


Abstract
Plasma pharmacokinetics, biodistribution, excretion, and metabolism of four modified 20-mer antisense oligonucleotides targeted to human intercellular adhesion molecule-1 mRNA have been characterized in rats and compared with a first-generation phosphorothioate oligodeoxynucleotide (PS ODN), ISIS 2302. The modified oligonucleotides contained 2′-O-(2-methoxyethyl) (2′-O-MOE) ribose sugar modifications on all or a portion of the nucleotides in the antisense sequence. The 2′-O-MOE-modified oligonucleotides were resistant to nuclease metabolism in both plasma and tissue. In general, plasma pharmacokinetics was not substantially altered by addition of the 2′-O-MOE modification to PS ODN. Thus, plasma clearance was dominated by distribution to tissues, broadly, with less than 10% of the administered dose excreted in urine or feces over 24 h. However, the 2′-O-MOE modification combined with the phosphodiester (PO) backbone exhibited 10-fold more rapid plasma clearance, with approximately 50% of the dose excreted in urine as intact oligonucleotide. Consistent with its rapid and extensive excretion, the PO 2′-O-MOE modification distributed to very few organs in any substantial amount with the exception of the kidney. Oligonucleotides that contained phosphorothioate backbones were highly bound to plasma proteins. Indeed, the primary characteristic that resulted in the most marked alterations in pharmacokinetics appeared to be the affinity and capacity of these compounds to bind plasma proteins. A balance of greater stability supplied by the 2′-O-MOE modification together with maintenance of plasma protein binding appears to be necessary to ensure favorable pharmacokinetics of this new generation of antisense oligonucleotides.
A number of antisense oligodeoxynucleotide compounds are currently being studied as potential therapeutics in a broad range of disease applications, including viral infectious disease, cancer, and various inflammatory diseases (Dorr and Kisner, 1998). Active clinical programs now exist for all of these diseases, with the first antisense drug approved in 1998 for antiviral treatment of cytomegalovirus infections of the retina (Stix, 1998). The predominant routes of administration for treatment of systemic diseases using antisense oligonucleotides to date are parenteral either by intravenous or subcutaneous injection. With few exceptions, the systemically administered oligonucleotides being studied today in the clinic are phosphorothioate oligodeoxynucleotides (PS ODNs).
The in vivo pharmacokinetics and metabolism of PS ODN antisense compounds have been well characterized in many species, including humans (Cossum et al., 1993; Sands et al., 1994; Agrawal et al., 1995;Geary et al., 1997b; Glover et al., 1997; Stevenson et al., 1999). PS ODNs are well absorbed from parenteral sites (Phillips et al., 1997), and display rapid distribution and prolonged elimination from tissue distribution sites (Geary et al., 1997a; Levin et al., 1998). PS ODNs distribute broadly in peripheral tissues with liver and kidney accumulating the highest concentrations. However, PS ODNs do not cross the blood-brain barrier and are not orally bioavailable (Nicklin et al., 1998). Eventual elimination from tissue is believed to be primarily a function of metabolism with little to no intact drug recovered in urine or feces at doses below 10 mg/kg.
We have previously reported in vivo pharmacokinetic properties of various novel chemically modified oligonucleotides (Crooke et al., 1996). A 2′-ribose modification of ribonucleotides (2′-O-methyl) exhibited improved in vitro stability with little to no change in overall tissue distribution. Mixed backbone sequences that begin to replace sulfur with oxygen in the bridging phosphate linkages have shown higher urine excretion and greater distribution to the kidney (Altmann et al., 1996). Other investigators have reported alterations in tissue distribution characteristics with backbone modifications that remove charge from the backbone, such as methylphosphonate modifications (DeLong et al., 1997) that show reduced tissue concentrations of oligonucleotide.
