


Abstract
Neuronal nicotinic receptors are ligand-gated ion channels of the central and peripheral central nervous system that regulate synaptic activity from both pre- and postsynaptic sites. The present study establishes the acute interaction of bupropion, an antidepressant agent that is also effective in nicotine dependence, with nicotine and nicotinic receptors using different in vivo and in vitro tests. Bupropion was found to block nicotine's antinociception (in two tests), motor effects, hypothermia, and convulsive effects with different potencies in the present investigation, suggesting that bupropion possesses some selectivity for neuronal nicotinic receptors underlying these various nicotinic effects. In addition, bupropion blocks nicotine activation of α3β2, α4β2, and α7 neuronal acetylcholine nicotinic receptors (nAChRs) with some degree of selectivity. It was ∼50 and 12 times more effective in blocking α3β2 and α4β2than α7. This functional blockade was noncompetitive, because it was insurmountable by increasing concentration of ACh in the nAChRs subtypes tested. Furthermore, bupropion at high concentration failed to displace brain [3H]nicotine binding sites, a site largely composed of α4β2 subunit combination. Given the observation that bupropion inhibition of α3β2 and α4β2receptors exhibits voltage-independence properties, bupropion may not be acting as an open channel blocker. These effects may explain in part bupropion's efficacy in nicotine dependence. Our present findings suggest that functional blockade of neuronal nAChRs are useful in nicotine dependence treatment.
Despite heightened education and prevention strategies, cigarette smoking remains a major health risk. Nicotine is believed to be the primary reason that people consume tobacco products. Indeed, substantial evidence now shows that nicotine is the addictive substance found in tobacco. Neuronal acetylcholine nicotinic receptors (nAChRs) are likely sites at which nicotine exerts its psychoactive and addictive effects. Currently, the treatment of nicotine dependence consists of using different forms of nicotine replacement therapy to assist in smoking cessation. These methods gradually replace nicotine in an attempt to reduce cravings and withdrawal in smokers. Additionally, bupropion (Zyban), an atypical antidepressant, has been approved for smoking cessation (Hurt et al., 1997). It is interesting to note that bupropion appears to work equally well in smokers with and without a past history of depression (Hayford et al., 1999), suggesting that bupropion's efficacy is not due to its antidepressant effect. However, mechanisms by which bupropion reduces nicotine intake are still unclear. The current presumed mechanism of action of bupropion involves modulation of dopaminergic and noradrenergic systems that have been implicated in addiction (Ascher et al., 1995). Indeed, bupropion is a relatively weak dopamine-reuptake inhibitor and inhibits the firing of locus coerulus noradrenergic neurons at high concentrations (Cooper et al., 1994). In addition, bupropion has been found to lack binding affinity for almost all of the major classes of neuronal receptors, including serotonergic, dopaminergic, β-adrenergic, α1- and α2-adrenergic receptors, and muscarinic cholinergic receptors (Ascher et al., 1995). Recently, we have reported that bupropion blocked nicotine-induced antinociception in the tail-flick test after systemic administration (Damaj et al., 1999b). Furthermore, bupropion has been found to be a functional inhibitor of nAChRs in both the muscle and the ganglia (Fryer and Lukas, 1999). The functional blockade of bupropion was found to be insurmountable by increasing agonist concentrations, suggesting noncompetitive inhibition of nAChRs function. Further studies are needed to examine roles that neuronal nAChRs may play as targets for bupropion. Such studies may hold promise for treating affective disorders and for understanding and treating addictive processes.
In the present study, we have examined the mechanisms of the bupropion-nicotine interaction after acute administration using in vitro and in vivo assays. For that, the potency of bupropion to block various pharmacological effects of nicotine (antinociception, hypothermia, seizures, and motor impairment) in animals was examined. A wide range of nicotinic effects is important to consider, because it is believed that various nicotinic receptor subtypes mediate different pharmacological effects of nicotine (Damaj et al., 1999a). Using the oocyte expression system, the effects of bupropion on the functional activity of the neuronal nAChRs α4β2, α3β2, and α7 expressed receptors, were also studied.
Materials and Methods
Animals
Male ICR mice (20–25 g) obtained from Harlan Laboratories (Indianapolis, IN) were used throughout the study. They were housed in groups of six and had free access to food and water. Animals were housed in an Association for Assessment of Laboratory Animal Care approved facility, and the study was approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University.
