


Abstract
Arylamine N-acetyltransferases (NATs; EC 2.3.1.5) catalyze both the N-acetylation and O-acetylation of arylamines and N-hydroxyarylamines. Humans possess two functional N-acetyltransferase genes, NAT1 and NAT2, as well as a nonfunctional pseudogene, NATP. Previous studies have identified Nat1 and Nat2 genes in the rat. In this study, we identified and characterized a third rat N-acetyltransferase gene (Nat3) consisting of a single open reading frame of 870 base pairs encoding a 290-amino acid protein, analogous to the previously identified human and rat N-acetyltransferase genes. Rat Nat3 nucleotide sequence was 77.2 and 75.9% identical to human NAT1 and NAT2, respectively. Rat Nat3 amino acid sequence was 68.6 and 67.2% identical to human NAT1 and NAT2, respectively. Rat Nat1, Nat2, and Nat3 were each cloned and recombinantly expressed in Escherichia coli. Recombinant rat Nat3 exhibited thermostability intermediate between recombinant rat Nat1 and Nat2. Recombinant rat Nat3 was functional and catalyzed the N-acetylation of several arylamine substrates, including 3-ethylaniline, 3,5-dimethylaniline, 5-aminosalicylic acid, 4-aminobiphenyl, 4,4′-methylenedianiline, 4,4′-methylenebis(2-chloroaniline), and 2-aminofluorene, and the O-acetylation of N-hydroxy-4-aminobiphenyl. The relative affinities of arylamine carcinogens such as 4-aminobiphenyl, N-hydroxy-4-aminobiphenyl, and 2-aminofluorene for N- and O-acetylation via recombinant rat Nat3 were comparable with recombinant rat Nat1 and higher than for recombinant rat Nat2. This study is the first to report a third arylamine N-acetyltransferase isozyme with significant functional capacity.
Arylamine N-acetyltransferases (NATs; EC 2.3.1.5) catalyze acetyl group transfer from acetyl-coenzyme A, or another acetyl donor, to the exocyclic amine of arylamines (N-acetylation) or the hydroxyl oxygen of N-hydroxylated arylamines (O-acetylation) (Hein, 1988). N-Acetyltransferases can bioactivate (O-acetylation) or deactivate (N-acetylation) arylamine procarcinogens such as 4-aminobiphenyl, a component of cigarette smoke (Patrianakos and Hoffman, 1979). NATs also metabolize various pharmaceutical drugs such as the antitubercular drug isoniazid, antibacterial sulfonamides, and the antiarrhythmic procainamide (Evans et al., 1960; Parker, 1969; Weber and Hein, 1985). Further insight into N-acetyltransferase function, expression, and regulation may predict individual incidences of pharmaceutical drug toxicities or individual tissue-specific cancer susceptibilities associated with environmental and/or occupational arylamine procarcinogen exposures.
The human genome contains two functional arylamine N-acetyltransferase loci, NAT1 and NAT2, coding for arylamine N-acetyltransferase enzymes that can be distinguished on the basis of substrate selectivity, structural stability, tissue-specific expression profile, and kinetic activity toward arylamine substrates (Blum et al., 1990; Hein et al., 1993; Boukouvala and Fakis, 2005). Humans also possess a nonfunctional pseudogene, NATP (Blum et al., 1990).
Rat models have been used for the study of arylamine N-acetyltransferases (Wick and Hanna, 1990; Hein et al., 1991, 1997; Grundmann et al., 2004; Lin et al., 2005; Zhang et al., 2006). Rat arylamine N-acetyltransferases are very similar in sequence and function to human N-acetyltransferases (Hein et al., 1997). Residues 125, 127, and 129 of the 290 amino acid protein determine substrate access to the active site, thereby influencing substrate selectivity (Goodfellow et al., 2000). Rat Nat2 and human NAT1 amino acid sequences both contain Phe125, Arg127, Tyr129, which is predictive of their similar arylamine substrate selectivities. Conversely, the human NAT2 protein contains Ser125, Ser127, Ser129, and the rat Nat1 protein contains Tyr125, Ser127, Tyr129, which is predictive of a weaker correlation between rat Nat1 and human NAT2 arylamine substrate selectivities. The same relationship is observed with regard to acetyl-donor substrate selectivities. The C-terminal undecapeptide, which is involved in controlling acetyl-coenzyme A hydrolysis (Mushtaq et al., 2002), is 100% identical when comparing rat Nat2 and human NAT1 but only 54% identical when comparing rat Nat1 and human NAT2. In terms of structural stability, human NAT2 is a more stable enzyme than human NAT1, and rat Nat1 is a more stable enzyme than rat Nat2, possibly suggesting structural similarities between the stable rat Nat1 and human NAT2, and the relatively unstable rat Nat2 and human NAT1 (Hein et al., 1993, 1997).
