


Abstract
CYP3A5 is the major CYP3A form in the human lung, and it is inducible by dexamethasone in the human A549 lung adenocarcinoma cell line. In the present study, we characterized the nature and mechanism of this induction process. The induction of CYP3A5 mRNA was assessed by quantitative reverse transcriptase-polymerase chain reaction in A549 cells. About 4-fold induction was detected by nanomolar concentrations of dexamethasone and also by budenoside and beclomethasone dipropionate, glucocorticoids used for the inhalation treatment of bronchial asthma, whereas the CYP3A4 inducers mifepristone (RU486), rifampicin, clotrimazole, and nifedipine were without effect. The glucocorticoid induction was blocked by the glucocorticoid receptor (GR) antagonist RU486. In transient transfection assays to A549 cells, CYP3A5 5′ regulatory region was activated by the dexamethasone treatment. In contrast, dexamethasone was unable to induce CYP3A5 transcription in GR-deficient COS-1 cells, but the induction could be achieved after GR cotransfection. The CYP3A5 expression was measured in alveolar macrophages from patients with respiratory diseases. The CYP3A5 expression level was decreased by smoking, but glucocorticoid therapy had no statistically significant effect. In conclusion, CYP3A5 is induced in the A549 cells by glucocorticoids through a GR-mediated pathway, whereas smoking may be able to depress CYP3A5 expression.
The lung is a major target organ for a multitude of inhaled chemical substances. Similar to the liver, which has efficient metabolic functions against xenobiotics entering the body, the lung also exhibits enzymatic defense mechanisms. Most attention has been focused on the cytochrome P450 (P450) enzyme system, which is responsible for the phase I metabolism of xenobiotics. Different P450 forms are expressed in various human pulmonary cell types. The major basis for pulmonary P450 research is that variations in the level of expression of this enzyme system are implicated as being partly responsible for the interindividual differences in susceptibility to lung cancer (Raunio et al., 1995; Bartsch et al., 2000).
Lung tissues activate several procarcinogens, such as the polycyclic aromatic hydrocarbon benzo[a]pyrene (B[a]P) and several N-nitrosamines, to form DNA adducts (Autrup, 1990). It has been shown that benzo[a]pyrene diol epoxide, the ultimate carcinogenic metabolite of B[a]P, causes lung cancer-specific mutations in the p53 tumor suppressor gene in human bronchial epithelial cells (Denissenko et al., 1996), thus providing a direct link between lung cancer and B[a]P exposure. CYP1A1 constitutes the major P450 enzyme activating B[a]P, but CYP3A enzymes may play a role in the second step by catalyzing activation of proximate metabolite B[a]P-7,8-diol (Shimada et al., 1989). Research on pulmonary P450 enzymes has been dominated by CYP1A1, which is induced by tobacco smoke in the human lung (McLemore et al., 1990; Anttila et al., 1991). Genetic polymorphisms of this gene have been linked with a higher risk for the development of lung cancer in the Japanese population (Kawajiri, 1999).
Human airway epithelial cells also contain CYP3A5 protein, which constitutes the major CYP3A form in the human lung and is localized to bronchial and alveolar epithelium (Kivisto et al., 1996; Anttila et al., 1997; Mace et al., 1998). CYP3A5 activates B[a]P dihydrodiols to DNA-binding metabolites (Roberts-Thomson et al., 1993). CYP3A5 also metabolizes steroid hormones, including glucocorticoid cortisol (Wrighton et al., 1990; Waxman et al., 1991; Kocarek et al., 1995).
CYP3A5 has been generally thought to be noninducible, but recent reports from our laboratory and others have given evidence for glucocorticoid induction of CYP3A5 (Schuetz et al., 1996; Hukkanen et al., 2000). The characterization of CYP3A5 regulation has been previously confused by the complex structure of the CYP3A gene locus. A recent report revealed that the originally cloned CYP3A5 5′ (Jounaidi et al., 1994) actually represent 5′ to the CYP3AP1 pseudogene, with an exon 1 similar to CYP3A5 (Finta and Zaphiropoulos, 2000). Therefore, the mechanisms behind the CYP3A5 induction by glucocorticoids need to be reanalyzed.
In our previous study, we detected the induction of CYP3A5 by dexamethasone in the human A549 lung adenocarcinoma cell line (Hukkanen et al., 2000). In the current study, we have further characterized the induction of CYP3A5 in human lung cells by glucocorticoids, including several compounds used for the treatment of asthma, and we demonstrate that CYP3A5 is induced in human lung-derived cells by a mechanism involving transcriptional induction mediated by the glucocorticoid receptor.
Materials and Methods
Patients.
Bronchoalveolar lavage samples were obtained from 32 patients (12 nonsmokers, 10 ex-smokers, and 10 smokers) during normal diagnostic procedures. All patients were examined due to a suspicion of lung cancer or parenchymal disease. The ex-smokers had stopped smoking 1 to 29 years before the procedure. Six patients were current glucocorticoid users, five had stopped their glucocorticoid use 3 days to 3 weeks before the procedure, and 21 patients had never used glucocorticoids. The study was approved by the local ethics committee, and the written informed consent for participation in this study was obtained from the patients.
