether GSH also could enhance the ability of verapamil to inhibit uptake of LTC4 into membrane vesicles. GSH alone (1 mM) did not inhibit [3H]LTC4transport, in agreement with results from earlier studies (Loe et al., 1996, 1998; Leier et al., 1996; Renes et al., 1999). However, a combination of verapamil (100 μM) and GSH (1 mM) inhibited LTC4 transport by >90% (Fig. 1). Similarly, verapamil alone had no effect on MRP1-mediated transport of either 17β-estradiol 17-(β-d-glucuronide) or vincristine, but in the presence of GSH, transport of these compounds was inhibited >80% (data not shown).

Effect of verapamil on [3H]LTC4 uptake by membrane vesicles from MRP1-transfected HeLa T14 cells. Membrane vesicles prepared from MRP1-transfected HeLa cells (T14) were incubated with 50 nM [3H]LTC4 in transport buffer for 30 s at 23°C with reducing agents (GSH, 2-mercaptoethanol, or DTT) or GSH derivatives (S-methyl GSH or GSSG) at the indicated concentrations and in the presence (hatched columns) or absence (solid columns) of 100 μM verapamil. Data were calculated as percentage of control uptake in the absence of verapamil and GSH (■). Verapamil by itself had no effect on LTC4 uptake (▩). Columns represent means of triplicate determinations (±S.E.) in a typical experiment. The control [3H]LTC4 uptake rate in this experiment was 126 ± 1 pmol mg−1min−1. 2-ME, 2-mercaptoethanol; S-MeGSH,S-methyl GSH.
We have previously shown that the nonreducing S-methyl derivative of GSH is able to support MRP1-mediated vincristine transport to steady-state levels that are ∼30% of those observed with GSH (Loe et al., 1998). It was therefore of interest to determine the effect of S-methyl GSH on LTC4transport, both in the presence and absence of verapamil. When membrane vesicles were incubated with S-methyl GSH (1 mM) alone, no inhibition was observed (Fig. 1). However, when S-methyl GSH was incubated together with verapamil, [3H]LTC4 transport was inhibited by ∼80%. In contrast, the sulfhydryl reducing agents DTT and 2-mercaptoethanol (5 mM) had no effect on the ability of verapamil to inhibit LTC4 transport. GSSG alone (50 μM) inhibited LTC4 transport by ∼30%, consistent with previous observations (Loe et al., 1998) and this inhibition was unaffected by addition of verapamil. Collectively, these results indicate that verapamil is capable of inhibiting MRP1-mediated LTC4 transport only in the presence of GSH or its methyl derivatives, and that the enhancing effect of GSH is not simply due to its sulfhydryl reducing properties.
[3H]Verapamil Uptake by Transfected HeLa Cells and Membrane Vesicles.
To determine whether verapamil itself might be a substrate for MRP1, [3H]verapamil accumulation was measured in intact control transfected C6 and MRP1-transfected T14 HeLa cells (Fig. 2A) and in inside-out membrane vesicles prepared from these cells (Fig.2B). Cellular accumulation of verapamil in both cell lines was rapid and reached a maximum of 72 to 82 pmol/106 cells between 5 and 30 min and then decreased to steady-state levels of 45 to 51 pmol/106 cells at 60 to 90 min (Fig. 2A). Thus, no differences in levels of cell-associated [3H]verapamil in intact control or MRP1-transfected HeLa cells were observed for up to 90 min. When [3H]verapamil uptake was measured in membrane vesicles prepared from MRP1-transfected T14 cells, very limited ATP-dependent uptake was observed in the first 3 min of incubation compared with uptake in the presence of AMP (Fig. 2B). Levels of vesicle-associated [3H]verapamil then declined to ∼4 pmol mg−1 between 3 and 10 min, which was not significantly different from steady-state levels found in the presence of AMP. Low level ATP-dependent transport observed at early time points also was observed in membrane vesicles prepared from vector-transfected control C6 cells (data not shown), indicating that uptake in the T14 vesicles is not mediated by MRP1. We and others have reported previously that transport of certain xenobiotics by MRP1 is significantly enhanced by the addition of physiological concentrations of GSH (Loe et al., 1996, 1997; Renes et al., 1999). However, the low-level uptake of [3H]verapamil by T14 membrane vesicles was similar in the presence and absence of 5 mM GSH (Fig. 2B). Collectively, these experiments suggest that verapamil itself is not actively transported by MRP1, nor is transport of verapamil stimulated by the addition of GSH.

