


Abstract
Many clinically important drug interactions occur due to inhibition of human liver cytochrome P450 3A (CYP3A) metabolism. The drug efflux pump P-glycoprotein (Pgp) can be an additional locus contributing to these drug interactions because there is overlap in drugs that are substrates for both proteins. We screened a number of CYP3A inhibitors (macrolide antibiotics, azole antifungals, and ergotpeptides) for their ability to interact with Pgp, compared with prototypical Pgp inhibitors. We used cell lines expressing human, mouse, and rat mdr1 genes. Pgp antagonism was defined by interactions of the drugs with four cell lines (LLC-PK1, L-MDR1, L-mdr1a, and L-mdr1b) using a microfluorometric calcein-AM assay and characterized for their inhibitor constant (Ki) toward calcein-AM. The compounds were further defined for their ability to inhibit MDR1 by their effect on vinblastine accumulation into L-MDR1 cells. Representative compounds from each class of drugs were further tested as Pgp substrates, defined by the ability of human Pgp or mouse mdr1a/Pgp to transport them across a polarized kidney epithelial cell in vitro. These same compounds were administered radiolabeled in vivo to mdr1a (+/+) and (−/−) mice and the distribution of radioactivity compared. The results are summarized as follows: 1) Some drug interactions with Pgp were substrate- and/or assay-dependent. 2) Ergot alkaloids were identified as a class of MDR1/Pgp chemosensitizers. 3) The Ergot alkaloids revealed species differences in the structure-activity relationships for inhibition of Pgp. Simultaneous inhibition of Pgp by many CYP3A inhibitors contributes to human variation in the extent of drug-drug interactions.
Drug-drug interactions are a major clinical problem. Because it has been estimated that up to 50% of drugs are metabolized by CYP3A, initial studies to understand the mechanism of these interactions examined the potential for drugs to effect CYP3A-mediated metabolism. The ability of drugs to act as inducers, inhibitors, or substrates for CYP3A was predictive of whether concurrent administration of these compounds with a known CYP3A substrate might lead to altered drug efficacy or toxicity. However, it is now appreciated that drug efflux, particularly by P-glycoprotein, also plays an important role in the disposition of many drugs. Like CYP3A, P-glycoprotein seems to have broad substrate specificities (Kim et al., 1999). We previously reported on the striking overlap in CYP3A and Pgp inducers (Beck, 1991). Others have surveyed 14 Pgp inhibitors (Wandel et al., 1999) and CYP3A substrates (Kim et al., 1999) for their ability to interact with Pgp. However, there has not been a comprehensive analysis of the potential of many known inhibitors of CYP3A to interact with Pgp.
The aims of this study were to improve our understanding of the basis for drug interactions involving what are currently defined as CYP3A inhibitors by defining their ability to interact with Pgp either as inhibitors or substrates. In addition, this analysis would identify any novel compounds that had potential to block Pgp function, and thus, that might serve as new multidrug-resistance modifiers. We chose a series of established CYP3A inhibitors (some of which are also CYP3A substrates) and compared the interactions with human MDR1 and the mouse ortholog mdr1a. We chose to compare two short-term assays of Pgp function to minimize any effects of metabolism or toxicity. In addition, because Pgp has multiple drug binding sites that differentially interact with Pgp substrates and inhibitors (Shapiro and Ling, 1997), by comparing different substrate (calcein-AM or vinblastine)-inhibitor pairs we increased the ability to predict inhibitory interactions of drugs with Pgp. Vinblastine was chosen because it interacts at two of the drug binding sites on Pgp and has equal affinity for both sites (Shapiro and Ling, 1997). If we assume that Pgp inhibitors (or substrates) binding at either of these sites will compete with vinblastine for transport we should be able to predict whether there will be a drug interaction. The calcein-AM assay was chosen because calcein-AM represents an attractive fast throughput assay applicable for large-scale screening of Pgp modulators. These studies form the foundation of studies in mdr1a (+/+) and (−/−) mice to assess the in vivo influence of Pgp modulators. We simultaneously compared the interactions of these drugs with rat mdr1b because this transporter is also abundantly expressed in rodent liver (Brown et al., 1993). The results of our studies identified ergotpeptides as an additional class of Pgp inhibitors, reveals the different structure-activity releationships by ergotpeptides for inhibition of human and rodent Pgp, and demonstrates that interactions of inhibitors with Pgp are substrate-dependent.
Materials and Methods
Drugs.
Calcein-AM was purchased from Molecular Probes (Eugene, OR), Transwell dishes (24.5 mm in diameter, 3.0-μm pore size; Costar no. 3414) and 96-well plates (Costar no. 3595) were from Fisher Scientific (Pittsburgh, PA), and [3H]vinblastine sulfate (11.7 Ci/mmol) was from Moravek (Brea, CA). Anti-mdr1 antibody was purchased from Calbiochem (San Diego, CA), 9,10-[9,10–3H(N)]-dihydro-α-ergocryptine (20.0 Ci/mmol) was from PerkinElmer Life Sciences (Boston, MA), [3H]reserpine (59.9 Ci/mmol) was from Dr. Shimon Schuldiner (Hebrew University, Jerusaem, Israel), and [3H]fluconazole (2.525 Ci/mmol) was from Dr. Dominique Sanglard (Centre Hospitalier Universitaire Vaudois, Lausanne, Switzerland). All ergot alkaloids were from Sigma/RBI (Natick, MA).
Cell Lines.
LLC-PK1 pig kidney epithelial cells were obtained from American Type Culture Collection (Rockville, MD) and cultured as described previously (Schinkel et al., 1995). LLC-PK1 derivative cell lines containing human MDR1 (L-MDR1) or mouse mdr1a (L-mdr1a) were generously provided by Dr. Alfred Schinkel (The Netherlands Cancer Institute, Amsterdam, The Netherlands) and cultured as described previously (Schinkel et al., 1995).
Generation of LLC-PK1 Cells Stably Overexpressing mdr1b.
Rat liver mdr1b (Brown et al., 1993) was obtained from Dr. Jeffrey Silverman (Sunesis Pharmaceuticals, San Francisco, CA) and subcloned into pcDNA3. LLC-PK1 cells were transfected with mdr1b and individual clones selected with 800 μg/ml G418 and 6.4 nM vincristine. No untransfected LLC-PK1 cells survived treatment with 6.4 nM vincristine.
Immunoblot Analysis of mdr1b Expression in Stably Transfected Cells.
L-mdr1b cell clones were sonicated and 10 μg of lysate was analyzed on 7.5% polyacrylamide gel electrophoresis gels immunoblotted using polyclonal rabbit anti-mdr1 antibody (Calbiochem). It was followed by anti-rabbit Ig secondary antibodies coupled with peroxidase and developed with the ECL detection system (Amersham Biosciences, Piscataway, NJ) following the manufacturer's instruction.