The bronchoalveolar macrophage samples were prepared as described earlier (Piipari et al., 2000). Briefly, bronchoalveolar lavage was performed under local anesthesia using a fiber optic bronchofiberoscope. Either a medial or lateral segment of the right middle lobe, or in a few cases, some other segment avoiding the tumor area, was lavaged with ten 20-ml aliquots of saline solution. Fresh lavage fluid was placed on ice, and a subset of cells was used for mRNA preparation.
Reagents.
Rifampicin, budesonide, beclomethasone dipropionate, dexamethasone, nifedipine, mifepristone (RU486), and clotrimazole were purchased from Sigma-Aldrich (St. Louis, MO).
Cell Culture.
The human lung adenocarcinoma cell line A549, originally from the American Type Culture Collection (Manassas, VA), was cultured in Ham's F-12 medium with l-glutamine (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen) and 10 μg/ml gentamicin (Invitrogen). The cells were cultured at 37°C in 5% CO2 and saturated humidity. Nearly confluent cells were incubated for 24 h in the presence of inducers in Ham's F-12 medium without fetal bovine serum. Control cultures contained the same concentration of vehicle (0.5% dimethyl sulfoxide). After the incubation, the cells were washed with phosphate-buffered saline (pH 7.4) suspended in the same buffer and centrifuged. The cell pellet was suspended in cell lysis buffer for mRNA extraction. The inducer concentrations used were not cytotoxic, judging from the cell morphology and the β-actin mRNA contents of the cells. HepG2 and COS-1 cells (American Type Culture Collection) were cultured in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum and 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen). Otherwise, culture conditions were similar to the A549 cells.
RT-PCR.
mRNA of A549 cells, alveolar macrophages, and human liver (positive control) was extracted with the QuickPrep Micro mRNA purification kit. mRNA (0.5 μg) was used for cDNA synthesis with the First-Strand synthesis kit. All reagents were from Amersham Biosciences AB (Uppsala, Sweden).
The PCR reactions contained 1 μl of cDNA (of the 15 μl of total cDNA), 2.0 U of DynaZyme DNA polymerase (Finnzymes, Helsinki, Finland), 5 μl of 10× DynaZyme reaction buffer, dNTP reaction mix (Finnzymes) at a final concentration of 200 μM, 50 pmol of each primer, and water to a final volume of 50 μl. Thirty-five PCR cycles of 1 min at 94°C, 1 min at 55–62°C, and 2 min at 72°C were performed. In every series of PCR reactions, there was a negative control containing an aliquot of cDNA synthesis reaction performed with heat-inactivated reverse transcriptase enzyme. All PCR reactions were performed at least three times. To exclude the chance of cross-hybridization with other sequences, each primer was compared with the European Molecular Biology Laboratory human gene bank using the FASTA program (Genetics Computer Group, Madison, WI). The primers [glucocorticoid receptor (GR), Se: GGAGTTTTCTTCTGGGTCCC, As: GAGAGCTTACATCTGGTCTC; pregnane X receptor (PXR), Se: AGCTGGAACCATGCTGACTT, As: AGGGGCGTAGCAAAGGGGTG; CAR, Se: AAGGAGCAAGAAGAGCTGATC, As: TCAGCTGCAGATCTCCTGGAG] were also designed to amplify regions containing at least one intron in the gene to exclude contamination of cDNA with genomic DNA. After the PCR, an 8-μl aliquot of each reaction mixture was electrophoresed in an agarose gel and stained with ethidium bromide. A visible band of the correct size was considered a sign of the specific mRNA being present in the sample.
Quantitative RT-PCR.
The total RNA from A549 cells for quantitative RT-PCR measurements was extracted with quanidium thiocyanate followed by centrifugation in cesium chloride. The first-strand cDNA was synthesized with the First-Strand synthesis kit (Amersham Biosciences AB) using 1 μg of RNA and pd(N)6 random hexadeoxynucleotides.
Quantitative PCR reactions were performed with an ABI 7700 sequence detection system using TaqMan chemistry (Applied Biosystems, Foster City, CA). The forward and reverse primers for CYP3A5 mRNA detection were AAGGAAGACTCACAGAACACAGTTGA and GGTTTCCACCGCCAAATTT, respectively. The 74-base pair amplicon was detected using the bifunctional fluorogenic probe 5′-Fam-AAGGAAAGTGGCGATGGACCTCATCC-Tamra-3′. The results were normalized to 18 S RNA quantified from the same samples using the forward and reverse primers TGGTTGCAAAGCTGAAACTTAAAG and AGTCAAATTAAGCCGCAGGC, respectively. The probe for the 18 S amplicon was 5′-Vic-CCTGGTGGTGCCCTTCCGTCA-Tamra-3′.
Plasmids.