Drugs
(−)-Nicotine was obtained from Aldrich Chemical Company, Inc. (Milwaukee, WI) and converted to the ditartrate salt as described byAceto et al. (1979). (−)-Epibatidine (hemi-oxalate salt) was supplied by Dr. S. Fletcher (Merck Sharp and Dohme, Essex, UK). Bupropion HCl was purchased from Research Biochemicals International (Natick, MA). Morphine was supplied by the National Institute on Drug Abuse (Washington, DC). All drugs were dissolved in physiological saline (0.9% sodium chloride) and given in a total volume of 1 ml/100 g of body weight for s.c. injections. All doses are expressed as the free base of the drug.
Intrathecal Injections
Intrathecal injections were performed free-hand between the L5 and L6 lumbar space in unanesthetized male mice according to the method of Hylden and Wilcox (1980). The injection was performed using a 30-gauge needle attached to a glass microsyringe. The injection volume in all cases was 5 μl. The accurate placement of the needle was evidenced by a quick “flick” of the mouse's tail. In protocols where two sequential injections were required in an animal, the flicking motion of the tail could be elicited with the subsequent injection.
Antinociceptive Tests
Tail-Flick Test.
Antinociception was assessed by the tail-flick method of D'Amour and Smith (1941) as modified by Dewey et al. (1970). A control response (2–4 s) was determined for each mouse before treatment, and a test latency was determined after drug administration. To minimize tissue damage, a maximum latency of 10 s was imposed. Antinociceptive response was calculated as percentage of maximum possible effect (%MPE), where %MPE = [(test − control)/(10 − control)] × 100. Groups of 8 to 12 animals were used for each dose and for each treatment. The mice were tested 5 min after either s.c. or intrathecal (i.t.) injections of nicotine. Antagonism studies were carried out by pretreating the mice with either saline or bupropion at different times before nicotine. The animals were tested 5 min after administration of nicotine.
Hot-Plate Test.
The method for this test is a modification of that described by Eddy and Leimback (1953) and Atwell and Jacobson (1978). Mice were placed into a 10-cm-wide glass cylinder on a hot-plate (Thermojust Apparatus) maintained at 55.0°C. Two control latencies at least 10 min apart were determined for each mouse. The normal latency (reaction time) was 6–10 s. Antinociceptive response was calculated as %MPE, as above. The reaction time was scored when the animal jumped or licked its paws. Eight mice per dose were injected s.c. with nicotine and tested 5 min after injection. Antagonism studies were carried out by pretreating the mice with either saline or bupropion at different times before nicotine. The animals were tested 5 min after administration of nicotine.
Behavioral Testing
Locomotor Activity.
Mice were placed into individual Omnitech photocell activity cages (28 × 16.5 cm) 5 min after s.c. administration of either 0.9% saline or nicotine. Interruptions of the photocell beams (two banks of eight cells each) were then recorded for the next 10 min. Data were expressed as number of photocell interruptions.
Body Temperature.
Rectal temperature was measured by a thermistor probe (inserted 24 mm) and digital thermometer (Yellow Springs Instrument Co., Yellow Springs, OH). Readings were taken just before and at 30 min after the s.c. injection of either saline or nicotine. The difference in rectal temperature before and after treatment was calculated for each mouse. The ambient temperature of the laboratory varied from 21–24°C from day to day.
Seizure Activity.
Following a s.c. injection of nicotine at a dose of 9 mg/kg, each animal was placed into a 30- × 30-cm Plexiglas cage and observed for 5 min. Whether a clonic seizure occurred within a 5-min time period was noted for each animal after s.c. administration of different drugs. This amount of time was chosen because seizures occur very quickly after nicotine administration. Results are expressed as the percentage of animals that seized. Antagonism studies were carried out by pretreating the mice s.c. with either saline or bupropion 15 min before nicotine.