The protein binding characteristics of unmodified phosphodiester and phosphorothioate ODNs have been characterized (Brown et al., 1994). Plasma protein binding of PS ODNs has been found to involve high-capacity binding to albumin and alpha-2 macroglobulin. Albumin binding is salt- and temperature-dependent and appears to be nonspecific in nature (Greig et al., 1995). It has been hypothesized that these prolific protein binding characteristics of PS ODNs are responsible for some of the nonspecific hemodynamic effects that have been shown to be concentration-dependent (Henry et al., 1997a,b; Levin, 1999). In addition, favorable pharmacokinetic characteristics such as rapid and extensive distribution of PS ODNs to tissue and very little loss by excretion following parenteral injection may also be a function of the plasma protein binding properties of these compounds.
PS ODNs are extensively metabolized in plasma and tissue by exonuclease digestion (Cummins et al., 1996; Gaus et al., 1997; Temsamani et al., 1997). Exonuclease chain-shortening of the antisense oligonucleotide results in a reduction in the amount of parent drug available for pharmacological action within the cell. Furthermore, clearance from cells and tissues is believed to be a function of metabolic rate. It has been hypothesized that the addition of 2′-alkoxy or alkyl moieties to the ribose sugar will stabilize oligonucleotides to nuclease degradation in vivo and that this in turn will enhance the biodistribution and pharmacokinetics of antisense compounds. To test this hypothesis, in vivo pharmacokinetic studies are required. The modified oligonucleotides used in this study contained 2′-O-(2-methoxyethyl) (2′-O-MOE) ribose sugar modifications on all or a portion of the nucleotides in the antisense sequence (Fig. 1). In this study, we report for the first time a thorough characterization of the pharmacokinetics of a series of compounds with partial and complete 2′-methoxyethyl ribose sugar modification of oligonucleotides in combination with both phosphorothioate and phosphodiester backbones. We also characterized the contribution of protein binding and nuclease stability to the in vivo pharmacokinetic characteristics of oligonucleotides.

Chemical structure for the 2′-O-(2-methoxyethyl)-modified oligonucleotides.
Materials and Methods
Antisense Oligonucleotides
Oligonucleotides were synthesized at Isis Pharmaceuticals, Inc., as previously described (Baker et al., 1997). Radiolabel was incorporated using 35S in ISIS 2302, ISIS 14725, and ISIS 11159 (all contain phosphorothioate backbone; Table1). The 35S was incorporated on the fifth nucleotide linkage in from the 5′ end to ensure prolonged stability of the radiolabel's association with the oligonucleotides. For ISIS 16952 (PO MOE), a nonexchangeable tritium label was incorporated in the 5′ methyl of the thymine base located three bases in from the 5′ end and ISIS 18268 (PO/PS alternating backbone MOE) oligonucleotide was labeled using a tritium label by heating in tritiated water and allowing the purine nucleobase to undergo tritium exchange (Graham et al., 1993; Table 1). All of the tested oligonucleotides were 20 bases in length and molecular weights ranged from 6368 to 7940 g/mol.
Oligonucleotide sequence and chemistry
Purification
All compounds were purified by reverse phase HPLC using a BondaPak HC18HA matrix (Waters Chromatography, Milford, MA). A three-component solvent system was used with 50 mM NaOAc (pH 7.2), H2O, and acetonitrile. Purification gradients were optimized for each compound before scale-up for the bulk purification. Bulk purification involved a final desalting of the oligonucleotides by HPLC. Final compounds used in this study were >90% full-length oligonucleotide. Greater than 97% of the radiolabel was associated with the full-length oligonucleotide portion.
ISIS 2302 is a PS ODN with a sequence complementary to the 3′ untranslated region of human ICAM-1 mRNA (Bennett et al., 1994). ISIS 14725 is a partially modified 2′-O-MOE-modified oligonucleotide of the same sequence (Table 1).
ISIS 11159 (PS, 2′-O-MOE), 16952 (PO, 2′-O-MOE), and ISIS 18268 (PS/PO alternating backbone, 2′-O-MOE) each contain identical sequences that are also targeted to human ICAM-1 mRNA. The sequence for these three oligonucleotides is complementary to the 5′ terminus site on the ICAM-1 message (Baker et al., 1997).