The CYP3A5 5′ −1355 to +40 and the CYP3AP1 5′ −1359 to +39 regions were amplified with PCR from human genomic DNA using Dynazyme EXT polymerase (Finnzymes). The numbering is based onJounaidi et al. (1994). The PCR products were cloned usingKpnI and NheI restriction sites to pGL3-Basic (Promega, Madison, WI) in front of the luciferase reporter gene. The identity of the constructs was verified by sequencing. The CYP3A5 5′ sequence amplified from liver M24 was found to be fully identical to the GenBank sequence (accession no. NG_000004). The corresponding sequence from liver M27 had two point mutations. These nucleotide changes had no effect on transcriptional activation. The GR expression plasmid and the pGRE2TATA-luc reporter plasmid were generously provided by Dr. Jorma Palvimo (Institute of Biomedicine, University of Helsinki, Finland).
Transfections.
The A549 cells were seeded to 24-well plates the day before transfection. The cells were transfected with LipofectAMINE 2000 transfection reagent (Invitrogen) according to the manufacturer's protocol, using 1 μg of plasmid DNA and 2.5 μl of transfection lipid and Opti-MEM I media (Invitrogen). Twenty-four hours after transfection, the media was replaced with serum-free Ham's F-12 medium, and the 10 μM dexamethasone or dimethyl sulfoxide was added when appropriate for 48 h. The HepG2 cells were transfected with Tfx-20 reagent (Promega) according to the manufacturer's protocol. The COS-1 cells were seeded to 24-well plates the day before transfection and transfected with LipofectAMINE 2000 transfection reagent, using 0.8 μg of reporter plasmid DNA, 0.2 μg of GR expression vector, and 3 μl of transfection lipid and Opti-MEM I media. The treatment was similar to A549 cells. To provide an internal control for transfection efficiency, a second reporter plasmid was transfected. For A549 and HepG2 cells, pSV-β-galactosidase plasmid (Promega) was used, and for COS-1 cells, pRL-TK plasmid (Promega) was used. The luciferase activities for A549 and HepG2 cells were measured using the luciferase assay system (Promega), and the β-galactosidase activities were measured with the luminescent β-galactosidase detection kit II (BD Biosciences Clontech, Palo Alto, CA). For COS-1 cells, the activities were measured using the Dual-luciferase reporter assay system (Promega).
Statistical Analyses.
Student's t test was used for comparison between two groups. Comparison of several groups was done with one-way ANOVA followed by Bonferroni's post hoc test. The results concerning the effects of glucocorticoid use and smoking on CYP3A5 mRNA levels in macrophages were analyzed with two-way ANOVA followed by Bonferroni's post hoc test.
Results
Induction of CYP3A5 in A549 Cells.
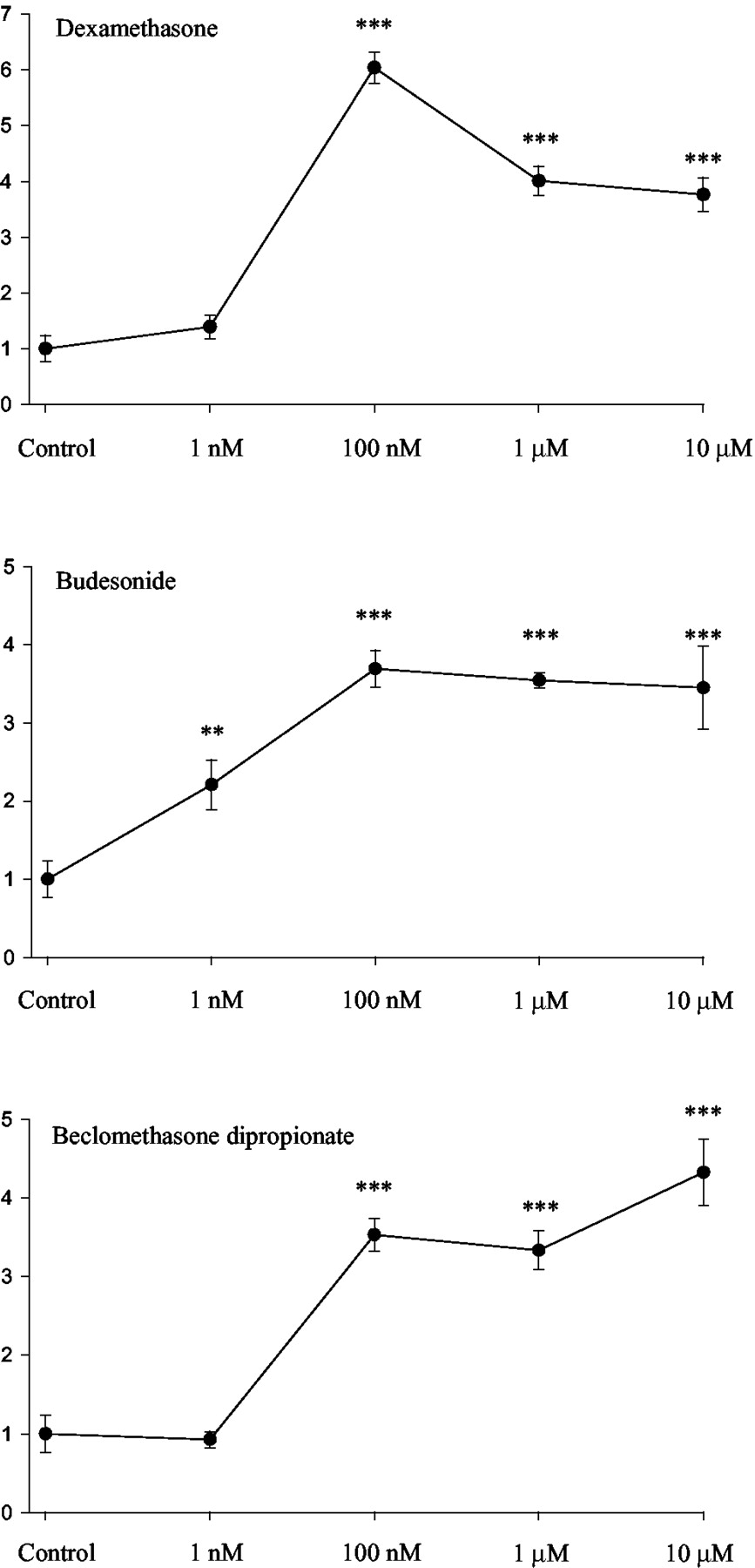