Time course of [3H]verapamil uptake by transfected HeLa cells and membrane vesicles. A, [3H]verapamil (1 μM) accumulation by intact control transfected C6 (○) or MRP1-transfected T14 (●) HeLa cells was determined over 90 min at 37°C. Results shown represent means of triplicate determinations (±S.E.) in a single experiment. B, [3H]verapamil (200 nM) uptake by T14 membrane vesicles was determined over 15 min at 37°C as described inExperimental Procedures. Closed symbols indicate ATP-dependent uptake in the presence (▴) or absence (▪) of 5 mM GSH; open symbols represent AMP-dependent uptake in the presence (▵) or absence (■) of GSH. Data points are means of triplicate determinations (±S.E.) in a typical experiment. VRP, verapamil.
Verapamil Stimulated [3H]GSH Transport in MRP1-Enriched Membrane Vesicles.
GSH by itself is not actively transported by MRP1-enriched membrane vesicles (Loe et al., 1996, 1998;Leier et al., 1996; Renes et al., 1999). However, in the presence of vincristine, and to a lesser extent other chemotherapeutic agents, MRP1-mediated ATP-dependent [3H]GSH uptake can be observed (Loe et al., 1998). To investigate whether verapamil might have a comparable stimulatory effect on GSH transport, a time course of [3H]GSH uptake (initial concentration 100 μM) in the presence and absence of verapamil (100 μM) was determined (Fig. 3A). In the absence of verapamil, [3H]GSH uptake into MRP1-enriched T14 membrane vesicles was similar in the presence of either 4 mM AMP or ATP and was ∼0.3 nmol mg−1 at 30 min (Fig. 3A). However, with the addition of 100 μM verapamil, ATP-dependent uptake of [3H]GSH increased linearly at a rate of 75 pmol mg−1 min−1 for 20 min to a maximum of ∼1.5 nmol mg−1. Net verapamil-stimulatable GSH uptake (in the presence of ATP) after subtraction of uptake in the presence of ATP alone was ∼1.2 nmol mg−1 at 20 min. Steady-state levels of 1.5 to 1.6 nmol mg−1 were maintained for up to 90 min. Verapamil-stimulated [3H]GSH uptake also was osmotically sensitive (Fig. 3B), confirming that the vesicle-associated increase in [3H]GSH represented transport into the vesicle lumen rather than surface binding. Verapamil-stimulatable ATP-dependent [3H]GSH uptake was measured in the presence of several MRP1-specific mAbs shown previously to inhibit transport of LTC4 and other MRP1 substrates (Loe et al., 1996, 1997, 1998; Renes et al., 1999; Hipfner et al., 1999b). Thus, three mAbs that recognize distinct conformation-dependent epitopes in the first (mAbs QCRL-2 and QCRL-3) and second (mAb QCRL-4) nucleotide-binding domains of MRP1 (Hipfner et al., 1999b) completely inhibited verapamil-stimulatable [3H]GSH uptake by T14 vesicles at 10 μg ml−1 (Fig. 3C). In contrast, mAb QCRL-1, which recognizes a linear epitope in the linker region of MRP1, had no effect. Finally, the stimulation of [3H]GSH transport by verapamil was shown to be concentration-dependent (Fig. 3D). Concentrations of verapamil <10 μM had no detectable effect on [3H]GSH uptake. However, as the verapamil concentration increased from 10 to 100 μM, [3H]GSH transport increased linearly to a maximum of ∼1.2 nmol mg−1 over 20 min at 200 to 300 μM verapamil. Transport levels then decreased with increasing verapamil concentrations until at 1 mM verapamil, stimulation of ATP-dependent transport of [3H]GSH was no longer observed.