Animal Studies
All animal studies were in compliance with published Unites States Department of Agriculture regulations and approved by an Institutional Animal Care and Use Committee. Sprague-Dawley male rats weighing between 200 and 250 g were randomly assigned to five treatment groups (one group for each compound tested).
Each group received a single intravenous bolus dose (3 mg/kg) of the tested oligonucleotide via the tail vein. Blood samples were collected at 2, 10, 20, 30, 60, and 90 min and at 2, 3, 4, 6, 8, 10, 12, and 24 h after single-dose administration to allow for pharmacokinetic analysis. Blood was taken via the orbital sinus under isoflurane anesthesia or at time of sacrifice via the abdominal aorta. Urine was collected over 24-h intervals following administration. Selected animals from each dose group (n = 3) were humanely euthanized at predetermined time points (1, 3, 8, and 24 h) after administration. Tissues were sampled at these time points to assess the biodistribution of the different oligonucleotides.
Bioanalytical Methods
Plasma and urine were mixed directly with liquid scintillation fluid. Tissues were digested in concentrated (35%) tetraethylammonium hydroxide aqueous solution (tetraethylammonium hydroxide digestion media).
The radioactivity content in each sample was measured by liquid scintillation spectroscopy. Each sample was counted for 5 min or to a 2 ς error value of 2%, whichever occurred first. All counts were converted to absolute radioactivity (dpm) by automatic chemiluminescence and quench correction. Samples having a radioactivity level of less than or equal to double background were considered below the limit of quantitation and considered zero for subsequent calculations.
Intact drug analysis was performed on plasma, urine, and tissues. Cold analysis was conducted using strong anion exchange-HPLC or quantitative capillary gel electrophoresis (Leeds et al., 1996); both separation methods used UV detection at 260 nm for quantification of oligonucleotide.
Plasma Protein Binding
Whole Plasma.
Ultrafiltration methods were used to quantitate unbound oligonucleotide over wide concentration ranges. Radiolabeled oligonucleotide was spiked at known concentrations to whole rat plasma and incubated at 37°C for 30 min. Following incubation, the plasma was transferred to Ultrafree (Waters) ultracentrifuge inserts (MWCO 30,000) and centrifuged at 5000 rpm (approximately 2000g) for 20 min. Unbound oligonucleotide was assayed using liquid scintillation counting and divided by the total counts in whole plasma to determine the unbound fraction. Fraction bound to protein was calculated simply by subtracting the unbound fraction from 100%.
Albumin Protein Binding Assays.
Mixed (50 nM), cold and hot, oligonucleotide was incubated at room temperature with increasing concentrations of albumin (fatty acid free serum albumin; Sigma Chemical, St. Louis, MO) for 1 h in phosphate-buffered saline buffer containing 10 mM EDTA and 0.005% Tween 80. After incubation, the samples were loaded onto low-binding, regenerated cellulose filter membranes with a molecular weight cut-off of 30,000 (Millipore). The samples were spun gently in a microfuge (NYCentrifuge 5415C; Eppendorf, Westbury, NY) at 3000 rpm (735g) for 3 to 6 min, collecting ∼20% of the loaded volume in the filtrate.
Aliquots from the filtrate and the initial (unfiltered) solutions were measured using a scintillation counter (model LS6000IC, Beckman, Fullerton, CA). The counts obtained in the filtrate aliquots represent the free (unbound) oligonucleotide and appropriate calculations are performed to obtain the concentration of free oligonucleotide. Further calculations yield the concentration of oligonucleotide bound to protein.
The equilibrium constant, Kd, was determined by fitting the data of fraction of oligonucleotide bound to protein concentration. Constants were determined from nonlinear regression analysis of a fraction of ODN bound (fbound) as a function of free albumin monomer concentration ([A]free). Concentration of albumin monomers in solution was calculated usingKd = 150 μM for monomer-dimer equilibrium (Kuznetsov et al., 1977; Zini et al., 1981).
Calculations and Statistics.