(−)-[3H]Nicotine Binding in Vitro
(−)-[3H]Nicotine binding assays in rat brain were performed in vitro according to the method of Scimeca and Martin (1988) with minor modifications. Tissue homogenate was prepared from whole rat brain (minus cerebellum) in 10 volumes of ice-cold 0.05 M sodium-potassium phosphate buffer (pH 7.4) and centrifuged (17,500g, 4°C) for 30 min. The pellet was then resuspended in 20 volumes of ice-cold glass-distilled water and allowed to remain on ice for 60 min before being centrifuged as before. The resulting pellet was then resuspended to a final tissue concentration of 10 mg/ml buffer. Membranes from whole brain (0.2 ml of final suspension) were incubated at 4°C for 2 h with phosphate buffer and [3H]nicotine at the indicated concentrations in a total volume of 1 ml. Nonspecific binding was determined in the presence of 100 μM unlabeled nicotine. The incubation was terminated by rapid filtration through a Whatman GF/C glass fiber filter (presoaked overnight in 0.1% poly(l-lysine) to reduce radioligand binding to the filters). Filters were washed twice with 3 ml of the buffer, and radioactivity on the filters was measured using a liquid scintillation spectrometer. The Bmax andKD values, obtained from Scatchard analysis, were determined via the KELL package of binding analysis programs for the Macintosh computer (Biosoft, Milltown, NJ). The ability of bupropion to displace 1.5 nM [3H]nicotine binding was determined in the presence of increasing concentrations of bupropion.
Oocyte Expression Studies
Oocyte Preparation.
Oocytes preparation was performed according to the method of Mirshahi and Woodward (1995) with minor modifications. Briefly, oocytes were isolated from female adult oocyte-positive Xenopus laevis frogs. Frogs were anesthetized in a 0.2% 3-aminobenzoic acid ethyl ester solution (Sigma Chemical Co., St. Louis, MO) for 30 min, and a fraction of the ovarian lobes was removed. The eggs were rinsed in Ca2+-free ND96 solution, treated with Collagenase type IA (Sigma) for 1 h to remove the follicle layer, and then rinsed again. Healthy stage V to VI oocytes were selected and maintained for up to 14 days after surgery in 0.5× L-15 media.
mRNA Preparation and Microinjection.
α4, α3, α7, and β2 rat subunits cDNA contained within a pcDNAIneo vector were kindly supplied by Dr. James Patrick (Baylor College of Medicine, Houston, TX). The template was linearized downstream of the coding sequence, and mRNA was synthesized using an in vitro transcription kit from Ambion (Austin, TX). The quantity and quality of message were determined via optical density (spectrophotometer Beckman Instruments Inc., Chaumburg, IL) and denaturing formaldehyde gel analysis. Oocytes were injected with either 51 ng (41 nl) of α4 and β2 or α3 and β2 mRNA, each mixed in a 1:1 ratio using a variable microinjector (Nanoject; Drummond Scientific Co., Broomall, PA). Oocytes were incubated in 0.5× L-15 media IA (Sigma) supplemented with penicillin, streptomycin, and gentamicin for 4 to 6 days at 19°C before recording.
Electrophysiological Recordings.
Oocytes were placed within a Plexiglas chamber (total volume 0.2 ml) and continually perfused (10 ml/min) with buffer consisting of 115 mM NaCl, 1.8 mM CaCl2, 2.5 mM KCl, 1.0 μM atropine and 10.0 mM HEPES at pH 7.2. Oocytes were impaled with two microelectrodes containing 3 M KCl (0.3–3 MΩ) and voltage-clamped at −70 mV using a Geneclamp amplifier (Axon Instruments Inc., Foster City, CA). Oocytes were stimulated for 10 s with various concentrations of acetylcholine (ACh) and nicotine using a six-port injection valve. Except where noted, applications were separated by 5-min periods of washout. Currents were filtered at 10 Hz and collected by a Macintosh Centris 650 with a 16-bit analog-to-digital interface board, and data were analyzed using Pulse Control voltage-clamp software running under the Igor Pro graphic platform (Wavemetrics, Lake Oswego, OR). Bupropion was applied at different concentrations, and concentration-response curves were normalized to the current induced by 1 μM (α4β2 receptors) or 10 μM (α3β2 receptors) of ACh. The normalizing concentration of ACh was applied before and after drug application to each oocyte to check for desensitization. Data were rejected if responses to the normalizing dose fell below 75% of the original response.
Statistical Analysis
Data were analyzed statistically by an ANOVA followed by the Fisher least significant difference multiple comparison test. The null hypothesis was rejected at the 0.05 level. IC50 (antagonist concentration 50%) and AD50 (antagonist dose 50%) values with 95% confidence limits were calculated by unweighted least-squares linear regression as described by Tallarida and Murray (1987).
Results
Binding Experiments
The Scatchard analysis of [3H]nicotine binding provided a KD of 1.3 ± 0.15 nM and a Bmax of 245 ± 46 fmol/mg of protein. Nicotine inhibited binding of [3H]nicotine to rat brain membranes with aKi value of 1.4 ± 0.20 nM. Bupropion at 1 and 10 μM concentrations did not displace [3H]nicotine binding.