CYP3A5 mRNA was measured by quantitative RT-PCR after exposure of the A549 cells for 24 h to well defined chemical inducers of CYP3A. CYP3A5 was induced only by the glucocorticoids dexamethasone, beclomethasone dipropionate, and budesonide (Fig. 1). CYP3A5 mRNA was not affected by treatment with typical inducers of CYP3A4, i.e., RU486, clotrimazole, rifampicin, or nifedipine. Induction by glucocorticoids was further characterized by dose-response analyses, which showed that ∼100 nM concentrations were sufficient for maximal induction (Fig.2).

Induction of CYP3A5 mRNA as assessed with quantitative RT-PCR. Cells were exposed for 24 h to 10 μM concentrations of the indicated chemicals. CLO, clotrimazole; RIF, rifampicin; NIFE, nifedipine; DEX, dexamethasone; BUD, budesonide; BECLO, beclomethasone dipropionate. The CYP3A5 mRNA levels were normalized against the 18 S RNA levels. Data are presented as means + S.E. of six experiments. ∗∗∗, difference to the vehicle control statistically significant (P < 0.001, one-way ANOVA).

Dose-response curves of CYP3A5 mRNA fold inductions over control as assessed with quantitative RT-PCR. Cells were exposed for 24 h to the indicated concentrations of glucocorticoids. Data are presented as means ± S.E. of six experiments. ∗∗,P < 0.01; ∗∗∗, P < 0.001 difference to the vehicle control statistically significant (one-way ANOVA).
CYP3A5 Glucocorticoid Induction Is Mediated by the Glucocorticoid Receptor.
To test the involvement of GR in the induction process of CYP3A5, a glucocorticoid receptor antagonist, RU486, was used. CYP3A5 induction by glucocorticoids was completely blocked by the RU486 (Fig. 3). Budenoside has the highest affinity for GR among the glucocorticoids tested (KD, ∼1 nM) (Boobis, 1998;Esmailpour et al., 1998). A 10-fold excess of RU486 with a similarKD (Cadepond et al., 1997) was able to block CYP3A5 induction by budenoside.

Effect of glucocorticoid receptor antagonist RU486 on CYP3A5 mRNA fold induction by glucocorticoids as assessed with quantitative RT-PCR. Cells were exposed for 24 h to a 1 μM concentration of glucocorticoids with/without a 10 μM concentration of RU486. DEX, dexamethasone; BUD, budesonide; BECLO, beclomethasone dipropionate. Data are presented as means + S.E. of six experiments. ∗∗∗, difference to the treatment with glucocorticoid alone statistically significant (P < 0.001, Student'st test).
Further, to exclude the involvement of nuclear receptors PXR and CAR that regulates induction of many P450 genes, including CYP3A4, RT-PCR with PXR, CAR, and GR primers were performed to detect the expression of mRNAs of these receptors. As illustrated in Fig.4, only GR mRNA was present. In separate experiments, a 24-h exposure to 10 μM dexamethasone, budenoside, or beclomethasone dipropionate did not induce PXR or CAR mRNA in A549 cells (data not shown).

Detection of PXR, CAR, and GR mRNAs in A549 cells and human liver with RT-PCR.
Regulation of CYP3A5 Transcription by Glucocorticoids.