Effect of verapamil on [3H]GSH transport by MRP1-enriched T14 membrane vesicles. A, time course of [3H]GSH uptake (initial concentration 100 μM) by T14 membrane vesicles was determined over 90 min at 37°C in the presence or absence of 100 μM verapamil. ○ indicate uptake in the presence of 4 mM AMP, whereas ● indicate uptake in the presence of 4 mM ATP. ▴ represent uptake in the presence of 4 mM ATP and 100 μM verapamil. B, osmotic sensitivity of verapamil stimulated ATP-dependent [3H]GSH transport by T14 membrane vesicles was determined in transport buffer containing different concentrations of sucrose. ○ indicate uptake in the presence of AMP, whereas ● indicate uptake in the presence of ATP. ▴ represent uptake in the presence of ATP and 100 μM verapamil. C, inhibition of verapamil stimulated ATP-dependent [3H]GSH transport by MRP1-specific mAbs. Membrane vesicles from MRP1-transfected T14 cells were incubated with [3H]GSH in transport buffer in the presence or absence of AMP or ATP and in the presence or absence of 100 μM verapamil for 20 min (■). Transport of [3H]GSH also was measured in the presence of ATP and verapamil after preincubation of membrane vesicles on ice for 1 h with the indicated MAbs (10 μg ml−1) (▪). D, concentration dependence of verapamil stimulated ATP-dependent [3H]GSH uptake. T14 membrane vesicles were incubated for 20 min at 37°C in the presence of 100 μM [3H]GSH, 10 mM DTT, and 4 mM ATP and the indicated concentrations (2–1000 μM) of verapamil. [3H]GSH uptake in the presence of ATP alone (0.45 ± 0.01 nmol mg−1 at 20 min) was subtracted from each data point.
Kinetic Parameters of Verapamil Stimulatable ATP-Dependent [3H]GSH Uptake by MRP1-Enriched T14 Membrane Vesicles.
[3H]GSH uptake by T14 membrane vesicles in the presence of verapamil was linear for up to 20 min (Fig.3A), which allowed us to determine the kinetic parameters for GSH transport in the presence of both verapamil and ATP. Thus, initial rates of ATP-dependent [3H]GSH uptake were measured at 100 μM verapamil and GSH concentrations ranging from 16 nM to 675 μM (Fig. 4A). A Lineweaver-Burk plot of the data yielded an apparentKm of 83 μM and aVmax of 55 pmol mg−1 min−1 (Fig. 4B).