Antinociception Studies
Antagonism of Nicotine's Effects.
Bupropion was evaluated for its ability to antagonize a 2.5 mg/kg dose of nicotine in the tail-flick procedure. As shown in Fig.1A, bupropion dose dependently blocked nicotine-induced antinociception with an AD50 of 2.4 mg/kg when given s.c. 10 min before nicotine.

Blockade of nicotine-induced antinociception in the tail-flick test by bupropion after s.c. injection. A, dose-response blockade of bupropion after s.c. administration in mice. Bupropion at different doses was administered s.c. 15 min before nicotine (2.0 mg/kg, s.c.), and mice were tested 5 min later. B, time course of bupropion effect on nicotine-induced antinociception (2 mg/kg) after s.c. administration of 25 mg/kg in mice. Each point represents the mean ± S.E. of 8 to 12 mice. *P < .05 compared with correspondent zero time point.
The duration of action of bupropion in the tail-flick test was dose-dependent with maximum blockade occurring between 5 and 15 min for the 5 mg/kg dose. The duration of action of bupropion antagonized nicotine for up to 4 h. Indeed, as illustrated in Fig. 1B, nicotine's effect recovered within 30 min after pretreatment with a dose of 5 mg/kg bupropion but was still significantly different from control 240 min after the dose of 25 mg/kg.
The blocking effect of bupropion in the tail-flick test was also seen after spinal administration. Indeed, after i.t. administration of bupropion, the antinociceptive effect of nicotine (20 μg/animal, i.t.) was significantly reduced in a dose-dependent manner (Fig.2) with an AD50 of 9 μg/animal when given 5 min before nicotine. By itself, bupropion (after s.c. and i.t. injections) did not cause antinociception at the indicated doses and times (Table1). Similar to the tail-flick test, bupropion dose dependently blocked nicotine-induced antinociception as measured by the hot-plate test, with an AD50 of 8 mg/kg when given s.c. 10 min before nicotine (Fig. 3B and Table2).
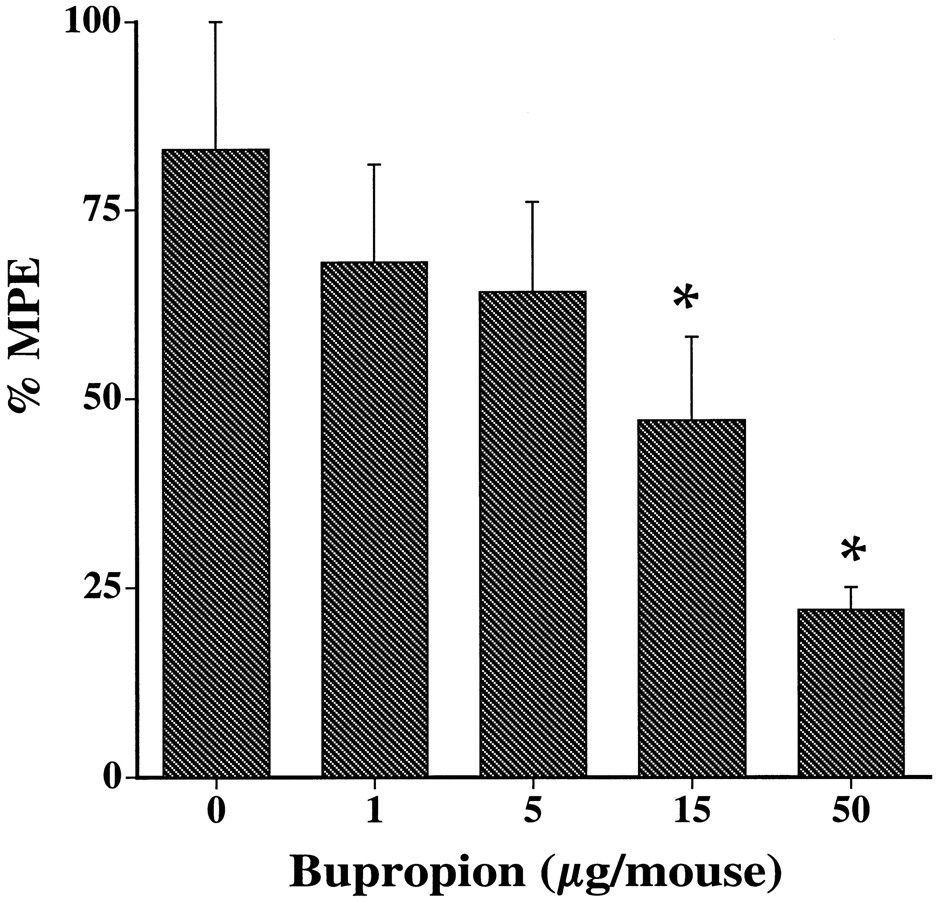
Blockade of nicotine-induced antinociception in the tail-flick test by bupropion after i.t. administration. Bupropion at different doses was administered i.t. 5 min before nicotine (20 μg/mouse, i.t.), and mice were tested 5 min later. Each point represents the mean ± S.E. of 8 to 12 mice. *P < .05 compared with correspondent zero time point.