To assess the role of transcriptional regulation in induction of CYP3A5 by glucocorticoids, a reporter gene construct containing the CYP3A5 5′ regulatory region and luciferase reporter gene was constructed. The characterization of CYP3A5 regulation has been previously confused by the complex structure of the CYP3A gene locus. The recent characterization of CYP3A locus (Finta and Zaphiropoulos, 2000) revealed that the originally cloned CYP3A5 5′ (Jounaidi et al., 1994) actually represents the 5′ area of the CYP3AP1 pseudogene, with an exon 1 similar to CYP3A5. The pseudogene 5′ region was found to have 89% sequence similarity to the CYP3A5 5′ region (Finta and Zaphiropoulos, 2000). The −1355 to +40 region of the CYP3A5 5′ and −1359 to +39 region of the CYP3AP1 pseudogene 5′ were amplified with PCR from genomic DNA from two individuals, one with a high level of CYP3A5 protein in the liver (M27) and another with very low level (M24). The liver CYP3A5 protein levels were determined by immunoblotting, using CYP3A5-specific antipeptide antibody (data not shown) as previously described (Hakkola et al., 2001).
The reporter gene constructs were transfected into A549 cells, and the cells were treated with dexamethasone or vehicle alone for 48 h. A construct with two copies of glucocorticoid response elements (GRE) in front of a TATA box was used as a positive control (pGRE2TATA-luc) (Moilanen et al., 1998). After treatment, the cells were harvested, and the luciferase activity was measured. The constructs with CYP3A5 5′ regulatory regions from the two individuals were much more actively transcribed than the construct with CYP3AP1 5′, indicating that the relatively few nucleotide differences between the CYP3A5 5′ and the CYP3AP1 pseudogene 5′ are critically important for the regulation of CYP3A5 gene expression (Fig.5). There was no difference in transcriptional activation by the constructs from the two different individuals. Dexamethasone treatment induced transcription of pGRE2TATA-luc construct 17-fold. CYP3A5 5′-luc was modestly activated 1.3- to 1.5-fold, indicating involvement of transcriptional mechanism in the induction by glucocorticoids (Fig. 5). Also, the hepatoma cell line HepG2 was transfected and treated in a similar manner. Results similar to the A549 cells were also obtained in the HepG2 cell line (data not shown), indicating that the glucocorticoid induction of CYP3A5 is not a unique characteristic of lung cells.

Transfection of the CYP3A5 5′ and the CYP3AP1 5′ luciferase reporter constructs to A549 cells. After transfection, the cells were treated with a 10 μM concentration of dexamethasone or vehicle only for 48 h. The values represent means + S.E. of four individual experiments. ∗, P < 0.05, ∗∗∗,P < 0.001 difference to the vehicle control statistically significant (Student's t test).
The significance of cellular content of GR for induction of CYP3A5 was studied next by overexpression studies. The GR expression plasmid was cotransfected together with CYP3A5 5′-luc plasmid or with control plasmid pGRE2TATA-luc to both A549 and HepG2 cells. Overexpression of GR increased basal expression level of pGRE2TATA-luc in both cell lines. However, the fold induction by dexamethasone treatment was increased only in HepG2 cells. In contrast, cotransfection of GR with CYP3A5 5′-luc increased the CYP3A5 5′ dexamethasone response from 1.3- to 3.1-fold and 1.2- to 3.2-fold in both A549 and HepG2, respectively (Fig.6).

a, transfection of the CYP3A5 5′ luciferase reporter construct to HepG2 cells with or without GR expression vector cotransfection. After transfection, the cells were treated with a 10 μM concentration of dexamethasone or vehicle only for 48 h. The values represent means + S.E. of four individual experiments. ∗∗∗, P < 0.001, effect of GR cotransfection on dexamethasone induction statistically significant (Student'st test). b, transfection of the CYP3A5 5′ luciferase reporter construct to A549 cells with or without GR expression vector cotransfection. After transfection, the cells were treated with a 10 μM concentration of dexamethasone or vehicle only for 48 h. The values represent means + S.E. of four individual experiments. ∗∗∗, P < 0.001, effect of GR cotransfection on dexamethasone induction statistically significant (Student'st test).
To confirm the contribution of the GR to induction mechanism, the CYP3A5 5′-luc reporter construct was transfected to the GR-deficient cell line COS-1, and the cells were treated with dexamethasone for 48 h. The pGRE2TATA-luc construct was transfected in a similar manner. Dexamethasone was unable to induce either CYP3A5 5′-luc or pGRE2TATA-luc in COS-1 cells. Cotransfection with GR-expression vector restored the dexamethasone induction for both the CYP3A5′-luc and the pGRE2TATA-luc (Fig.7), indicating that the GR is both necessary and sufficient for CYP3A5 induction by dexamethasone.