Calcein-AM Fluorometry Assay.
This was performed as described previously (Tiberghien and Loor, 1996). LLC-PK1, L-MDR1, L-mdr1b, and L-mdr1a cells were cultured in Costar 96-well plates purchased from Fisher Scientific on day 0 at 100,000 cells/well in phenol-free medium. We carried out the inhibitor studies at theKm value of calcein-AM for Pgp in L-MDR1 cells (determined by Dr. Ryan Yates, personal communication, to be ∼1 μM). On day 1, medium was removed and the well washed once with 200 μl of Hanks' buffer (Invitrogen, Carlsbad, CA). Hanks' buffer (100 μl) with or without 2× reverser was added and the cells incubated for 30 min at 37°C. Then 100 μl of Hanks' buffer containing calcein-AM (2 μM in DMSO) (Molecular Probes) was added to reach a final calcein-AM plate concentration of 1 μM, and the microplates were analyzed with a fluorescence microplate reader (Cytofluor 2350; Millipore Corporation, Bedford, MA) with excitation and emission wavelengths set at 485 and 530 nm, respectively (calcein excitation, 494 nm; emission, 517 nm). The plate was scanned at 3-min intervals repeated 11 times over 30 min at 25°C. For each drug, simultaneous treatment of LLC-PK1 cells allowed determination of whether there were nonspecific effects of modulators on, for example, calcein fluorescence or esterase activity. Each data point was determined by averaging at least two independent experiments using three wells per cell line per treatment. The data were fitted using a modified form of the Michaelis-Menten equation (Lan et al., 1996).
[3H]Vinblastine Accumulation in LLC-PK1 and L-MDR1 Cells.
To assess drug uptake we used a modification of the procedure described previously (Schuetz and Schuetz, 1993). Briefly, cultured cells were placed in media containing 2 μM [3H] and unlabeled vinblastine in the presence or absence of various concentrations of inhibitor and incubated at 37°C with 5% CO2 for 1 h. Individual dishes were washed three times with ice-cold phosphate-buffered saline, cells scraped to harvest, resuspended in phosphate-buffered saline, sonicated, and analyzed for radioactivity using a scintillation counter. Each data point was assayed in duplicate and the experiment repeated three times. The Ki was calculated using a modified form of the Michaelis-Menten equation (Schuetz and Schuetz, 1993).
Cell Culture and Transport Assays.
Transport assays were performed as described previously (Schinkel et al., 1995). Briefly, cells were plated on day 0 at 2 × 106cells/well containing 2 ml of medium in each compartment of the Transwell dish. On day 1 or 2 medium was changed. On day 3 (cell density ranged from 3.3 to 3.9 × 106cells/well) cells were washed and the assay started at time 0 by adding radiolabeled drug to either the apical or basal compartment. For investigation of effect of the inhibitor on [3H]vinblastine transport, cells were preincubated in both compartments with 20 μM inhibitor for 1 h before the transport experiment. After 1 h, the medium was replaced with fresh medium with inhibitor (both compartments) and radiolabeled drug in either the apical or basal compartment and transport experiments commenced. At 1, 2, 3, and 4 h, 50-μl aliquots were sampled from the opposite compartment, counted, and expressed as the percentage of radioactivity appearing in the opposite compartment relative to radioactivity added at time 0. The quality of the cell monolayers was determined by routinely measuring before the experiment the transepithelial electrical resistance that normally ranged from 100 to 250 ohm · cm2. Translocation of [3H]vinblastine was used as a positive control in each experiment. The radiolabeled drug was always added at a 2 or 5 μM final concentration containing ∼0.25 μCi/ml.
Drug Distribution Studies.
Male mdr1a (+/+) and (−/−) mice (∼12 weeks of age) in an FVB background were obtained from Taconic Farms (Germantown, NY). Three age-matched mdr1a(+/+) mice and three mdr1a (−/−) mice were compared for each drug. For oral gavage animals were dosed with the following formulations such that 100 μl of drug was administered per 10 g of body weight. Radiolabeled drugs were added to the drug stocks. Reserpine (100 μg/ml) was dissolved in 10% (v/v) DMSO in corn oil and dosed to 1.0 mg/kg and animals received 0.1 μCi of [3H]reserpine per gram of body weight. Mice were sacrificed 4 h later. Fluconazole (100 μg/ml) was dissolved in 1% (v/v) ethanol in normal saline and dosed to 1.0 mg/kg and animals received 0.1 μCi of [3H]fluconazole per gram of body weight. Mice were sacrificed 4 h later. Dihydroergocryptine (100 μg/ml) was dissolved in 10% (v/v) DMSO in normal saline and dosed to 1.0 mg/kg and animals received 0.1 μCi of [3H]dihydroergocryptine per gram of body weight. Mice were sacrificed 4 h later. Animals were anesthetized with metofane and blood obtained by cardiac puncture into heparanized tubes. Tissues were removed and flash frozen. Tissues were weighed and homogenized in 4% (wt/vol) bovine serum albumin, and 200-μl aliquots analyzed by liquid scintillation counting. Results were calculated as nanograms of drug per gram of tissue. The statistical significance of differences between total radioactivity levels in tissues ofmdr1a (+/+) versus (−/−) mice was determined using the student's unpaired two-tailed t test.
Results
Characterization of LLC Cell Lines Stably Expressing mdr1b
Expression of mdr1b Protein.