Effect of bupropion pretreatment on different pharmacological measures after s.c. administration in mice

Blockade of various nicotine's behavioral effects by bupropion after s.c. injection in mice. A, nicotine-induced hypothermia (1 mg/kg, s.c.); B, nicotine-induced antinociception in the hot-plate test (0.75 mg/kg, s.c.); C, nicotine-induced hypomotility (1 mg/kg, s.c.); and D, nicotine-induced seizures (9 mg/kg, s.c.). Bupropion at different doses was administered s.c. 15 min before nicotine, and mice were tested 5 min later except for hypothermia (measure after 30 min). Each point represents the mean ± S.E. of 8 to 12 mice. *P < .05 compared with correspondent zero time point.
Summary of the blockade potency of bupropion on different pharmacological actions of nicotine after s.c. and i.t. administration in mice
Effects of Bupropion on Other Antinociceptive Agents.
The effects of bupropion on the antinociceptive effects of morphine and epibatidine, a very potent nicotinic agonist, were investigated after s.c. administration in the tail-flick test. As shown in Table3, bupropion at a dose five times higher than its AD50 for nicotine blockade failed to significantly block the effect of an active dose of morphine in the tail-flick test. However, bupropion completely blocked the effect of an active dose of (−)-epibatidine. Thus, the effect of bupropion appears not to be generalized to non-nicotinic analgesic substances, because it did not block the effects of morphine administration.
Effect of bupropion pretreatment on morphine and (−)-epibatidine-induced antinociception after s.c. injection in mice using the tail-flick test
Behavioral Interaction of Nicotine and Bupropion
To further characterize bupropion/nicotine interactions, additional experiments were conducted to determine whether bupropion would attenuate other nicotine effects in a dose-responsive manner. Pretreatment with bupropion blocked the effect of a s.c. dose of 1 mg/kg nicotine on body temperature. Indeed, bupropion significantly blocked nicotine's hypothermic effects with an AD50 of 8.5 mg/kg (Fig. 3A). In addition, bupropion blocked nicotine-induced hypomotility with an AD50 of 4 mg/kg (Fig. 3C). Bupropion was also effective in antagonizing nicotine-induced seizures in mice with an AD50 of 4.5 mg/kg (Fig. 3D). Furthermore, by itself, bupropion did not significantly induce seizure behavior nor alter mice locomotor activity and body temperature at the indicated doses and times (Table 1).
Interaction of Bupropion with Expressed Neuronal Nicotinic Receptors
Acute Effect of Bupropion on nAChRs Function.
Because bupropion blocked nicotine's behavioral effects, its potency as a blocker at various neuronal nicotinic receptors was investigated. Bupropion at 50 μM elicited little current when applied for 10 s to oocytes expressing the α4β2, α7, or α3β2 subunit combinations (data not shown). Although it did not activate these expressed receptors, bupropion antagonized the effects of ACh in a concentration-related manner. Although relatively weak when co-applied with 1 μM ACh (IC50 = 55 μM) at the α4β2 receptor subtype (Fig. 4A), the potency of bupropion was increased about 7-fold by pre-exposure (20 s) to the receptor before addition of the agonist (Fig. 4B). Similar results were observed with α3β2 receptors (data not shown). In addition, bupropion displayed differential potency in blocking the various nicotinic receptors with the α3β2 receptor being the most sensitive. Indeed, the concentration of bupropion that blocked 50% of the nicotinic current was determined to be 1.3 (0.7–2.2) and 8 (5.2–11.7) μM for α3β2 and α4β2 receptors, respectively (Fig. 5A). α7 receptors were the least sensitive to bupropion blockade with an estimated IC50 of 60 μM (Figs. 5B and 6).