Multiexponential equations were used to describe the pharmacokinetic plasma profiles. Best-fit nonlinear regression analysis was performed using WINNONLIN (version 1.1). Area under the plasma concentration-time curve (AUC) was performed using the linear trapezoidal rule and extrapolated to infinity by dividing the last plasma concentration by the slope of the terminal phase (Clast/λterm). Statistical moment analysis was used to calculate mean residence time (MRT) and volume of distribution at steady state (Vss):
Results
Plasma Pharmacokinetics and Metabolism.
Plasma pharmacokinetics for the oligonucleotides were best described by biexponential equations and were characterized by rapid distribution to tissue and broad uptake in many tissues for all phosphorothioate oligonucleotides tested independent of sugar chemistry (Fig.2). However, remarkable differences in metabolism were seen in plasma. ISIS 2302 (PS ODN) was rapidly metabolized, such that approximately 50% of the circulating oligonucleotide was intact parent compound by 30 min after intravenous administration. However, for both ISIS 14725 (3′ partial MOE modified 2302 sequence) and ISIS 11159, little to no measurable metabolites were seen in the plasma using either HPLC or capillary gel electrophoresis methods.
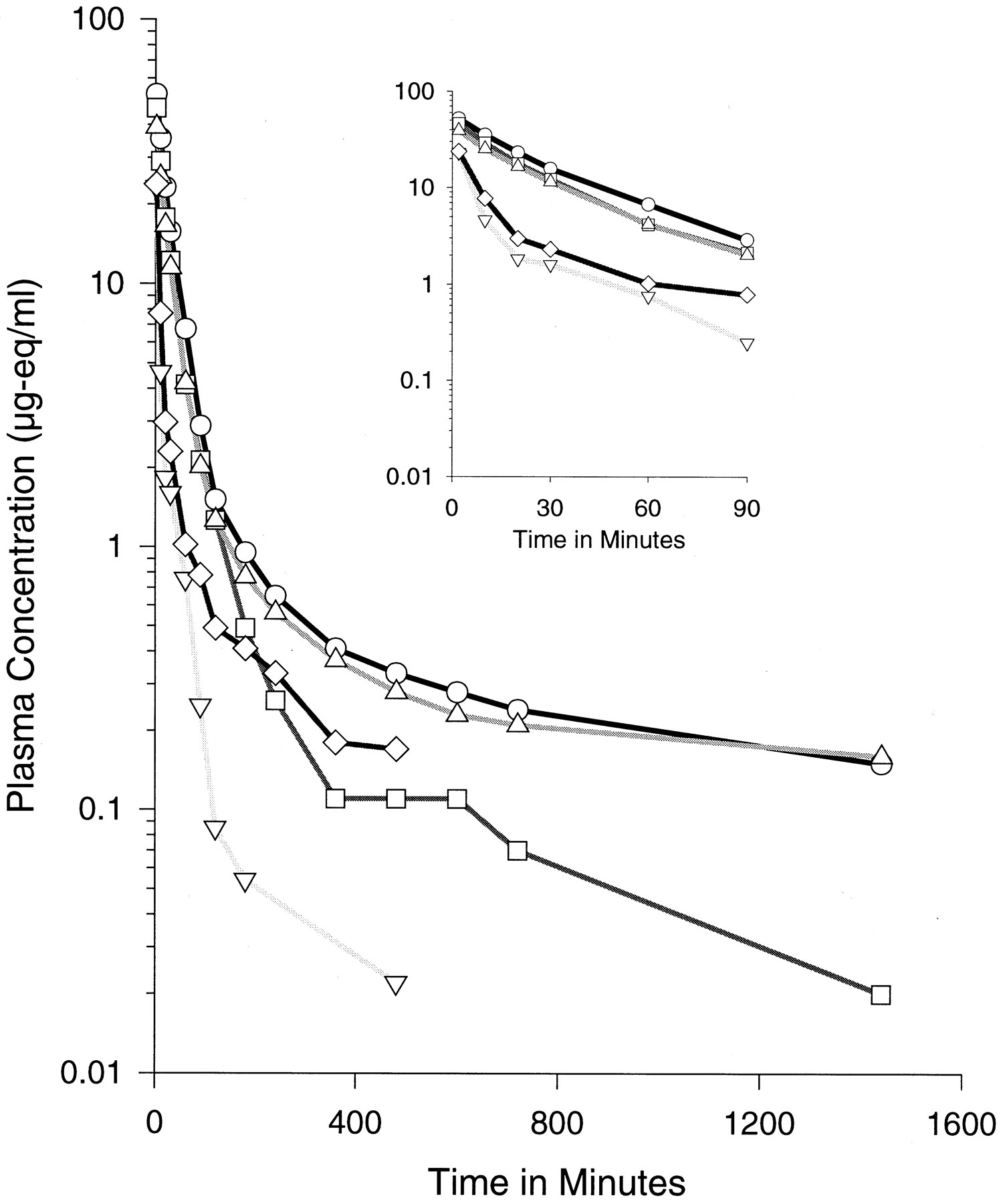
Plasma oligonucleotide equivalent concentration-time profiles following bolus intravenous injection of 3-mg/kg dose. Each point represents the average of three animals. The symbols represent different chemistries: ○, ISIS 2302; ▵ ISIS 14725; ■, ISIS 11159; ▿, ISIS 16952; ⋄, ISIS 18268. The inset figure provides an expanded view of the early portion of the curves from time 0 to 90 min.
Initial distribution half-lives for the three phosphorothioate oligonucleotides were similar and rapid with half-lives of approximately 17, 15, and 11 min for ISIS 2302, 14725, and 11159, respectively (Table 2). Terminal half-lives of radiolabel equivalents were similar for ISIS 2302 and ISIS 14725 (3.2 h). However, the terminal half-life of ISIS 11159 was somewhat faster (1.3 h). Overall, plasma clearance was similar for the PS ODN, ISIS 2302, and the partially modified ISIS 14725, but was somewhat more rapid for the fully modified ISIS 11159.
Plasma pharmacokinetic parameter estimates for various chemically modified oligonucleotides (both 2′-ribose and backbone modified)