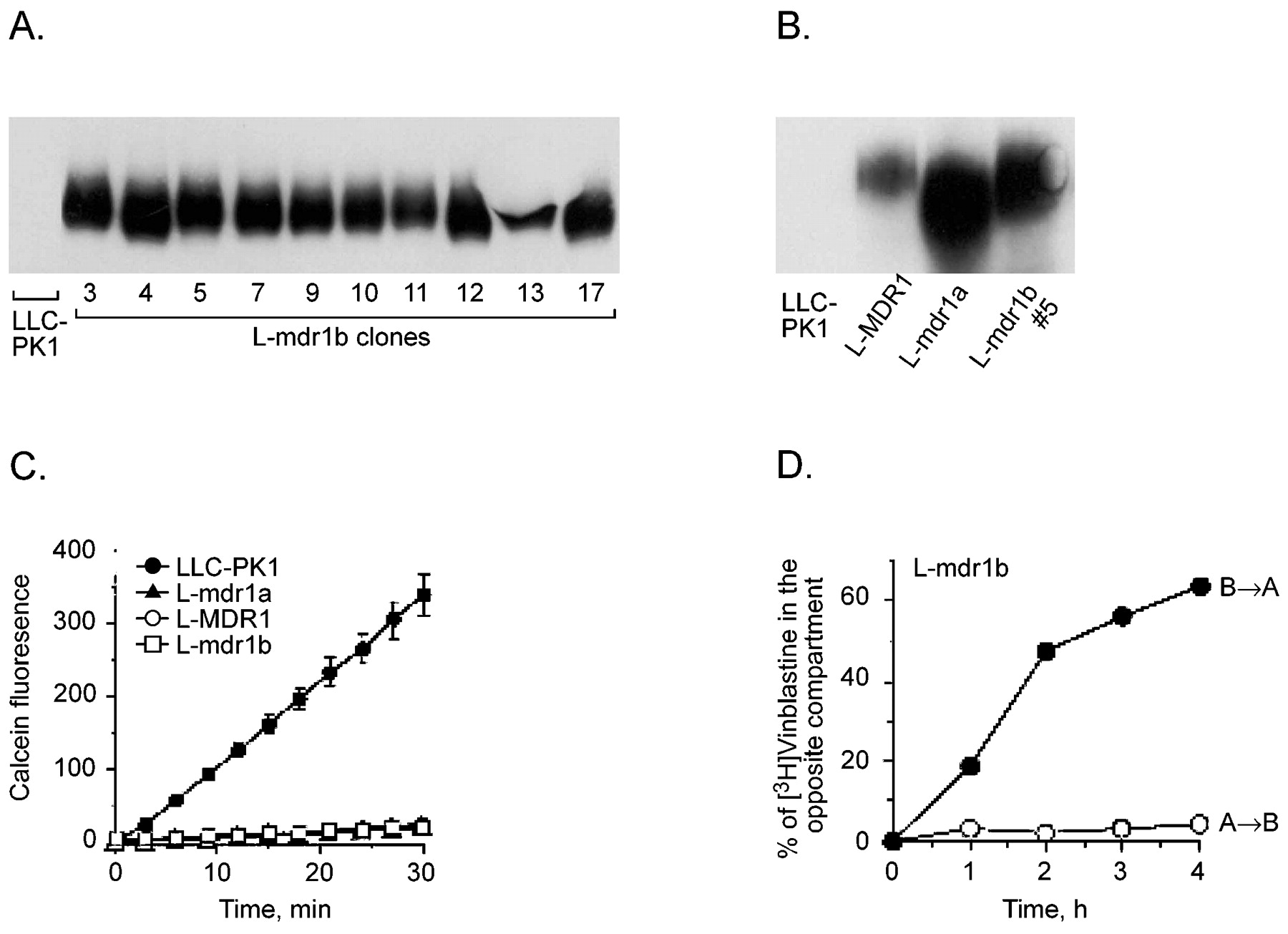
The expression of mdr1b was determined among 10 L-mdr1b clones by immunoblot analysis with anti-MDR1 antibody (Fig. 1A). Mdr1b was readily detectable in each of the clones but undetectable in LLC-PK1 parental cells. Pgp protein was robustly expressed in each cell line used in this study: L-MDR1 (human), L-mdr1a (mouse), and L-mdr1b (rat) (Fig. 1B).

Characterization of L-mdr1b cell lines. A, 20 μg of total lysate from individual L-mdr1b clones (3–17) was compared with LLC-PK1 cells on immunoblots developed with anti-MDR1 IgG. B, 20 μg of total lysate from LLC-PK1, L-MDR1, L-mdr1a, and L-mdr1b clone 5 was compared on immunoblots with anti-MDR1 IgG. C, calcein-AM uptake into LLC-PK1 (●), L-MDR1 (○), L-mdr1a (▴), and mdr1b (■) cells. Calcein-AM at 1 μM was added at time 0 and fluorescence was measured every 3 min. Each data represent the mean ± the standard deviation of four independent measurements. D, transepithelial transport of [3H]vinblastine (2 μM) in L-mdr1b cell clone 11. Open symbols reveal the apical-to-basal transport, and closed symbols show the basal-to-apical transport of [3H]vinblastine. Each data point represents single measurements.
Calcein Accumulation.
mdr1b function was characterized with a calcein-AM microfluorometric assay that takes advantage of the fact that calcein-AM, a nonfluorescent substrate for Pgp, is cleaved by cytosolic esterases inside the cell into the fluorescent product calcein that is not a Pgp substrate. Previous studies have demonstrated that there is a good correlation between the level of Pgp expression and intracellular calcein signal (Liminga et al., 1994; Tiberghien and Loor, 1996). The uptake of calcein-AM into LLC-PK1 cells and derivative cells expressing MDR1, mdr1a, and mdr1b was linear over time at 0.5, 1, or 1.5 μM (data not shown). Addition of 1 μM calcein-AM resulted in a time-dependent increase in calcein fluorescence in LLC-PK1 cells (9.4 ± 0.39 fluorescence units/min), whereas comparatively, L-mdr1a and L-MDR1 cells showed significantly reduced uptake of calcein-AM and intracellular calcein fluorescence (0.84 ± 0.21 and 0.88 ± 0.1 fluorescence units/min, respectively) (Fig. 1C). Each of the mdr1b cell lines was capable of preventing uptake of calcein-AM (range among 10 independent clones from 0.51 to 2.29 fluorescence units/min).
Vinblastine Transport.
We evaluated the ability of mdr1b/Pgp to alter the translocation of vinblastine using polarized LLC-PK1 cells or L-mdr1b clone #11. In LLC-PK1 cells the rate of vinblastine movement is the same in either direction (apical to basal and basal to apical; data not shown). Compared with LLC-PK1 cells the rate of apical-to-basal movement of vinblastine was decreased, and the rate of basal-to-apical flux was increased in L-mdr1b cells (Fig. 1D).
Effect of Prototypical CYP3A and Pgp Inhibitors on Calcein-AM Accumulation in L-MDR1, Mouse L-mdr1a, and rat L-mdr1b Cells, and on [3H]Vinblastine Accumulation in L-MDR1 Cells
The inhibitor constant (Ki) for drugs that interfere with CYP3A-mediated metabolism of CsA in human hepatocytes has been established previously (Pichard et al., 1990) (Table 1). We analyzed the ability of these same drugs to block Pgp-mediated efflux using the microfluorometric calcein-AM assay (Liminga et al., 1994; Tiberghien and Loor, 1996). Because calcein-AM is effluxed by Pgp, coincubation with a Pgp inhibitor will block Pgp efflux of calcein-AM and result in a greater calcein fluorescence. The ability of three reported Pgp and CYP3A inhibitors to restore calcein-AM uptake to cells containing Pgp was first determined. MDR1 and mdr1b Pgps showed the same rank order of inhibition by these three Pgp modulators (Table 1). The potency of inhibition did not seem directly related to the modulators partition coefficient.