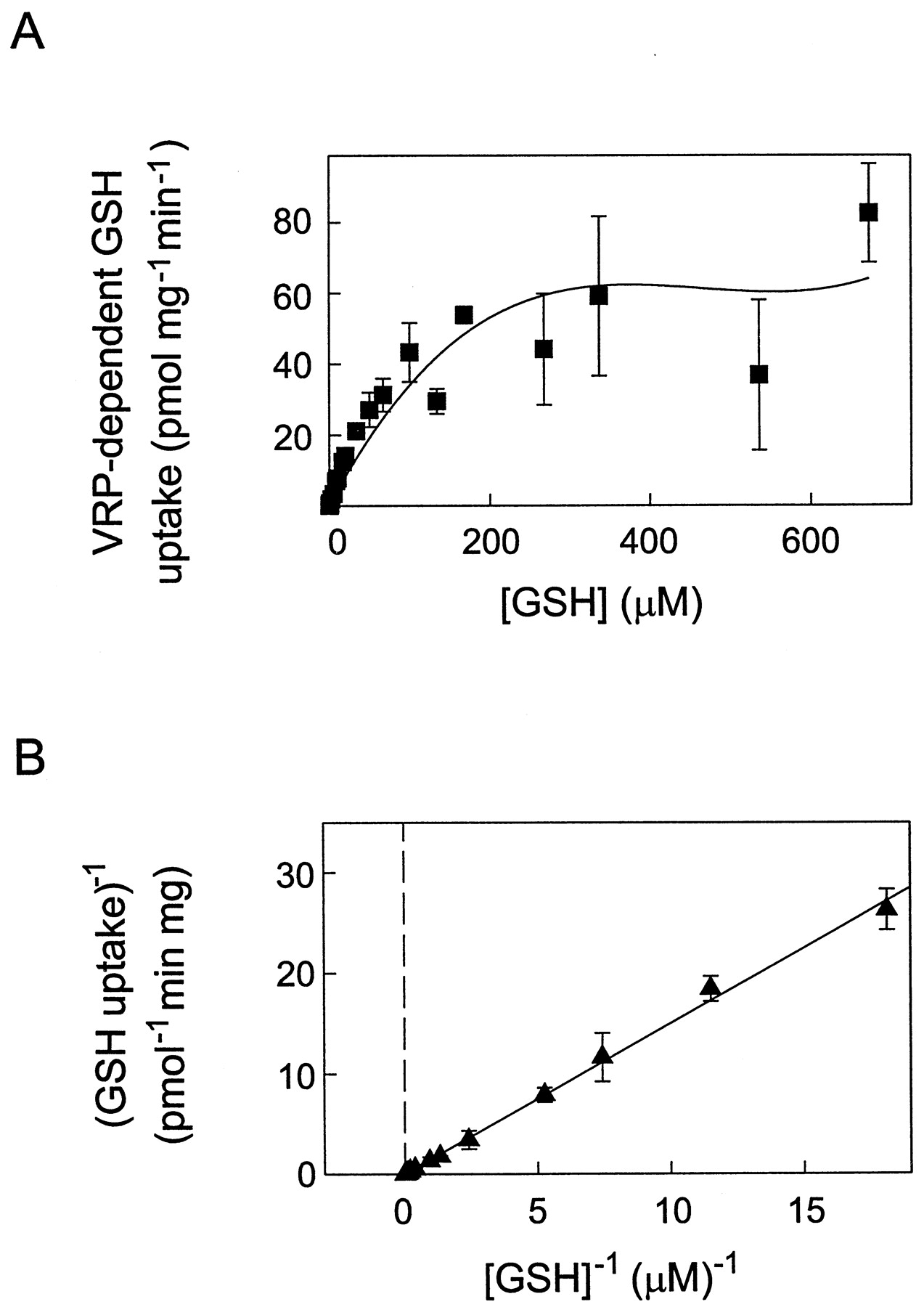
Michaelis-Menten and Lineweaver-Burk analyses of verapamil-stimulatable ATP-dependent [3H]GSH uptake by MRP1-enriched T14 membrane vesicles. A, [3H]GSH uptake by T14 membrane vesicles was measured at GSH concentrations ranging from 16 nM to 675 μM for 20 min at 37°C in the presence of 4 mM ATP, 10 mM DTT, and 100 μM verapamil. Data shown represent verapamil-stimulated ATP-dependent [3H]GSH uptake, determined by subtracting uptake values obtained in the presence of ATP alone from those values obtained in the presence of ATP and verapamil. Data were plotted as Vo versus [S] to confirm that the appropriate concentration range was selected to observe both zero-order and first-order rate kinetics. B, kinetic parameters were determined from regression analysis of the Lineweaver-Burk transformation of the data shown in A. The calculatedKm and Vmax for verapamil-stimulated ATP-dependent [3H]GSH uptake were 83 μM and 55 pmol mg−1 min−1, respectively. Data points in all cases represent means of three to six replicate determinations (±S.D.).
Characterization of Verapamil and GSH Inhibition of [3H]LTC4 Transport by MRP1-Enriched T14 Vesicles.
To further investigate the observation that GSH enhances the ability of verapamil to inhibit MRP1-mediated LTC4 uptake, we examined the concentration dependence of the inhibition with respect to both verapamil and GSH at a single substrate concentration (50 nM LTC4). In agreement with previous reports, GSH alone was a poor inhibitor of [3H]LTC4 transport with an IC50 of ∼12 mM (Fig.5A). However, this IC50 was reduced >100-fold to 100 μM in the presence of verapamil (100 μM), a concentration at which GSH alone does not significantly inhibit LTC4 transport (Fig. 5B). Similarly, in the presence of 5 mM GSH, the IC50 for verapamil was 3 μM, whereas in the absence of GSH, verapamil did not inhibit [3H]LTC4 transport, even at concentrations up to 500 μM.