Initial distribution half-life for the phosphodiester (PO) oligonucleotide (ISIS 16952) was much faster (approximately 2.5 min) compared with the modified phosphorothioate oligonucleotides. A more rapid terminal elimination phase was also apparent compared with those modified with phosphorothioate chemistries. In fact, overall plasma clearance of ISIS 16952 was nearly 10-fold more rapid than ISIS 2302. Clearance of this PO-modified oligonucleotide appeared to be primarily a function of excretion in urine and rapid distribution primarily to the kidney.
When the oligonucleotide backbone was modified to contain alternating diester (PO) and phosphorothioate (PS) linkages (ISIS 18268), there was a less rapid initial distribution with a half-life of approximately 5 min. Furthermore, the apparent terminal elimination half-life of the radiolabel equivalents for this compound was restored to greater than 1 h (1.7 h), similar to ISIS 11159, the fully phosphorothioate-modified sequence. Plasma clearance for radiolabel equivalents of ISIS 18268 was still more rapid than the fully phosphorothioate-modified oligonucleotides, but was approximately one-third the clearance of the full phosphodiester oligonucleotide (54 versus 158 ml/h).
Vss was similar among ISIS 2302, ISIS 14725, and ISIS 11159 (PS MOE), but was considerably higher for ISIS 16952 (PO MOE) and ISIS 18268 (PO/PS MOE). This observation is likely due to more rapid distribution to tissue and added rapid filtration in the kidney for ISIS 16952 and ISIS 18268. For all the compounds tested the Vss was higher than total plasma volume of the rat, indicative of the extravascular distribution of the oligonucleotides.
Tissue Distribution and Metabolism.
All of the PS oligonucleotides distributed rapidly and broadly to many tissues throughout the rat (Table 3). Liver and kidney were sites of highest tissue concentrations, consistent with previous oligonucleotides studied. Differentiation in distribution was noticeable in the liver where highest concentrations were seen for the unmodified oligodeoxynucleotide ISIS 2302. 2′-O-MOE-modified oligonucleotides distributed to a somewhat lesser extent to this organ. The fully modified PS oligonucleotide (ISIS 11159) distributed to a greater extent to the kidney than either ISIS 2302 or ISIS 14725 (partially modified).