Ki for reversal of accumulation of vinblastine or calcein-AM in L-MDR1, L-mdr1a, and L-mdr1b cells compared with LLC-PK1 parent cells
A more traditional assay of measuring Pgp inhibition is to analyze the effect of drugs on the accumulation of [3H]vinblastine. TheKi for reserpine and cyclosporin A blocking Pgp uptake of vinblastine in L-MDR1 cells was very similar to the Ki for enhancing vinblastine or daunomycin accumulation in P388 cells expressing MDR1 (Lan et al., 1996). For most drugs, lower concentrations of reversal agent were required to inhibit the uptake of the radiolabeled probe [3H]VBL, compared with the fluorescent probe calcein-AM, similar to another report comparing Pgp functional assays (Bosch et al., 1997).
Macrolide Antibiotics as Pgp Inhibitors.
The macrolide antibiotics triacetyloleandomycin and erthryomycin are potent inhibitors of CYP3A (Table 1). Preincubation with 1, 10, or 100 μM erythromycin had no effect on calcein fluorescence in LLC-PK1, L-MDR1, L-mdr1a, or L-mdr1b cells (Table 1). Treatment with 50 and 100 μM TAO slightly restored calcein retention in MDR1 cells to 15.1 and 25.1% of the calcein signal in LLC cells. TheKi values of erythromycin and troleandomycin for MDR1 were over 1000 and 483.3 μM, respectively (Table 1). MDR1 and mdr1a showed similar sensitivity to TAO, whereas TAO failed to inhibit mdr1b. In contrast to its inability to enhance calcein-AM uptake in L-MDR1 cells, erythromycin increased [3H]VBL accumulation with aKi of 38 (Table 1). This observation seems to be consistent with the report that Pgp substrates are unable to increase calcein-AM uptake (Tiberghien and Loor, 1996) because we have shown that erythromycin is transported by Pgp (Schuetz et al., 1998).
Azole Antifungals as Pgp Inhibitors.
Comparison of the interactions of clotrimazole, miconazole, ketoconazole, and fluconazole with the various cell lines revealed a rank order ofKi values in which KCZ > clotrimazole > miconazole > FCZ (Table 1). Fluconazole was incapable of blocking Pgps effect on calcein-AM at concentrations of 10, 50, or 100 μM, with the highest concentration tested representing 10 times the Ki reported for fluconazole inhibition of CYP3A4-mediated metabolism of midazolam (Gibbs et al., 1999) or CsA (Pichard et al., 1990). There was good correlation between the ability of azole antifungals to enhance vinblastine and calcein-AM accumulation (r2 = 0.999).
Dopaminergics as Pgp Inhibitors.
Structure-activity relationship among ergot peptides in their ability to block Pgpfunction. The ergot peptides interact with CYP3A (Peyronneau et al., 1994). The ergot peptides selected are amides formed between lysergic acid, or bromolysergic acid, and cyclic tripeptides all containing a proline residue. The chemical structure of the ergot peptides is shown in Table 2. To explore the structure-activity relationship among the ergot alkaloids we started with ergometrine, the lysergic acid derivative lacking any cyclic tripeptide and found it weakly capable of restoring calcein-AM uptake in L-MDR1 and mdr1a with estimatedKi values of 115 and 117 μM. In contrast, almost 10 times more ergometrine was needed to effectively inhibit mdr1b. Similarly, ergometrine was the least potent ergot alkaloid at restoring vinblastine accumulation.
Structure activity relationship of ergot and dihydroergot alkaloids interactions with P-glycoprotein

A simple substitution of the hydrogen at the R1 position (ergocornine) to a bromine [bromocriptine (Br)] greatly increased the ability of the ergot peptide to inhibit MDR1 or mdr1a Pgp and restore accumulation of either calcein-AM or vinblastine. For MDR1, bromocriptine had aKi (2.81 μM) that was 50 times lower than ergocornine (105) in affecting calcein-AM accumulation. It should be noted that this bromine R1 substitution had very little effect on mdr1b inhibition (ergocornine Ki = 19 μM versus bromocriptine Ki = 6.4 μM).
Increasing the apparent length of the R2 (5′ position) side chain yielded better inhibitors of MDR1 and mdr1a, unlike mdr1b. For instance, the rank order of Ki values for MDR1 (with calcein-AM) ranged from α-ergocryptine with the longest R2 substitution (methyl-propyl) (Ki = 12.2 μM) > ergocristine with a phenyl-methyl (Ki = 42.8 μM) > ergocornine with the smallest pseudopeptide moiety R2 = CH(CH3)2 (methyl-ethyl) (Ki = 105.2 μM) and may be due to the increased lipophilicity of ergocristine compared with ergocornine. Similarly, increasing the length of the R2 substitution increased the potency of ergot alkaloids in inhibiting MDR1 and restoring vinblastine accumulation.
Modification at R3 (2′ position) also effected the potency of the ergot peptides to interact with MDR1 and mdr1a but not mdr1b, and this was seen with either vinblastine or calcein-AM as substrates. For example, with calcein-AM as a substrate, modification from a CH(CH3)2 (methyl-ethyl) (ergocristine) to a CH3 (methyl) (ergotamine) decreased the potency of inhibition from 42.8 to 98.9 μM for MDR1 and from 39.4 to 188 μM for mdr1a.
Reduction of the double bond at position 9,10 of lysergic acid to yield the respective dihydro-ergopeptides severely altered the inhibitory concentrations for MDR1, mdr1a, and mdr1b with calcein-AM as a substrate. Conversion of ergocristine to dihydroergocristine required 3- to 10-fold greater drug to inhibit the Pgps. It should be noted that this loss in inhibitory potency is not due to a decrease in lipophilicity because for the most part the reduction in the 9,10 double bond of lysergic acid increases lipophilicity. These studies reveal that mdr1b has different structural requirements for inhibition by ergot peptides compared with MDR1 and mdr1a, which for the most part behave similarly. Interestingly, when vinblastine was the substrate the potency of ergocristine and ergocryptine and the dihydro congeners to each inhibit MDR1 were similar. Only the dihydro modification of ergotamine resulted in a significant loss of potency toward MDR1.
However, for the ergot alkaloids two of the dihydroergots (dihydroergocriptine and dihydrogocristine) enhanced accumulation of VBL while having little effect on calcein-AM (Ki > 360). This result suggested that these two ergot alkaloids might be substrates for Pgp.
Ergot peptides have previously been analyzed for their affinity for cytochromes P450 3A. Microsomes from yeast producing hPCN1 (CYP3A4) were incubated with ergopeptide alkaloids and substrate binding to CYP3A measured by difference visible spectroscopy.Ki values (micromolar concentration) were reported for many ergot alkaloids (Peyronneau et al., 1994). A separate report (Pichard et al., 1990) determined theKi for bromocriptine, ergotamine, and dihyroergotamine to inhibit CYP3A4 metabolism of cyclosporin A (Table2).
Correlation between Inhibition of Human and Rodent mdr1.