Effect of verapamil and GSH on [3H]LTC4 transport by MRP1-enriched T14 membrane vesicles. A, ATP-dependent [3H]LTC4 transport (at an initial substrate concentration of 50 nM) was determined at concentrations of GSH ranging from 20 μM to 50 mM in the presence (▪) or absence (●) of 100 μM verapamil. Control [3H]LTC4 uptake rates in the absence and presence of verapamil were 115 ± 13 and 73 ± 4 pmol mg−1 min−1, respectively. B, ATP-dependent [3H]LTC4 transport was determined at various concentrations of verapamil ranging from 0.2 μM to 1 mM in the presence (▴) or absence (▾) of 5 mM GSH. Control [3H]LTC4 uptake rates in the absence and presence of 5 mM GSH were 114 ± 1 and 71 ± 1 pmol mg−1 min−1, respectively. C, concentration dependence of LTC4 (12 nM-1 μM) transport was determined in the presence of 5 mM GSH and verapamil (0 μM, ●; 3 μM, ■; and 6 μM, ▪). Double-reciprocal plots were generated and an apparent Ki (verapamil) of 1.2 μM was calculated from the apparent Km (157 nM) andVmax (532 pmol mg−1min−1) in the presence of 5 mM GSH and the indicated concentrations of verapamil. D, concentration dependence of LTC4 (12.5 nM-1 μM) transport was determined in the presence of 100 μM verapamil and GSH (0 μM, ▾; 200 μM, ⋄; 400 μM, ♦). Double-reciprocal plots were generated and an apparentKi (GSH) of 77 ± 6 μM was calculated from the apparent Km (97 nM) andVmax (532 pmol mg−1min−1) in the presence of 100 μM verapamil and the indicated concentrations of GSH. Data points in all cases represent means (±S.E.) of triplicate determinations in a typical experiment. VRP, verapamil.
The mode of inhibition of [3H]LTC4 transport by the combination of verapamil and GSH was further investigated by measuring LTC4 transport (at substrate concentrations ranging from 12.5 nM to 1 μM) in the presence of a constant concentration of GSH (5 mM) and three concentrations of verapamil (0, 3, and 6 μM). Similar experiments were carried out at a constant concentration of verapamil (100 μM) and three concentrations of GSH (0, 200, and 400 μM). Lineweaver-Burk analyses of the data indicated that verapamil and GSH together behave as competitive inhibitors of LTC4 transport, with apparentKi values of 1.2 ± 0.1 μM (verapamil) (Fig. 5C) and 77 ± 6 μM (GSH) (Fig. 5D).
Experiments similar to those described above were carried out to determine the [3H]LTC4transport inhibition characteristics of verapamil andS-methyl GSH. We found that S-methyl GSH alone was a relatively poor inhibitor of LTC4transport, with an IC50 of ∼3 mM. This value was reduced ∼15-fold to 0.2 mM in the presence of 100 μM verapamil. Similarly, although verapamil alone did not inhibit [3H]LTC4 transport, the IC50 for verapamil in the presence of 1 mMS-methyl GSH was ∼15 μM. The combination of verapamil and S-methyl GSH also showed competitive inhibition of LTC4 transport (data not shown).
Discussion
Verapamil is a diphenylalkylamine derivative that exhibits a myriad of biological activities that include its inhibitory effects on several ion channel proteins. One of the most clinically important interactions is with the voltage-gated L-type Ca2+ channel where verapamil binds to the open conformation of the pore-forming α1-subunit (Striessnig et al., 1990). Verapamil also has been shown to circumvent multidrug resistance associated with overexpression of P-glycoprotein in cultured tumor cell lines and in in vivo xenograft model systems (Tsuruo et al., 1981; Yin et al., 1989). However, this activity generally requires substantially higher concentrations of verapamil than are needed for ion channel inhibition, which in turn leads to cardiovascular toxicity when this compound is used as a chemosensitizer in vivo. In cultured tumor cells, chemosensitization has been attributed to the ability of verapamil to restore drug accumulation by binding to P-glycoprotein, thus preventing binding and efflux of cytotoxic drugs. Furthermore, verapamil itself is a substrate for P-glycoprotein and is a potent stimulator of the ATPase activity associated with this protein (Shapiro and Ling, 1995; Sharom, 1997). However, cells overexpressing P-glycoprotein are not cross resistant to verapamil (Sharom, 1997). Thus, it has been proposed that verapamil, and some other chemosensitizers of P-glycoprotein-mediated resistance, spontaneously equilibrate or flip-flop between lipid bilayers and thus are cycled in a futile manner and not actually transported (Eytan et al., 1996; Sharom, 1997).