Oligonucleotide equivalent concentrations (μg-eq/g of tissue) in tissues collected 24 h after intravenous injection at a dose of 3 mg/kg
However, the largest variance in tissue distribution was seen for the PO 2′-O-MOE-modified oligonucleotide ISIS 16952. Only traces of radioactivity associated with this oligonucleotide were observed in any tissue other than the kidney. Approximately 40% of the administered drug was recovered in kidney 24 h after administration. The addition of 10 sulfurs to the backbone (ISIS 18268 alternating PO/PS backbone chemistry) reduced the amount of oligonucleotide distributed to the kidney and partially restored distribution to the peripheral tissues.
The unmodified PS ODN ISIS 2302 was substantially degraded in tissue by 24 h with only 25 to 30% of the total measurable oligonucleotide in tissue identified as parent 20-mer (Fig.3). The fully 2′-O-MOE-modified oligonucleotides (either PS or PO backbones) appeared to be completely resistant to nuclease degradation even in tissue. The partially modified 2′-O-MOE oligonucleotide ISIS 14725 was partially protected from exonuclease metabolism and, thus, more slowly degraded in most tissues with fewer measurable metabolites apparent at 24 h (∼50% intact).

Capillary gel electrophoresis representative electropherograms of liver and kidney tissue extracts illustrate the extent of oligonucleotide metabolism associated with the various chemistry modifications. Tissues assayed were collected 24 h after bolus intravenous injection at 3-mg/kg dose.
Urinary Excretion.
Although 32% of the radioactivity associated with the PS ODN ISIS 2302 was excreted in the urine, less than 1% of the radiolabel was associated with oligonucleotide as measured by HPLC (Table4). For the more nuclease-resistant chemistries, less 35S was recovered in the urine by 24 h after administration (11.2–16.5%) and more of the label was associated with oligonucleotide. This observation is presumably due to the more stable nature of the 2′-O-MOE-modified chemistry. For the PO 2′-O-MOE compound ISIS 16952, the majority of the excreted radiolabel was associated with parent compound or nearly intact oligonucleotide metabolite(s). Nearly half (45%) of the administered PO oligonucleotide compound was rapidly excreted in the urine. Urinary excretion of the PO 2′-O-MOE oligonucleotide was greatly reduced (from 45 to 16%) by incorporation of 10 alternating phosphorothioate links in the same sequence and sugar chemistry (ISIS 18268). Once again, most of the radioactivity collected in urine was associated with intact ISIS 18268.
Urine excretion
Protein Binding.
Differences in plasma protein binding appear to explain the differences in pharmacokinetic behavior. ISIS 2302 is greater than 99% bound to plasma proteins, and this binding does not appear to saturate upon increasing concentration up to 70 μg/ml in rat whole plasma. It is this large extent of binding to plasma proteins that likely prevents glomerular filtration of the phosphorothioate oligonucleotides. Modification with the 2′-O-MOE appears to lessen the affinity of binding to albumin (Fig.4). Comparison ofKd values for ISIS 2302 and ISIS 14725 demonstrate that eight 2′-O-MOE modifications at the 3′ terminus can reduce affinity. However, 2′-O-MOE modification does not significantly alter the extent of binding to plasma proteins for the PS MOE modifications (Table 5).

Fraction of oligonucleotide bound to albumin plotted as a function of free albumin concentration. Each symbol represents the average of five separate measurements (n = 5). The symbols represent different chemistries: ▪, ISIS 2302; ♦, ISIS 14725; ▴, ISIS 11159; ×, ISIS 18268; ∗, ISIS 16952.
Plasma protein binding
The greatest impact on binding affinity appears to be related to the presence or absence of phosphodiester linkages in the backbone. The phosphodiester (PO) 2′-O-MOE (ISIS 16952) had both a large decrease in affinity to albumin and decreased extent of binding (capacity) to plasma proteins overall. The reduced binding to plasma proteins likely explains the remarkable increase in urine excretion. Indeed, plasma clearance and urine excretion are well correlated to the plasma protein binding characteristics of these compounds (Fig.5).