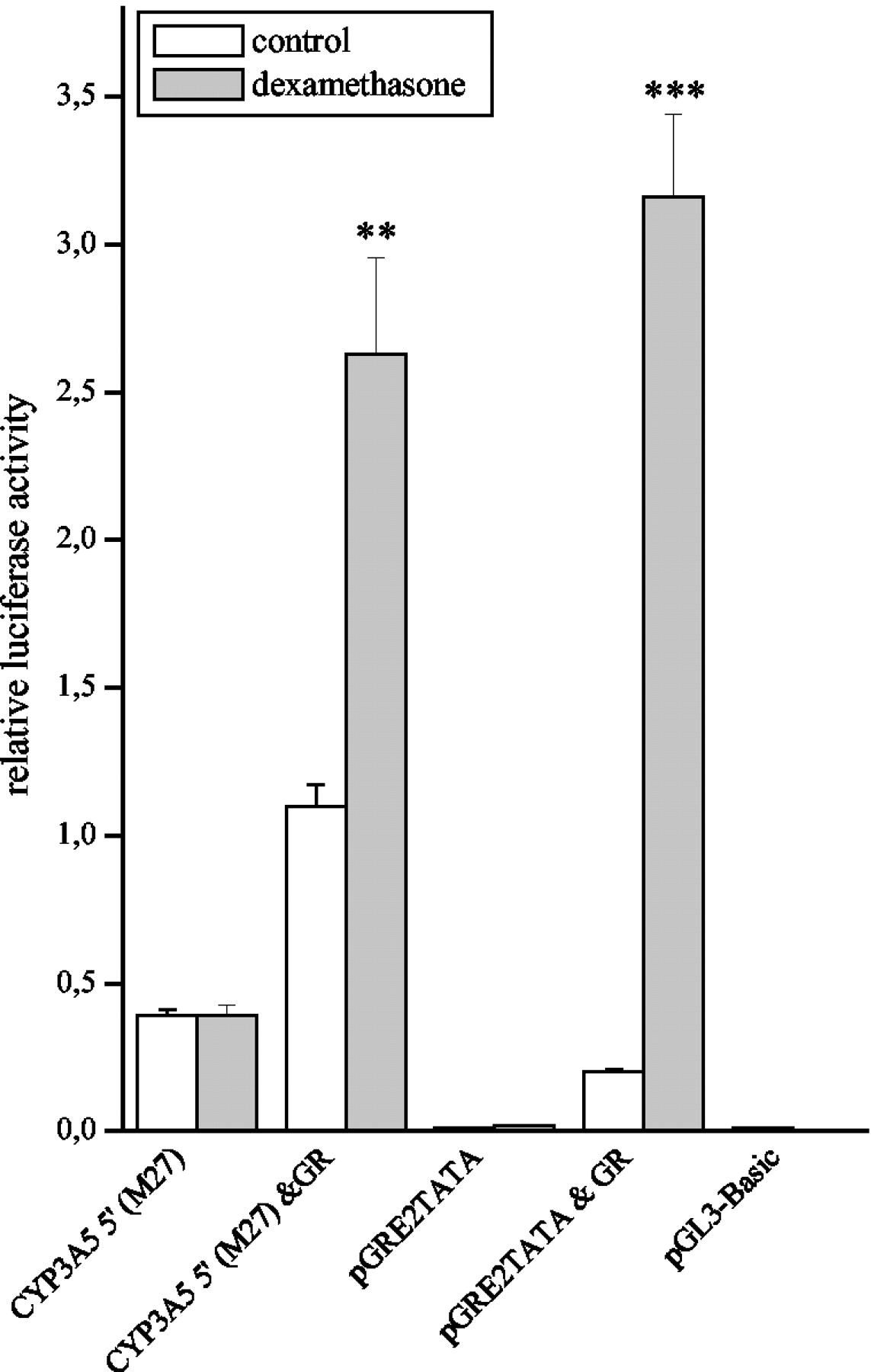
Transfection of the CYP3A5 5′ luciferase reporter construct to COS-1 cells with or without GR expression vector cotransfection. After transfection, the cells were treated with a 10 μM concentration of dexamethasone or vehicle only for 48 h. The values represent means + S.E. of four individual experiments. ∗∗,P < 0.01), ∗∗∗, P < 0.001 difference to the vehicle control statistically significant (Student's t test).
Effect of Inhaled Glucocorticoids and Smoking on the Alveolar Macrophage CYP3A5 mRNA Levels.
To test the hypothesis that inhaled glucocorticoids affect the level of CYP3A5 mRNA of alveolar macrophages in patients with respiratory diseases in vivo, CYP3A5 mRNA was measured with quantitative RT-PCR from 32 macrophage samples. In two-way ANOVA, the CYP3A5 expression level was dependent on smoking status (F = 4.028, P = 0.03) but not on glucocorticoid use (F = 0.214, P = 0.809). Tobacco smoking and glucocorticoid use were statistically not dependent on each other (F = 1.048 andP = 0.365). Smoking decreased the CYP3A5 level by 93% compared with nonsmokers (Bonferroni's post hoc test,P = 0.039) (Fig. 8). Ex-smokers' CYP3A5 level was 88% of nonsmokers' CYP3A5 level (P = 0.106) (Fig.8). The use of glucocorticoids increased the CYP3A5 level by 45% in nonsmokers (P = 0.438). Sex and age were tested as confounding factors, but they did not influence the results.