The effect of verapamil on the resistance of MRP1-overexpressing cells is at best modest and is usually observed at even higher concentrations than are required to modulate P-glycoprotein-mediated resistance (Cole et al., 1989, 1994; Barrand et al., 1993). We have previously shown that verapamil enhances vincristine accumulation to a very limited extent in resistant MRP1-transfected HeLa cells and that a similar enhancement is observed in control-transfected HeLa cells (Cole et al., 1994). These observations suggest that any effect of this agent on the drug sensitivity of tumor cells cannot be solely attributable to a direct inhibition of MRP1 transport activity. Consistent with this conclusion, we have now shown that verapamil is a very poor inhibitor of MRP1-mediated transport activity in membrane vesicles with significant levels of inhibition observed at only at extremely high concentrations. Other investigators have reported that verapamil is a relatively poor inhibitor of the organic anion transport activities of the MRP1-related yeast cadmium resistance factor (YCF1) and the mammalian hepatocanalicular cyclic multispecific organic anion transporter (cMOAT)/MRP2. Thus, the IC50 for inhibition of ATP-dependent uptake of S-(2,4-dinitrophenyl) glutathione by YCF1 and cMOAT/MRP2 is >100 μM verapamil (Li et al., 1996) (A. Morikawa, Y. Goto, H. Suzuki, T. Hirohashi, and Y. Sugiyama, unpublished observations). This apparent difference in sensitivity of the MRP-related ABC proteins and P-glycoprotein-related ABC proteins to inhibition by verapamil is perhaps not surprising given the structural and substrate specificity differences displayed by these two subclasses of transporters (Hipfner et al., 1999a). It is also consistent with the ability of a verapamil photoaffinity analog to label the 170-kDa P-glycoprotein but not the 190-kDa MRP1 in membrane preparations from human leukemia cells expressing elevated levels of these proteins (McGrath et al., 1989). Finally, in contrast to its potent ability to stimulate P-glycoprotein ATPase activity (Shapiro and Ling, 1995; Sharom, 1997), we have observed no stimulation of MRP1 ATPase activity by verapamil in either the presence or absence of GSH in purified reconstituted preparations (unpublished observations).
In previous studies, we showed that inhibition of LTC4 transport in MRP1-enriched membrane vesicles by vincristine and certain other unconjugated xenobiotics could be stimulated significantly by GSH (Loe et al., 1996, 1997, 1998). We have now shown that GSH as well as its nonreducing S-methyl derivative also can markedly enhance verapamil inhibition of MRP1-mediated LTC4 transport. Similarly, 17β-estradiol 17-(β-d-glucuronide) and vincristine transport by MRP1 is inhibited by verapamil only in the presence of GSH (data not shown). In contrast, although GSH andS-methyl GSH can stimulate direct uptake of vincristine by MRP1 (Loe et al., 1998; Renes et al., 1999), these agents did not stimulate verapamil uptake. The ability of GSH to stimulate vincristine but not verapamil transport is in agreement with the finding that intact MRP1-transfected mammalian cells accumulate reduced levels of the former but not the latter drug (Fig. 2) (Cole et al., 1994; Zaman et al., 1995). These observations suggest that these two hydrophobic molecules interact with MRP1 in different ways despite the fact that they both show competitive inhibition of LTC4transport in the presence of GSH. The requirement for physiological concentrations of GSH to show inhibition of LTC4transport by verapamil suggests that verapamil binding to MRP1 is also dependent on the presence of GSH (or its nonreducingS-methyl derivative). Consistent with this idea has been the demonstration that MRP1 vanadate-induced trapping of nucleotide by anticancer drugs is stimulated by GSH (Taguchi et al., 1997). In view of these findings, it would be of interest to determine whether labeling of MRP1 with a verapamil photoaffinity analog also could be detected in the presence of GSH.