For the azole antifungals there was good correlation between inhibition of MDR1 and mdr1a and between MDR1 and mdr1b (r2 = 0.998 and 0.789, respectively) and between mdr1a versus mdr1b (r2 = 0.789). For the ergot alkaloids inhibition of MDR1 was strongly correlated to inhibition of mdr1b (r2= 0.923) but not to inhibition of mdr1a (r2 = 0.199). However, if the dihydroergot alkaloids were left out of the correlational analysis the association for MDR1 and mdr1a inhibition improved (r2 = 0.682). For all ergot alkaloids the correlations were calculated based on theKi values that could actually be measured; therefore, ergometrine was not included in the MDR1 versus mdr1b calculation.
Correlation between a Compound's Lipophilicity and Ability to Inhibit Pgp.
It has been suggested that the octanol/water partition coefficient is highly correlated with the ability of some drugs to interact with Pgp (Lampidis et al., 1997). Therefore, we compared Ki values for these drugs to block Pgp-mediated efflux of calcein on L-MDR1 cells with the octanol/water partition coefficient. When we combined all of the compounds in these studies and compared their lipid solubility with their Ki for Pgp inhibition of calcein-AM or vinblastine accumulation (r2 = 0.02), there was no correlation. However, among the azole antifungals there was a good correlation between lipophilicity and inhibition of MDR1/Pgp measured by calcein-AM (r2 = 0.957) or vinblastine (r2 = 0.952) assays. Among the class of ergot alkaloids, the inhibitory potency Pgp inhibitors was positively correlated to lipophilicity. The correlation for inhibition of MDR1 by all ergot alkaloids tested by calcein-AM assay and the octanol/water partition coefficient wasr2 = 0.422. However, excluding the dihydroergot peptides (that showed no efficacy as Pgp blockers), we observed a high correlation between lipophilicity and inhibition of MDR1/Pgp function as measured by intracellular calcein (r2 = 0.913) (Fig.2). This result is consistent with other reports that among structurally related compounds there is a significant correlation between a compound's lipid solubility and its ability to interact with Pgp as either an inhibitor (Lampidis et al., 1997; Khan et al., 1998) or as a substrate (Stein, 1997).
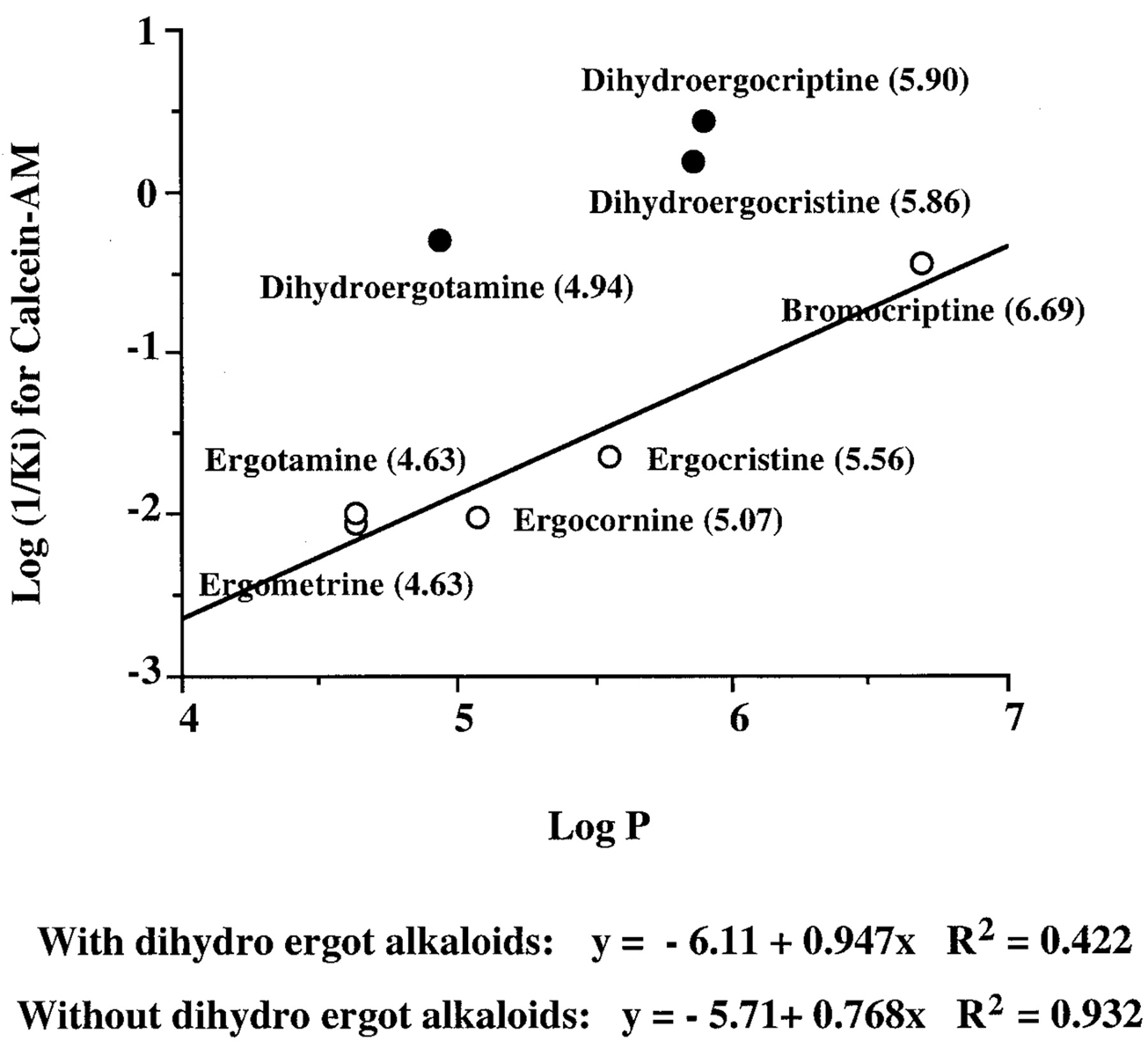
Correlation of MDR1-modulating activity (Ki) and lipophilicity. A, inhibition of MDR1 (expressed as log (1/Ki) values of modulators determined in calcein-AM uptake assays) and log P values. The value in each of the parentheses represents log P.
Inhibition of L-MDR1-mediated Transport of [3H]Vinblastine across Cells Cultured in Transwell.