Correlation of plasma clearance (top) and urine excretion (bottom) to the bound fraction of oligonucleotide to whole plasma.
Discussion
The 2′-O-(2-methoxyethyl) modification of the ribose provides additional improvements to oligonucleotide biological stability and affinity of binding to its ultimate mRNA target (Freier and Altmann, 1997; Cook, 1998). Recently, a number of 2′ modifications at the pentofuranose sugar moiety have been reported that offer the dual advantages of high nuclease resistance and enhanced binding affinity to its complementary sense sequence (Manoharan, 1999). Most second-generation antisense compounds currently undergoing clinical trials are PS “gapmer” oligonucleotides that contain 2′-modified ribonucleotides on one or both ends with a 2′-deoxy (2′-H) segment in the center as required to maintain RNase H activity. Since 2′ modifications such as 2′-O-MOE provide sufficient nuclease resistance even with a PO backbone, use of these modifications may reduce nonspecific protein binding properties characteristic of oligonucleotides with PS linkages.
It is clear from this study that nuclease degradation of the oligonucleotides is almost completely blocked by the 2′-O-MOE modification. Protection was substantially better when the 3′ end of the oligonucleotide was modified and protection was essentially complete when the oligonucleotide was uniformly modified. A recent structural study explains the unique antisense properties of 2′-O-MOE modification (Teplova et al., 1999). The extended preorganization of the 2′-O-methoxyethyl side chain into an RNA-like conformation coupled with extensive hydration may account for less protein binding, high RNA binding affinity, and high nuclease resistance. Similar improvements in stability have been reported for 2′-O-methyl hybrid oligonucleotides (Crooke et al., 1996;Zhang et al., 1996) blocked on both the 3′ and 5′ end.
The stabilization of these modified oligodeoxynucleotides in plasma and tissue will likely improve their ability to elicit an antisense response and are likely to greatly decrease their elimination rate from tissues and cells. Dosing regimens used in the clinic involve either every other day or continuous i.v. infusion over several weeks for phosphorothioate oligodeoxynucleotides (Glover et al., 1997; Stevenson et al., 1999; Yuen et al., 1999). The greater stability of this second-generation chemistry should allow for less frequent dosing and may obviate the need for continuous infusions. We recently reported improved intestinal permeability for 2′-O-MOE-modified antisense oligonucleotides (Khatsenko et al., 2000). Improved stability in the gastrointestinal tract before absorption coupled with improved permeability may provide opportunities for oral bioavailability of antisense oligonucleotides.
From this study, the impact of altering the ribose sugar chemistry in combination with the backbone chemistry of oligonucleotides on in vivo pharmacokinetics is clear. Marked changes in distribution of oligonucleotide are not seen with the addition of 2′-O-MOE modification alone, either partially or in full. Rather, some subtle shifting of distribution away from liver and to kidney is seen as the 2′-O-MOE modification is increased. However, removal of sulfur from the backbone in combination with the 2′-O-MOE modification causes a remarkable shift of distribution away from most systemic tissues other than kidney. In addition, renal excretion is increased as sulfur is removed from the backbone. The mechanism for this shift in distribution and renal excretion appears to be related to a loss in plasma protein binding affinity and capacity.
The plasma protein binding properties of phosphorothioate oligonucleotides have been known for some time (Brown et al., 1994). The phosphorothioate modification provides not only improved stability for oligodeoxynucleotides compared with phosphodiester (DNA) but also much improved distribution and pharmacokinetics overall. Indeed, no other modification has exhibited such remarkable organ, suborgan, and intracellular distribution (Graham et al., 1998; Butler et al., 2000). It is apparent from this study that the protein binding properties of these oligonucleotides are critical to the distribution and ultimately the elimination kinetics of oligonucleotides.