In contrast to the differing ability of GSH to stimulate uptake of vincristine and verapamil in MRP1-enriched membrane vesicles, both of these xenobiotics stimulate GSH uptake by MRP1 (Fig. 3). Stimulation of GSH uptake by verapamil, as shown previously for vincristine (Loe et al., 1998), is concentration dependent and can be attributed to active transport by MRP1 on the basis of its dependence on ATP, its saturability and osmotic sensitivity, and its inhibition by several MRP1-specific mAbs (Fig. 3). Verapamil appears to be a more potent stimulator of GSH transport than vincristine because uptake levels of GSH in MRP1-enriched T14 membrane vesicles in the presence of 100 μM drug differ by ∼6-fold (1.2 nmol GSH mg−1 20 min−1 versus 0.19 nmol GSH mg−1 20 min−1). Moreover, steady-state levels of verapamil-stimulatable GSH transport were achieved much more rapidly than steady-state levels of vincristine-stimulated transport. Our data are consistent with our previous model proposing a bipartite binding site on MRP1 for hydrophobic and anionic moieties that allows for binding of conjugated organic anions such as LTC4 as well as cooperative binding of drug and GSH (Loe et al., 1996). For the amphipathic drug vincristine, transport of both drug and GSH is observed. For a more hydrophobic drug such as verapamil, it may enter into futile recycling in the membrane (Eytan et al., 1996) so that only GSH transport is observed. Alternatively, subsequent to binding to MRP1, verapamil may be released on the cis side of the membrane without undergoing translocation across the bilayer. Further investigation is necessary to discriminate between these and other possibilities.
Both high- and low-affinity transport systems for reduced GSH have been kinetically characterized in several tissues, most extensively in the liver. However, the precise role(s) of ABC transporters in GSH homeostasis is not entirely clear. Our finding that MRP1 actively transports GSH in the presence of verapamil with relatively high affinity (Km = 83 μM) appears to contrast with recent studies of GSH transport by other MRP-related proteins. Thus, ATP-dependent GSH uptake into secretory vesicles by the yeast YCF1 protein has a reported Kmof ∼15 mM (Rebbeor et al., 1998). The active efflux of GSH by the more closely related mammalian cMOAT/MRP2 also has been described as a low-affinity process (Paulusma et al., 1999). In neither case, however, was GSH transport measured in the presence of verapamil. Moreover, although MRP1 and its most closely related proteins share several common substrates, the relative affinities of the different MRP-related ABC transporter proteins for some of these substrates appear to vary markedly (Keppler et al., 1997; Suzuki and Sugiyama, 1998). The structural basis of this variation in substrate affinities and specificities is currently the subject of intense investigation.
A role for MRP1 in cellular GSH homeostasis has been proposed previously on the basis of studies in mice in which the mrp1gene has been disrupted (Rappa et al., 1999). Thus, certain tissues in homozygous knock-out mrp1(−/−) mice displayed a tendency to contain higher intracellular concentrations of GSH than the same tissues in wild-typemrp1(+/+) mice. Moreover, exposure ofmrp1(+/+) embryonic stem cells to sodium arsenite or etoposide stimulated GSH efflux to a greater extent than inmrp1(−/−) stem cells. Because we and others have demonstrated that GSH alone is not actively transported by MRP1 in membrane vesicles, these observations suggest that MRP1-mediated export of GSH in the absence of an exogenous stimulus probably occurs in association with an endogenous metabolite(s), the identity of which is presently unknown (Zaman et al., 1995; Rappa et al., 1999; Hipfner et al., 1999a).