Box plot of CYP3A5 mRNA levels in alveolar macrophages of smokers, ex-smokers, and nonsmokers as measured with quantitative RT-PCR. The medians, percentiles, and extreme cases (ο) are presented. Difference to nonsmokers was tested with two-way ANOVA followed by Bonferroni's post hoc test: smokers, P= 0.039; ex-smokers, P = 0.106.
Discussion
CYP3A5 is the main CYP3A form in the human lung, and it is localized to bronchial, bronchiolar, and alveolar epithelium as well as alveolar macrophages (Kivisto et al., 1995, 1996; Anttila et al., 1997). Compared with CYP3A4, the CYP3A5 enzyme shows roughly the same substrate preference pattern but lower turnover rates (Aoyama et al., 1989; Wrighton et al., 1990; Yamazaki et al., 1995). However, CYP3A5 cannot metabolize some CYP3A4 substrates, such as erythromycin and quinidine (Wrighton et al., 1990).
We have previously shown CYP3A5 to be induced by the synthetic glucocorticoid dexamethasone in the human A549 lung adenocarcinoma cell line, indicating that this cell line is suitable for mechanistic studies of CYP3A5 pulmonary regulation (Hukkanen et al., 2000). In the current study, we demonstrate that besides dexamethasone, beclomethasone dipropionate and budesonide, which are inhaled glucocorticoids commonly used for the treatment of asthma, also induce CYP3A5 mRNA about 4-fold in A549 cells.
The glucocorticoid induction of CYP3A5 in human lung has several implications on the physiological, pharmacological, and toxicological significance of CYP3A5. The induction of CYP3A5 by low concentrations of glucocorticoids suggests that CYP3A5 could have some physiological roles in maintaining the steroid hormone balance in lung, since CYP3A5 is active in the metabolism of steroid hormones (Waxman et al., 1991), including endogenous glucocorticoid cortisol (Wrighton et al., 1990). Thus, the local concentration of endogenous glucocorticoids might be regulated by negative feedback involving induction of CYP3A5. Maximal induction was reached at concentrations as low as ∼100 nM, suggesting that CYP3A5 could be induced in vivo in patients using these inhaled glucocorticoids. The inhaled glucocorticoid budesonide is also metabolized by CYP3A (Jonsson et al., 1995), indicating that budesonide possibly autoinduces its own metabolism in human lung. Furthermore, the CYP3A forms are of importance in the metabolism of inhaled xenobiotics, including tobacco-derived procarcinogens, such as PAHs and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (Hecht, 1999). CYP3A5 is active in the metabolism of the benzo[a]pyrene proximate carcinogen benzo[a]pyrene-7,8-diol (Roberts-Thomson et al., 1993; Shou et al., 1994). This is reflected in the positive correlation between PAH-DNA adducts and CYP3A5 levels in alveolar macrophages (Piipari et al., 2000). Also, in human pulmonary microsomes, the last step of benzo[a]pyrene activation is stimulated by α-naphthoflavone, a CYP3A activator (Shimada et al., 1989, 1992). If CYP3A5 is induced in human lung in vivo by glucocorticoids, it could have a modulating effect on the individual susceptibility to lung cancer in patients using inhaled or systemic glucocorticoids.
The mechanism of dexamethasone induction of CYP3A4 has been a target of numerous recent studies, and the role for both GR and PXR receptors has been suggested. The recent investigation by Pascussi et al. (2001)suggested dual regulation of CYP3A4 by dexamethasone. The lower, nanomolar concentration acts indirectly through GR by up-regulating expression of PXR, which in turn, possibly ligand independently, moderately activates CYP3A4 transcription. The higher micromolar concentrations would bind to and activate the PXR, resulting in more extensive activation of CYP3A4 transcription. In the current study, we show that the glucocorticoid induction of CYP3A5 is regulated differently from CYP3A4. Several lines of evidence support the notion that CYP3A5 induction by glucocorticoids is mediated by direct action of GR and not through PXR. The induction takes place in an A549 lung adenocarcinoma cell line expressing GR but not the other CYP3A-regulating receptors, PXR or CAR. The induction is achieved with relatively low nanomolar concentrations of dexamethasone that are known to result in activation of human GR but not human PXR (Pascussi et al., 2001). The GR antagonist RU486 was able to inhibit glucocorticoid induction of CYP3A5. Finally, dexamethasone was unable to activate CYP3A5 transcription in the GR-deficient cell line COS-1, but the activation could be achieved after GR cotransfection.