It is known that distribution to peripheral tissues and ultimate uptake into the cells of target organs is critical to the success of antisense therapy for a wide range of diseases. It is apparent from this work that plasma protein binding is required to maintain broad distribution of oligonucleotides. It follows that maintenance and, perhaps, control of plasma protein binding, is a critical element in the rational design of oligonucleotides. Recent data indicate that phosphorothioate oligonucleotides may shuttle from low-affinity, high-capacity binding in circulating blood to higher affinity, lower capacity sites in some tissues (S. Crooke, unpublished data). Indeed, this shuttling process may be involved in the uptake of oligonucleotides into cells in vivo and ultimately be involved in their intracellular disposition (Lorenz et al., 2000).
The critical link between protein binding and useful pharmacokinetics for this class of compound also has a potential negative component. Drugs that bind plasma proteins have the capacity to interfere with other drugs that also bind to similar proteins. At least one publication suggests there may be an interaction between phosphorothioate oligonucleotides and aspirin (Agrawal et al., 1998), for example. Although it can be argued that the heroic doses used in the published investigation are not realistic to the clinic, this report indicates the importance of a thorough understanding of the plasma protein binding characteristics of oligonucleotides. In addition, because phosphorothioate oligonucleotides bind to many proteins, the potential for protein interactions is great and may manifest itself in the form of unwanted side effects and nonsequence-dependent toxicities. Indeed, interaction with proteins in plasma that are involved with hemostasis has been reported for phosphorothioate oligodeoxynucleotides (Henry et al., 1994; Sheehan and Lan, 1998). In monkeys, high doses of phosphorothioate oligodeoxynucleotides have been shown to activate the alternate complement pathway (Henry et al., 1997a). These acute toxicities have been shown to be clearly phosphorothioate oligodeoxynucleotide plasma concentration-dependent (Levin et al., 1998). Nevertheless, at clinically relevant doses, the more severe and dose-limiting effects of phosphorothioates has not been realized (Glover et al., 1997; Stevenson et al., 1999; Yuen et al., 1999).
It is clear that the protein binding properties of oligonucleotides appear to play a critical role in the in vivo distribution and ultimate elimination of these compounds, whereas stability of the oligonucleotide may play a critical role in improving the residence time of these compounds in cells. Indeed, the single most remarkable change in pharmacokinetic behavior of the oligonucleotide analogs reported here is linked to the backbone chemistry, which has been shown to effect both affinity and capacity of binding to plasma proteins. The 2′-O-MOE chemistry provides remarkable protection from nuclease degradation and will likely slow clearance of oligonucleotide from tissue and ultimately the target cells. It is apparent from these data that a balance of improved stability and the maintenance of plasma protein binding appear to be necessary for the ultimate usefulness of these new oligonucleotide modifications in vivo.
Acknowledgments
This work would not have been possible without the dedicated efforts of the staff and personnel at ClinTrials BioResearch (Montreal, Canada) with special thanks to Sylvie Duscharme. We also gratefully acknowledge the enlightening scientific discussions with Drs. Stanley Crooke, Frank Bennett, and Dan Cook. The superb technical assistance of Keith Bunker and Jon Fitchett is also acknowledged. We also gratefully acknowledge the superb administrative assistance provided by Karen Keyer.
Footnotes
-
Send reprint requests to: Richard S. Geary, Ph.D., Isis Pharmaceuticals, Inc., 2292 Faraday Ave., Carlsbad, CA 92008. E-mail:rgeary{at}isisph.com
- Abbreviations:
- PS ODN
- phosphorothioate oligodeoxynucleotide
- 2′-O-methyl
- 2′-ribose modification of ribonucleotides
- ODN
- oligodeoxynucleotide
- 2′-O-MOE
- 2′-O-(2-methoxyethyl)
- PO MOE
- phosphodiester 2′-O-(2-methoxyethyl)
- ICAM-1
- intercellular adhesion molecule-1
- AUC
- areas under the plasma concentration-time curve
- MRT
- mean residence time
- Vss
- volume of distribution at steady state
- PO
- phosphodiester
- PS
- phosphorothioate
- Received September 27, 2000.
- Accepted November 30, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}