We further compared representative and potent inhibitors identified in the calcein-AM assay for their capacity to inhibit Pgp-mediated transport of [3H]vinblastine across L-MDR1 cells cultured in Transwell dishes. Addition of 20 μM inhibitor to apical and basal compartments before addition of [3H]vinblastine demonstrated that reserpine and cyclosporin A, inhibited [3H]vinblastine transport in both directions, enhancing apical-to-basal flux and decreasing basal-to-apical transport across L-MDR1 cells (Fig. 3, A and B). Ketoconazole enhanced [3H]vinblastine apical-to-basolateral movement up to 8.4%, but was less effective on basal-to-apical flux of [3H]vinblastine across L-MDR1 cells (Fig. 3C). Fluconazole had no effect on flux of [3H]vinblastine in either direction (Fig. 3D). Bromocriptine enhanced [3H]vinblastine apical-to-basal transport up to 10.2% and decreased basal-to-apical transport up to 28.9% across L-MDR1 cells (Fig. 3E). In contrast, dihydroergocryptine had no effect on flux of [3H]vinblastine in either direction (Fig. 3F), consistent with its poor ability to inhibit calcein-AM transport (Table2).
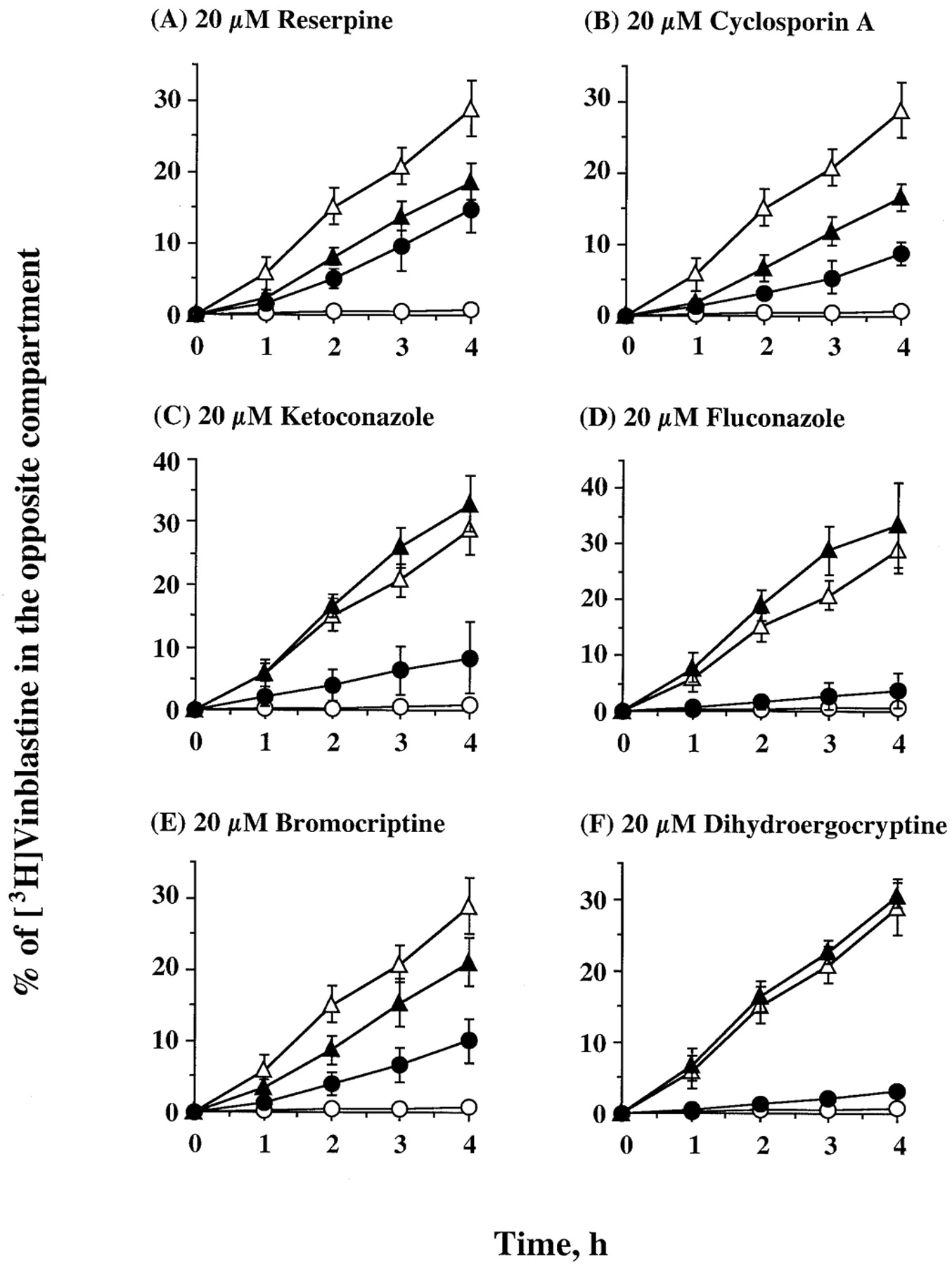
Effect of reserpine (A), cyclosporin A (B), ketoconazole (C), fluconazole (D), bromocriptine (E), and dihydroergocryptine (F) on the transepithelial transport of [3H]vinblastine across L-MDR1 cell monolayers. Cells were preloaded in both compartments for 1 h with 20 μM reagents and the assay started by adding [3H]vinblastine to either the apical or basal compartment and sampling the opposite compartment at the indicated times. The apical-to-basal transport (●) and basal-to-apical transport (▴) in the presence of 20 μM reversal reagents were compared with the same movement in the absence of reversal reagents (open symbols). Each data point represents the mean ± standard deviation of five to eight independent measurements.
Evaluation of Drugs as Pgp Substrates
We selected representative compounds from each class of drugs [macrolide antibiotics (erythromycin), azole antifungals (fluconazole), classical Pgp inhibitor (reserpine), and ergot alkaloid (dihydroergocriptine)] for further study as Pgp substrates in vitro and in vivo. We previously demonstrated that the representative macrolide antibiotic erythromycin is Pgp-transported (Schuetz et al., 1998). Compared with translocation in the parent LLC-PK1 cells, the rate of apical-to-basal flux of [3H]reserpine was diminished, whereas basal-to-apical flux was enhanced 1.5- to 2-fold in both L-MDR1 cells and L-mdr1a cells compared with LLC-PK1 cells (Fig. 4). These results demonstrated that reserpine is a substrate for mouse and human mdr1. There was a small difference in the rate of transepithelial translocation of the azole antifungal [3H]fluconazole in either direction across the epithelia of L-mdr1a cell compared with LLC-PK1 cell, whereas flux of fluconazole in either direction was no different between L-MDR1 and LLC-PK1 cells (Fig. 4). Last, we tested [3H]dihydroergocryptine, an ergot alkaloid for its ability to be Pgp-translocated. L-MDR1 and L-mdr1a exhibited markedly greater basal-to-apical transport and significantly diminished (30-fold) apical-to-basal transport compared with the parent LLC-PK1 cells (Fig. 4). This result indicates that dihydroergocryptine is a substrate for human MDR1 and mouse mdr1a Pgp.