Identification of a third mouse Nat locus by Kelly and Sim (1994) suggested a third Nat locus in other rodents. Land et al. (1996) published southern blot analyses for rat Nat1 and Nat2 that suggested the presence of a third rat N-acetyltransferase gene. Recently, a “rattus norvegicus similar to mouse Nat3” (original title) open reading frame (ORF) sequence (XM_224762) was predicted by automated computational analysis of annotated genomic sequence NW_047470 using gene prediction method GNOMON (National Center for Biotechnology Information, Bethesda, MD). The genomic sequence for this entry originated from rat strain BN/SsNHsd/MCW. According to National Center for Biotechnology Information Map Viewer (Wheeler et al., 2005) and BLAT from University of California Santa Cruz Genome Bioinformatics (Kent, 2002), the rat Nats are located within approximately 75 kb and map to 16p14 on rat chromosome 16. The order of the rat Nat genes on chromosome 16 from 5′ to 3′ is Nat1-Nat2-Nat3, from centromere to telomere, with approximately 10 kb between Nat1 and Nat2 and approximately 60 kb between Nat2 and Nat3. This is similar to the arrangement of mouse Nat3 relative to mouse Nat1 and Nat2 on mouse chromosome 8 (Boukouvala and Fakis, 2005). However, mouse Nat3 has extremely low catalytic activity, and N-acetylated only a few arylamine substrates (Fretland et al., 1997; Estrada-Rodgers et al., 1998). Because the predicted rat Nat3 gene is highly homologous to mouse Nat3 in both nucleotide and predicted amino acid sequence, 91.0 and 87.9% identical, respectively, rat Nat3 may also have very low activity compared with rat Nat1 and Nat2.
The rat Nat3 ORF from GenBank entry XM_224762 was predicted from a large-scale genome project and could contain sequencing errors. In addition, since the Brown Norway (BN) inbred rat strain is genetically closer to wild rats than it is to other inbred rat strains, the predicted sequence may not be identical to the Nat3 ORF found in other common inbred rat strains (Canzian, 1997; Thomas et al., 2003). The aims of this study included complete sequencing of the putative rat Nat3 gene from three inbred rat strains, cloning the putative rat Nat3 ORF into a prokaryotic expression vector, and recombinant expression of the rat Nat3 gene in a high expressing bacterial system to characterize Nat3 catalytic activity, substrate selectivity, and intrinsic stability. Rat Nat1 and Nat2 were also analyzed in the same system in parallel for comparison to rat Nat3.
Materials and Methods
Cloning and Sequencing. Rat Nat1 and Nat2 were cloned into pKK223-3 prokaryotic expression vector as described previously (Doll and Hein, 1995). The 870-base pair Nat3 ORF was amplified from Fisher 344 (F344), Sprague-Dawley (SPRD), and Wistar Kyoto (WKY) genomic DNA by duplicate independent PCR reactions. As a control, these duplicate PCR reactions were carried through the following cloning and sequencing steps in parallel. Gene-specific Nat3 PCR primers (Table 1) were designed based on the predicted Nat3 sequence from GenBank accession number XM_224762 with restriction sites XmaI and PstI designed into the primer to facilitate directional cloning into the expression vector. The Nat3 ORF was cloned into pcDNA3.1 vector by TA cloning (Invitrogen, Carlsbad, CA), and the Nat3 ORF insert was completely sequenced (top and bottom strands) using commercially available pcDNA3.1-specific primers T7 (forward) and BGH (reverse) (Invitrogen), and six Nat3-specific forward and reverse primers (Table 1). Sequencing reactions were performed using BigDye Terminator version 1.1 cycle sequencing kit (Applied Biosystems, Foster City, CA) and analyzed with an ABI Prism 310 genetic analyzer (Applied Biosystems). Sequencing data were aligned to verify sequence using SeqMan II version 5.03 from DNAStar Inc. (Madison, WI).
Rat N-acetyltransferase 3 PCR and sequencing primers
Subcloning and Expression. The rat Nat3 ORF insert was subcloned into the pKK223-3 bacterial expression vector (Pharmacia-LKB Biotechnology, Piscataway, NJ) in duplicate using XmaI and PstI restriction enzymes. Duplicate clones were carried through the expression steps as a control. The 5′ and 3′ ligation sites were verified by sequencing. JM105 strain Escherichia coli were made competent and transformed with the Nat3/pKK223-3 plasmid using previously described methods (Chung and Miller, 1988; Chung et al., 1989). JM105 competent E. coli were transformed with an empty pKK223-3 vector as a negative control.
Recombinant Nat1, Nat2, Nat3, and vector were expressed in JM105 E. coli as described previously (Doll and Hein, 1995). Total bacterial lysate protein concentration was determined for each expression (Bradford, 1976).