It is interesting to note that the Kmfor GSH transport in the presence of verapamil is similar to that found previously for oxidized GSSG (40–90 μM) (Leier et al., 1996) although the Vmax values for GSH and GSSG differ significantly in membrane vesicles expressing comparable levels of MRP1 [55 pmol min−1mg−1 versus 500 pmol min−1 mg−1]. Maintenance of low cellular GSSG levels and high GSH levels is important to regulate the redox potential of the cell. Under conditions of oxidative stress, when the rate of GSH oxidation exceeds the rate of GSSG reduction by GSH reductase, the GSSG transporting activity of MRP1 may play a crucial role in preventing GSSG from reacting with protein thiol groups. The wide tissue distribution of MRP1 and relatively high affinity and capacity for GSSG make it well suited for this function.
In contrast to GSSG, GSH concentrations in most cells under normal circumstances are in the millimolar range, suggesting that GSH transport systems with a low Km, such as reported for MRP2/cMOAT, might be more important for regulating cellular GSH levels. However, tissue distribution and membrane localization also are relevant considerations when attributing the relative physiological significance of various GSH transport systems. For example, MRP2/cMOAT is expressed at very low levels in all tissues except the liver where it is expressed at much higher levels than MRP1 (Paulusma et al., 1999). Moreover, the former protein is localized on apical (canalicular) membranes, whereas the latter protein is present in basolateral membranes (Evers et al., 1996). Hyperbilirubinemic rats in which MRP2/cMOAT is absent display a defect in biliary GSH excretion (Lu et al., 1996), and verapamil has been shown to stimulate biliary GSH efflux in a rat liver perfusion model (Karwinski et al., 1996). These observations suggest an essential role for the MRP2/cMOAT transporter in biliary GSH transport, although the physiological function of this process remains uncertain.
The physiological significance of the localization of MRP1 to basolateral membranes of polarized cells is not clear. Recently, MRP1 expressed in transfected polarized canine kidney cells was shown to increase GSH excretion across the basolateral membrane 4-fold, a process that was inhibited by depletion of ATP (Paulusma et al., 1999). Cysteine availability is often rate limiting in GSH biosynthesis and can be increased by ectoenzyme-mediated metabolism of plasma GSH thought to be primarily derived from the liver. It is possible that xenobiotics may stimulate MRP1-mediated GSH transport across hepatic basolateral membranes in response to signals for increased cysteine requirements for GSH biosynthesis elsewhere. Another somewhat more abundantly expressed MRP-related protein, MRP3, also is localized to basolateral hepatocyte membranes (Konig et al., 1999) but whether MRP3 is capable of GSH transport that can be stimulated by verapamil or any other xenobiotic is not yet known.
In summary, we have shown that verapamil is a poor inhibitor of MRP1-mediated LTC4 transport but strongly stimulates GSH transport by this protein. Thus, our data together with that of other investigators suggest that when verapamil is observed to enhance the drug sensitivity of MRP1-overexpressing cells, it is more likely the result of GSH efflux than the result of an increase in drug accumulation caused by inhibition of MRP1 itself (Cole et al., 1994;Versantvoort et al., 1995; Grech et al., 1998). This might explain, at least in part, the variable chemosensitizing activity of verapamil observed in different MRP1-overexpressing cell lines because GSH levels in these cell lines are known to vary markedly (Lautier et al., 1996).
Acknowledgments
We thank Libby Eastman and Kathy Sparks for able technical assistance and Maureen Rogers for skillful assistance in the preparation of the manuscript.
Footnotes
-
Send reprint requests to: Susan P. C. Cole, Ph.D., Cancer Research Laboratories, Room 328, Botterell Hall, Queen's University, Kingston, Ontario, Canada, K7L 3N6. E-mail:coles{at}post.queensu.ca
-
↵1 This study was supported by Grant MT-10519 from the Medical Research Council of Canada.
-
↵2 R.G.D. is the Stauffer Research Professor of Queen's University.
-
↵3 S.P.C.C. is a Senior Scientist of Cancer Care Ontario.
- Abbreviations:
- MRP1
- multidrug resistance protein 1
- ABC
- ATP-binding cassette
- GSH
- reduced glutathione
- LTC4
- leukotriene C4
- GSSG
- glutathione disulfide
- DTT
- dithiothreitol
- mAb
- monoclonal antibody
- cMOAT
- cyclic multispecific organic anion transporter
- Received November 8, 1999.
- Accepted January 19, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}