Transepithelial transport of compounds by MDR1/Pgp. The apical-to-basal transport and basal-to-apical movement of [3H]fluconazole, (FCZ), [3H]dihydroergocryptine (DECP), and [3H]reserpine (2 μM) across LLC-PK1 cells (○), L-MDR1 cells (▴), and L-mdr1a cell (▪) monolayers. Each data point represents the mean ± the standard deviation of four independent measurements.
Drug Distribution Studies
To extend these findings in vivo, we determined the influence ofmdr1a on the oral absorption and tissue distribution of some of these drugs in mice nullizygous for mdr1a. We expected to find that the mdr1a (−/−) mice, compared withmdr1a (+/+) mice, would achieve higher tissue concentrations of any xenobiotic whose translocation was influenced by Pgp, particularly in the brain.
Mice received a single oral dose of [3H]reserpine (1.0 mg/kg) 4 h before sacrifice. The plasma level of reserpine was not different in the wild-type compared with mdr1a (−/−) mice. The ratio of total radioactivity was 1.5- to 2-fold higher in liver, heart, kidney, lung, and spleen of knockout mice compared with wild-type mice (Table3). Likewise, the [3H]reserpine was almost 3-fold higher in the brains of the mdr1a (−/−) mice, although this difference did not reach statistical significance because of interanimal variation. These results demonstrate that reserpine disposition is influenced bymdr1a Pgp. In a separate study we treated mdr1a(−/−) and (+/+) mice orally with 5 mg/kg reserpine for 24 h and observed only in the mdr1a (−/−) mice pronounced lethargy, a sedative side effect most likely attributable to the centrally acting side effects of this drug.
Tissue levels of radioactivity in mdr1a (+/+) and (−/−) mice 4 h after oral gavage
The absence of mdr1a had no effect on the plasma concentration of [3H]fluconazole 4 h after a single oral treatment (1.0 mg/kg). Furthermore, the tissue concentrations of fluconazole except liver were not different betweenmdr1a (+/+) and (−/−) mice (Table 3). Indeed, there was 2-fold greater total radioactivity in the livers of the wild-type mice, compared with the mdr1a knockouts. Thus, fluconazole disposition is not influenced by mdr1a Pgp.
The tissue distribution of the dopaminergic dihydroergocryptine was assessed 4 h after an oral dose of 1.0 mg/kg. The 2-fold higher plasma concentration of [3H]dihydroergocryptine in mdr1a (−/−) mice supports the concept of intestinal Pgp limiting oral dihydroergocryptine absorption (Table 3). Dihydroergocryptine concentration of kidney in mdr1a (−/−) mice was higher (4-fold) compared with wild-type mice, with other mdr1a (−/−) tissues showing 1.5- to 2-fold greater radioactivity than (+/+) mice, which may reflect the increased plasma concentration of drug.
Discussion
To determine how drugs can best be used to maximize their efficacy and minimize their toxicity requires knowledge of all of the dynamic processes involved in drug disposition. Given the potential overlap in substrates, inducers, and inhibitors of Pgp and CYP3A4 it becomes important to understand the extent to which each component of drug detoxification may participate in a drug interaction. Toward this goal we characterized the ability of representative CYP3A4 inhibitors to interact with human and rodent Pgp using two different Pgp substrates. The rank order and absolute Ki values of drugs inhibiting calcein-AM versus vinblastine uptake showed incomplete overlap. One factor that could contribute to the disparity in the effect of Pgp modulators is that vinblastine and calcein-AM are binding to distinct sites on Pgp (Shapiro and Ling, 1997; Shapiro et al., 1999). Thus, the effect of inhibitors on Pgp may be substrate-dependent, reflecting the differential extent to which calcein-AM or vinblastine interacts with either of the Pgp binding sites and the differential extent to which the inhibitors compete at these or other inhibitory sites. The knowledge of how calcein AM interacts at one or both Pgp binding sites may be informative in enhancing our understanding of how chemicals interact with Pgp because some of the compounds tested were also Pgp substrates (e.g., erythromycin and dihydroergocryptine) but could not enhance calcein-AM accumulation; whereas other compounds were Pgp substrates (e.g., cylosporin A and reserpine) and could enhance calcein-AM accumulation. Cumulatively, these results suggest the need to select more than one Pgp substrate when screening for drug interactions, because the extent of inhibition may vary depending on the substrate chosen.
If the inhibitors (and substrates) interact with mdr1-like Pgps in each species in a similar manner then the rank order of inhibitors would be expected to be similar. Some inhibitors were equally potent for the three transporters, whereas some drugs showed no relationship in their ability to interact with the three transporters. Because the amino acid sequences of the three mdr1 transporters are not identical, the substrate-inhibitor transporter interactions could vary. Indeed, it is well documented that amino acid changes in Pgp can affect Pgp function. Nevertheless, it is important to compare the ability of Pgp modulators to interact with rodent and human Pgp because the effects of Pgp modulators are frequently screened in rodents and rodent xenograft models. Additionally, because the mouse mdr1b and rat mdr1b share 92% identity, building a larger database of information regarding the structural features of chemicals important for interaction with the various mdr1 proteins (Pgp pharmacophores) will ultimately aid in pinpointing the amino acid residues in Pgp important for interaction with drugs (Ekins et al., 2002).
There is a growing body of knowledge of structural and functional features that make an effective Pgp modulator. Structural features important for ergot alkaloid inhibition of Pgp may be important determinants for the activity of other compounds. The cyclic tripeptide moiety of ergot alkaloids was important for the interaction of these drugs with Pgp as the absence of the cyclic tripeptide in ergometrine rendered it relatively incapable of blocking Pgp function. The most potent ergot peptide, bromocriptine, increased the accumulation of calcein to a level comparable with that observed in the parental LLC cells. The position and functionality of R1, the bromine in bromocriptine (Ki = 2.81) compared with the hydrogen in ergocornine (Ki = 105.2) markedly enhanced the efficacy of drugs as Pgp modulators. Reduction of the double bond at carbons 9,10 and introduction of hydroxyl groups decreased any ability to restore calcein-AM accumulation. In contrast, dihydroergocriptine and dihydroergocristine were effective at restoring vinblastine accumulation. A possible explanation for this discrepancy is that it has previously been observed that Pgp substrates do not enhance calcein-AM uptake in Pgp-overexpressing cells (Tiberghien and Loor, 1996). Interestingly, our results showing that Pgp interacts with ergot alkaloids may also explain the “hepatic, nonmonoamine dihydroergocriptine binding sites” previously identified in syriam hamster liver membranes (Korneyev and Cincotta, 1996). Finally, the identification of ergot alkaloids as Pgp inhibitors adds these chemicals to a growing list of paseudopeptides that interact with Pgp (e.g., CsA, rapamycin, FK506, PSC833, and pristinamycin IA; Phung-Ba et al., 1995) and support the notion that endogenous peptides or pseudopeptide compounds interact with this transporter (Oude Elferink and Zadina, 2001).