Effects of different mode of bupropion application on ACh-induced currents in the α4β2 nicotinic receptor subtype expressed in oocyte. A, example of current elicited by 1 μM ACh in the absence or when co-applied with different concentrations of bupropion in an oocyte expressing α4β2 receptor. ACh or bupropion was applied as a 10-s pulse, and changes in current from baseline values were measured for a total of 1 min. B, responses of neuronal α4β2 receptor to 1 μM ACh in the absence and presence of different concentrations of bupropion when it was preapplied at the tested concentration for 20 s then followed by a co-application of bupropion and ACh. Each application was separated by a 5-min washout. Oocytes were held at −70 mV. ACh “after” represents the current activated by an application of ACh after bupropion washout.

Concentration dependence of bupropion inhibition of nAChR subtypes. A, responses of neuronal α3β2 receptor to 10 μM ACh in the absence and presence of different concentrations of bupropion. B, concentration dependence of bupropion inhibition of α7 receptor subtype. Responses of up to 250 μM ACh in the absence and presence of different concentrations of bupropion were recorded. ACh or bupropion was applied as a 10-s pulse, and changes in current from baseline values were measured for a total of 1 min. Each application was separated by a 5-min washout. Oocytes were held at −70 mV. ACh “after” represents the current activated by an application of ACh after bupropion washout.

Concentration-response relationship of bupropion's inhibition of α4β2(●), α3β2 (■), and α7 (▴) nAChR subtypes in oocytes. Also shown is a bupropion concentration-response curve (○) in α4β2 receptors without preapplication of the drug. Each point represents the mean ± S.E. of 8 to 12 recordings.
Mechanisms of nAChRs Functional Blockade.
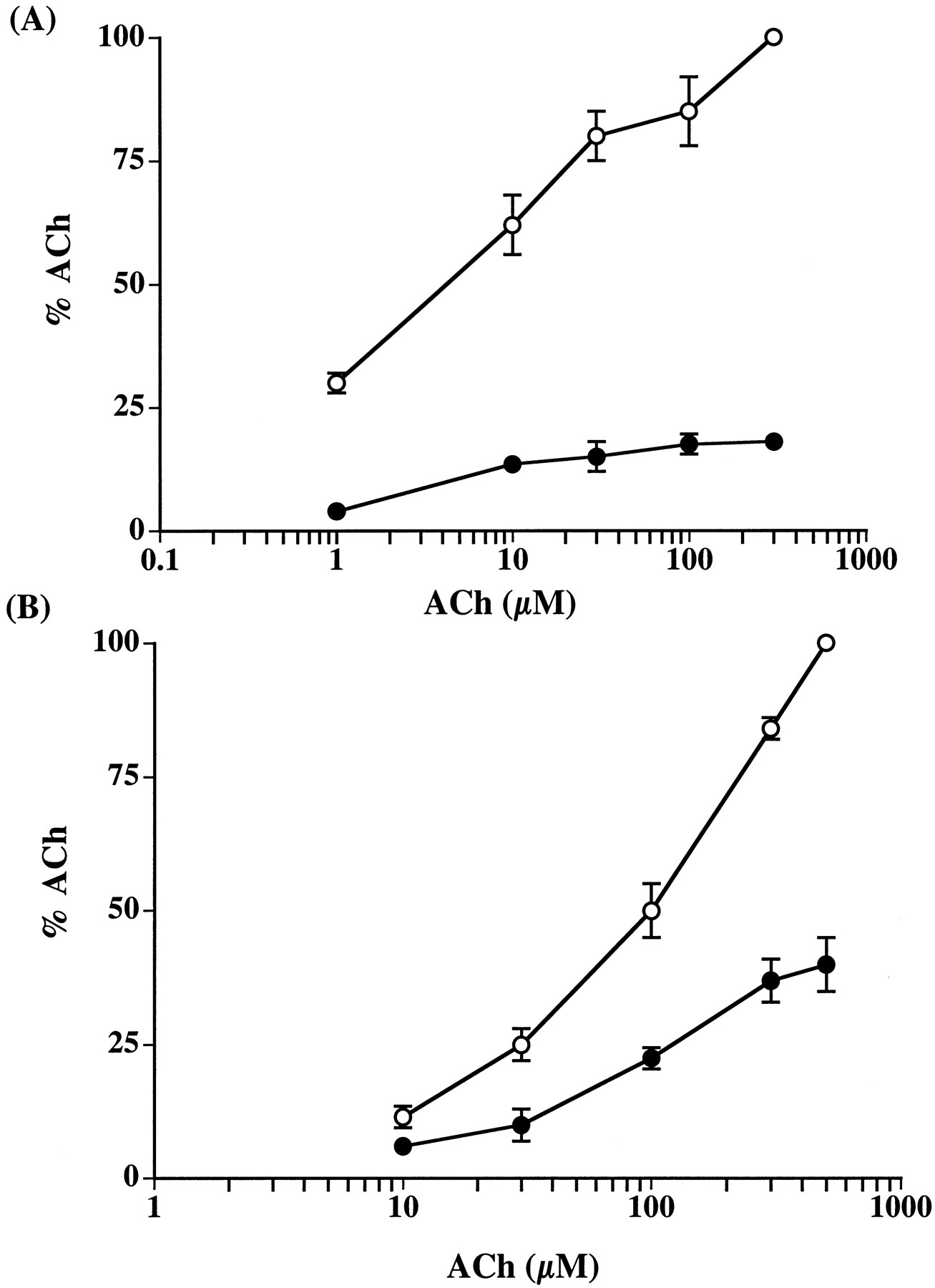
The effect of bupropion on α3β2 and α4β2 receptors was reversible after 5-min washout period (Figs. 4 and 5). Similar results were also obtained with α7 receptors (Fig. 5). ACh dose-response profiles were obtained either alone or in the presence of bupropion at concentrations near its IC84 values for α3β2 and α4β2 receptor subtypes to explore mechanisms of inhibition. Bupropion functional blockade was insurmountable by increasing concentration of ACh in both receptor subtypes (Fig. 7, A and B), suggesting that bupropion acts noncompetitively to inhibit the function of nAChR. Furthermore, as shown in Fig. 8, bupropion functional blockade was voltage-independent in both α3β2 and α4β2 receptor subtypes.