The dexamethasone induction of CYP3A5 was found to involve transcriptional activation based on transfection experiments with CYP3A5 5′-luciferase reporter constructs. However, the luciferase activities were increased relatively modestly, 1.3- to 1.5-fold by dexamethasone treatment. Overexpression of GR in A459 and HepG2 cells increased dexamethasone induction of two copies of classical GRE elements containing pGRE2TATA-luc construct only in HepG2 cells but not in A549 cells. This suggests that A549 cells contain a sufficient amount of GR for full glucocorticoid induction, whereas HepG2 cells do not. However, the CYP3A5 5′-luc dexamethasone response was efficiently increased in both cell lines by GR overexpression. This may signal that CYP3A5 induction by glucocorticoids is mediated by low-affinity GRE in the CYP3A5 5′. CYP3AP1 has been previously shown to contain an atypical, functional glucocorticoid-responsive element (Schuetz et al., 1996). The corresponding sequence region in CYP3A5 5′ is similar, but not identical, to the CYP3AP1 glucocorticoid response area. Therefore, this region could play a role in the glucocorticoid induction of CYP3A5, but this needs to be confirmed in further studies. Interestingly, dexamethasone and other tested glucocorticoids induced CYP3A5 mRNA 4-fold in A549 cells without any GR supplementation, suggesting that in the natural chromatin structure, the CYP3A5 5′ affinity to GR may be higher than in the context of the plasmid. Alternatively, it is possible that additional regulatory elements may be present in the gene regions not included in the studied reporter construct, or that post-transcriptional mechanisms could contribute to the induction.
We tested the hypothesis that CYP3A5 would be induced by inhaled glucocorticoids in alveolar macrophages of patients with respiratory diseases in vivo. However, the differences in CYP3A5 expression in alveolar macrophages between nonsmoking current glucocorticoid users, ex-users, and nonusers were insignificant (a 45% increase of CYP3A5 in glucocorticoid users compared with nonusers, P = 0.438). This could be due to several possible reasons: 1) the observed induction of CYP3A5 in A549 cells does not occur in the corresponding cell type in vivo; 2) glucocorticoids do not induce CYP3A5 in alveolar macrophages; 3) inhaled doses of glucocorticoids do not reach alveolar macrophages in sufficient amounts; or 4) endogenous glucocorticoid levels activate CYP3A5 transcription, reducing the effect of exogenous glucocorticoid administration. A study with bronchial epithelial samples from glucocorticoid users and controls would be needed to find out whether CYP3A5 is induced in human lung tissue in vivo.
Cigarette smoking had a markedly decreasing effect on CYP3A5 mRNA levels in alveolar macrophages. CYP3A5 expression in current smokers was only 7% of the CYP3A5 level in nonsmokers. This is in agreement with a recent study that demonstrated that smoking decreases CYP3A5 protein levels in alveolar macrophages (Piipari et al., 2000). Importantly, it was also shown that CYP3A5 protein levels correlate positively with PAH-DNA adducts in macrophages of smokers when the data were normalized by the number of cigarettes smoked per day. However, down-regulation of pulmonary CYP3A5 by cigarette smoking may reduce its contribution to smoking-induced lung cancer. The mechanism of this repression is unknown. Treatment of the A549 cells with 2,3,7,8-tetrachlorodibenzo-p-dioxin had no effect on CYP3A5 expression (Hukkanen et al., 2000).
In conclusion, the present study indicates that CYP3A5 is induced by glucocorticoids, including the compounds used for the treatment of asthma at concentrations that could be achieved in vivo. The glucocorticoid induction of CYP3A5 is regulated differently than CYP3A4, is mediated by GR, and involves transcriptional regulation of the CYP3A5 gene. In vivo, cigarette smoking represses CYP3A5 expression in alveolar macrophages by currently unknown mechanism(s). The pulmonary CYP3A5 is therefore under the regulation of environmental and medical substances, adding another level to the variation primarily determined by genetic factors.
Acknowledgments
The excellent technical assistance of Päivi Kylli and Ritva Tauriainen is gratefully acknowledged. We thank Dr. Jorma Palvimo for providing the GR expression plasmid and the pGRE2TATA-luc reporter plasmid and Dr. Mika Ilves for help with quantitative RT-PCR.
Footnotes
-
This work was funded in part by grants to J.Hu. from the Finnish Cultural Foundation, the Northern Finland Cancer Society, the Finnish Drug Research Foundation, the Finnish Cancer Foundation, the Foundation of University Pharmacy, the Research and Science Foundation of Farmos, and the Emil Aaltonen Foundation and to O.P. from TEKES (Technology Development Center, Finland).
-
DOI: 10.1124/jpet.102.038208
- Abbreviations:
- P450
- cytochrome P450
- B[a]P
- benzo[a]pyrene
- CAR
- constitutively active receptor
- GR
- glucocorticoid receptor
- PAH
- polycyclic aromatic hydrocarbon
- PXR
- pregnane X receptor
- RT-PCR
- reverse transcriptase-polymerase chain reaction
- RU486
- mifepristone
- PCR
- polymerase chain reaction
- Tamra
- 5-carboxytetramethylrhodamine
- ANOVA
- analysis of variance
- GRE
- glucocorticoid response elements
- Received May 2, 2002.
- Accepted November 6, 2002.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}