Azole antifungals are involved in many drug-drug interactions (Albengres et al., 1998). The rank order of these drugs as effective Pgp inhibitors was KCZ > CTZ > miconazole. Inhibition of Pgp by KCZ is consistent with other reports that this azole antifungal can block Pgp and can enhance the oral bioavailability of Pgp substrates (Salphati and Benet, 1998; Zhang et al., 1998). Fluconazole was only weakly capable of interacting with Pgp as an inhibitor. This finding is consistent with the decreased propensity for drug-drug interactions with fluconazole compared with other antifungals such as KCZ, an avid Pgp inhibitor. Fluconazole is also less potent as a CYP3A4 inhibitor (Ki = 9.21 μM) compared with ketoconazole (Ki = 26.7 nM) (Gibbs et al., 1999). A species difference in FCZ transport was noted; FCZ was a mouse mdr1a substrate but was not a substrate for human MDR1.
Another azole antifungal agent, itraconazole (ITZ), is a substrate for mdr1a Pgp. This evidence came in part from studies demonstrating a higher concentration of ITZ in the brains and plasma of mdr1a (−/−) compared with (+/+) mice (Miyama et al., 1998). However, these authors found that the concentration of ITZ in the livers of the mdr1a (+/+) mice were actually higher than in (−/−) mice. This result is strikingly similar to the enhanced liver concentration of [3H]FCZ in the livers of the mdr1a (+/+) compared with (−/−) mice. This result was all the more surprising because fluconazole seemed to be a weak substrate for mouse mdr1a in Transwell transport experiments (Fig. 4). Because azole antifungals bind avidly to CYP3A proteins, we compared CYP3A expression in the livers of these mdr1a (+/+) and (−/−) mice treated with FCZ but found no difference in CYP3A expression between the mdr1a genotypes (F. Schuetz, unpublished observation). We favor the idea that in the absence of mdr1a, another hepatic transporter is up-regulated and decreases the liver concentration of FCZ in mdr1a (−/−) mice. This transporter is unlikely to be mdr1b because mdr1b Pgp levels are also elevated in kidneys of mdr1a (−/−) mice (Schinkel et al., 1994), yet we observed no difference in kidney FCZ disposition in mice with and without mdr1a. In other species such as Candida dubliniensismultidrug transporters involved in fluconzole transport have been identified. The same C. dubliniensis transporter also interacts with methotrexate. Because several of the mammalian multidrug-resistance proteins have been shown to affect the intracellular disposition of methotrexate it is possible that, by analogy with Candida, one of these multidrug-resistance proteins participates in hepatic efflux of fluconazole.
The overlapping tissue distribution of CYP3A and Pgp and broad spectrum of drugs that interact with both proteins present particular challenges to drug absorption and delivery to the systemic circulation. Moreover, the interaction of coadministered drugs with CYP3A and Pgp in the gut leads to major drug-drug interactions. Given the potential overlap in substrates, inducers, and inhibitors of Pgp and CYP3A4 it becomes important to understand the relative contribution of CYP3A and Pgp to specific drug interactions. Most drugs that inhibit CYP3A also inhibited Pgp. These results suggest that many drugs that are CYP3A inhibitors also inhibit Pgp and that Pgp plays an important role in drug interactions with ergot alkaloids and macrolide antibiotics. Moreover, because there are some significant differences in theKi for these drugs with Pgp versus CYP3A, this will contribute to interindividual variability in the magnitude of inhibitory drug-drug interactions.
Clinically important drug interactions have been reported with concurrent oral administration of ergot alkaloids, erythromycin, or TAO (Campana et al., 1996) and other drugs (von Rosenteil and Adam, 1995;Campana et al., 1996). For example, a transient increase in CsA bioavailability and synergism in prevention of autoimmune disease in rats has been noted previously when long-acting bromocriptine microcapsules were administered concurrently (Niedhart, 1996); and erythromycin increases the plasma concentration of α-dihydroergocryptine in humans (de Mey et al., 2001). These drug interactions are thought to be mediated by CYP3A because ergotamine and dihydroergotamine were proposed as CYP3A substrates (Pichard et al., 1990), and ergots such as ergotamine, dihydroergotamine, and bromocriptine are potent inhibitors of CYP3A-mediated metabolism (Pichard et al., 1990). Drug interactions with macrolide antibiotics are attributed to inhibition of CYP3A (Thummel and Wilkinson, 1998). For drugs such as erythromycin the apparentKi for inhibition of MDR1-Pgp (37 μM) is in the same range as CYP3A inhibition (16–194 μM), and thus Pgp clearly contributes to many drug interactions with erythromycin. On the other hand, the Ki for inhibition of Pgp by some compounds (such as the azole antifungals) was markedly higher than the concentration required to inhibit CYP3A. For example, the estimated MDR1 Ki for fluconazole was 400 to 1000 μM, whereas the Kifor CYP3A is 1.3 to 63 μM. The extent of Pgp inhibition, and the extent to which it participates in drug-drug interactions, will thus be directly linked to the extent to which these relevant concentrations of fluconazole can be achieved in the small intestine or even in plasma. Intriguingly, TAO inhibits CYP3A with aKi of ∼10 μM (Pichard et al., 1990), whereas the Ki for MDR1 inhibition was ∼9-fold higher. Presumably this differential inhibition is due to the very different nature of inhibition of these proteins, i.e., TAO inhibits CYP3A4 by forming a stable cytochrome P450-iron-metabolite complex. In total, these results suggest that although some systemic drug interactions involve both the CYP3A4 and Pgp locus (e.g., erythromycin and ketoconazole) other systemic drug interactions (e.g., FCZ and TAO) may predominantly involve CYP3A4.
Acknowledgments
We gratefully acknowledge Dr. Shimon Schuldiner (Alexander Silberman Institute of Life Sciences, Hebrew University, Jerusalem, Israel) for the [3H]reserpine.
Footnotes
-
This study was supported by National Institutes of Health Grants ES08658 and P30 CA21745 and a Cancer center support grant, and the American Lebanese Syrian Associated Charities.
-
DOI: 10.1124/jpet.102.037549
- Abbreviations:
- Pgp
- p-glycoprotein
- MDR/mdr
- multidrug resistance
- AM
- acetoxymethyl ester
- DMSO
- dimethyl sulfoxide
- TAO
- triacetyloleandomycin
- VBL
- vinblastine
- KCZ
- ketoconazole
- FCZ
- fluconazole
- CsA
- cyclosporin A
- ITZ
- itraconazole
- Received April 15, 2002.
- Accepted June 4, 2002.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}