Bupropion noncompetitive functional blockade of nAChR subtypes. Concentration-response curves for ACh-induced current in nAChRs at different concentrations of test drug. Currents were measured in the presence of (○) ACh alone or in the presence of (●) ACh with 25 μM bupropion at the (A) α4β2 receptors or (B) with 10 μM bupropion at the α3β2 receptors. Data were normalized to a 300 μM (α4β2) or 500 μM (α3β2) control response. Each data point represents mean ± S.E. of five to seven recordings.

Voltage independence of bupropion inhibition of (●) α4β2 and (■) α3β2 nAChRs. ACh was applied in the presence of either 1 μM (α3β2) or 10 μM (α4β2) bupropion after a brief equilibrium period at the specified holding potential. Each data point represents mean ± S.E. of five to seven recordings.
Discussion
The primary findings of this study are that bupropion is a blocker of nicotinic behavioral effects and a potent inhibitor of neuronal nAChRs subtypes. This functional blockade is noncompetitive, but the precise mechanism at the nAChRs subtypes studied is still not clear and needs further electrophysiological study.
Here we present the conclusion, supported by our recent report (Damaj et al., 1999b), that bupropion inhibits nicotine's effects after systemic and central administration. Indeed, bupropion was found to block nicotine's antinociception (in two tests), motor effects, hypothermia, and convulsive effects with different potencies in the present investigation. The fact that bupropion was most potent in blocking nicotine-induced antinociception in the tail-flick test, followed by the hot-plate and the hypothermic effects, and was least potent in blocking the motor and convulsive effects suggest that bupropion possesses some selectivity for neuronal nicotinic receptors underlying these various nicotinic effects. The time course of nicotine blockade by bupropion correlates well with plasma profile and half-life values of bupropion in the mouse. The effect of bupropion dissipates after 30 to 60 min and >240 min after the doses of 5 and 25 mg/kg, respectively. This time course is consistent with the ∼20-min half-life of bupropion after i.v. injection of 10 mg/kg (Butz et al., 1982) and the ∼70-min half-life after an i.p. dose of 40 mg/kg (Suckow et al., 1986) in mice. Our data obtained after spinal injection in the tail-flick and inhibition studies in expressed nAChRs suggest that unchanged bupropion is involved in the blockade of functional blockade of nicotine and its receptors. The metabolism of bupropion after spinal injection (5 min after injection) and oocytes application (10 s application time) is probably very minimal and slow in these conditions. However, the participation of bupropion metabolites in its antagonistic effects cannot be excluded, because bupropion major metabolites were active in behavioral tests (Martin et al., 1990).
Bupropion blocks nicotine activation of α3β2, α4β2, and α7 neuronal nAChRs with some degree of selectivity. It was ∼50 and 12 times more effective in blocking α3β2 and α4β2 than α7.
This functional blockade is noncompetitive, because it was insurmountable by increasing concentration of ACh in the nAChRs subtypes tested. In addition, bupropion at high concentration failed to displace brain [3H]nicotine binding sites, a site largely composed of α4β2 subunit combination. Given the observation that bupropion inhibition of α3β2 and α4β2 receptors exhibits voltage-independent properties, bupropion may not be acting as an open channel blocker.
The pronounced effects that bupropion has on nAChRs in vitro is most likely related to nicotine's effects in vivo. We can notice some relationship between bupropion's blockade α4β2 receptors and nicotine's effects in the analgesic tests. Indeed, the antinociceptive response of nicotine in these tests was reported to involve the α4β2 nicotinic receptor subtype (Damaj et al., 1998; Marubio et al., 1999). However, the correlation between the in vivo antagonistic effects of bupropion and its functional blockade at different neuronal nAChRs is difficult to assess from our results.
But how relevant is this functional blockade observed in vivo and in vitro to bupropion's therapeutic effects? The range of bupropion doses used in blocking the different behavioral effects of nicotine is similar to that of its activity in the different antidepressant behavioral tests (Martin et al., 1990). In addition, bupropion potency as a nicotinic antagonist is lower than its in vivo potency in blocking striatal dopamine uptake in mice (Stathis et al., 1995). Serum concentrations of bupropion in humans can rise to a peak near 0.5 to 1 μM after oral administration compound (Findlay et al., 1981; Hysu et al., 1997). Our data indicate that bupropion inhibits particular nAChRs subtypes within this concentration range, suggesting the possibility of neuronal nAChRs mediating bupropion's effects. In addition, other studies have found that plasma levels of its major metabolite, hydroxybupropion, reach 10 to 100 times the concentration of the parent compound (Findlay et al., 1981; Welch et al., 1987; Hysu et al., 1997). Given the extensive metabolism of bupropion in humans and the apparent clinical activity of hydroxybupropion (Martin et al., 1990) as well as its long half-life, we can hypothesize that inhibition of certain nAChR subtypes is involved, in part, in the antidepressant effects of bupropion. It will be important, however, to test the effects of different bupropion metabolites on neuronal nAChRs. Moreover, bupropion's therapeutic use in nicotine dependence may also relate to its activity as an antagonist at nAChRs. Indeed, mecamylamine, a classical nicotinic antagonist, is effective as a smoking cessation aid when used in combination with a nicotine patch (Rose et al., 1994). The relatively high sensitivity of bupropion on α3β2 subtype is very important, because α3 subunits are located in catecholamine-rich brain regions implicated in pleasure and reward (Lukas, 1998). Hence, these effects may explain, in part, bupropion's efficacy in nicotine dependence. Our findings along with those of Fryer and Lukas (1999), suggest that functional blockade of neuronal nAChRs is useful in nicotine dependence treatment. However, the involvement of dopamine and norepinephrine systems in bupropion's therapeutic effects cannot be excluded.
In conclusion, bupropion does block peripheral and central effects of nicotine, apparently through functional inhibition of neuronal nAChRs. These findings contribute to our search for receptors involved in drug dependence and in the discovery of new pharmacological agents for depression and nicotine dependence.
Acknowledgments
We greatly appreciate the technical assistance of Dr. Tie Han and Gray Patrick.
Footnotes
-
Send reprint requests to: Dr. M. Imad Damaj, Department of Pharmacology and Toxicology, Medical College of Virginia, Virginia Commonwealth University, Box 980613, Richmond, VA 23298-0613. E-mail:mdamaj{at}hsc.vcu.edu
-
↵1 This work was supported by National Institute on Drug Abuse Grant DA-05274.
- Abbreviations:
- nAChR
- acetylcholine nicotinic receptor
- ACh
- acetylcholine
- %MPE
- maximum possible effect
- i.t.
- intrathecal
- AD50
- antagonist dose 50%
- Received March 20, 2000.
- Accepted June 26, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}