Western Blotting. A 19-residue peptide (N-RVFGIRLETKLVPKCGNWL-C) was designed for Nat3-selective polyclonal antiserum production. The peptide matches C-terminal residues 269 to 287 of the deduced rat Nat3 amino acid sequence and differs from the same regions of rat Nat1 and Nat2 by nine and 10 residues, respectively. Peptide synthesis and polyclonal antiserum production were accomplished by Invitrogen.
Bacterial lysates were diluted to 1 mg/ml in homogenization buffer (20 mM sodium phosphate, 1 mM EDTA, 1 mM dithiothreitol, and protease inhibitor cocktail, pH 7.4) and Laemmli loading buffer (5% 2-mercaptoethanol) (Bio-Rad, Hercules, CA), boiled for 5 min, electrophoresed through 12% Tris/glycine PAGEr Duramide gels (Cambrex, East Rutherford, NJ) in 1× Tris/glycine/SDS running buffer (Bio-Rad), and transferred to Amersham Hybond-ECL nitrocellulose membrane with a TE 70 semidry transfer unit (GE Healthcare) in 1× Tris/glycine transfer buffer (Bio-Rad). The membrane was stained with Ponceau S solution to verify equal protein loading. After stain removal, membranes were blocked overnight at 4°C with Blocker Blotto in Tris-buffered saline (Pierce Chemical, Rockford, IL), then incubated with primary antiserum in blocking buffer plus 0.05% Tween 20 at room temperature for 2 h, and with goat anti-rabbit horseradish peroxidase-conjugated secondary antibody in blocking buffer plus 0.05% Tween 20 at RT for 1 h. Tris-buffered saline washing steps (3 × 10 min) were included between the primary and secondary antibody incubations and after the secondary antibody incubation. Membranes were incubated with Amersham ECL Plus chemiluminescent working solution (GE Healthcare), exposed to Kodak Bio-Max film (Eastman Kodak, Rochester, NY), and developed using a Kodak X-OMAT 2000A processor.
Enzyme Activity Assays. Bacterial lysates expressing recombinant Nat1, Nat2, Nat3, or vector (negative control) were assayed for N-acetylation of 3-ethylaniline (3EA), 3,5-dimethylaniline (35DMA), o-toluidine (OT), 2,6-dimethylaniline (26DMA), 5-aminosalicylic acid (5AS), p-aminobenzoic acid (PABA), p-aminoacetanilide (PAA), 3-amino-4-methoxyacetanilide (3A4MAA), N-(p-aminobenzoyl)glutamate (pABG), procainamide (PA), 4-aminobiphenyl (ABP), 4,4′-methylenedianiline (MDA), 4,4′-methylenebis(2-chloroaniline) (MOCA), 2-aminofluorene (AF), isoniazid (INH), and sulfamethazine (SMZ) and for O-acetylation of N-hydroxy-4-aminobiphenyl (N-OH-ABP).
Bacterial lysates were assayed for N-acetylation of PABA, pABG, ABP, AF, SMZ, MDA, and MOCA by measurement of N-acetylated product. For initial velocity measurements, lysates (in triplicate) were incubated with 1 mM acetyl-coenzyme A and substrate (100 μM MDA, AF, ABP, MOCA; 200 μM pABG, 300 μm PABA, SMZ) for 10 min at 37°C, and the reaction was stopped upon addition of 1/10 volume of 1 M acetic acid. Control reactions substituted water for acetyl coenzyme A. N-Acetylated products were separated and measured using a Beckman System Gold high-performance liquid chromatography (HPLC) system. HPLC methods for measuring the N-acetylation of PABA, ABP, AF, SMZ, and MDA have been described previously (Leff et al., 1999; Fretland et al., 2002; Zhang et al., 2006). The HPLC separation for the N-acetylation of MOCA was identical to the MDA method, with mono-acetyl-MOCA eluting at 16.5 min. pABG and N-acetyl-pABG product were separated by HPLC with a mobile phase of 20 mM sodium perchlorate, pH 2.5, and acetonitrile. Elution was conducted with a linear gradient from 0% acetonitrile/100% sodium perchlorate to 50% acetonitrile/50% sodium perchlorate over 5 min at a flow rate of 2 ml/min. Under these conditions, N-acetyl-pABG eluted at 9.5 min.
Bacterial lysates were assayed for N-acetylation of PAA, 3EA, 35DMA, OT, 26DMA, 3A4MAA, INH, PA, and 5AS as described above and then analyzed using a colorimetric assay (300 μm substrate) as described previously (De Angelis et al., 1998). In brief, to a 300-μl reaction, 600 μl of 3.2 M guanidine HCl/0.1 M sodium phosphate, pH 6.8, and 0.1 ml of 2 mM 5,5′-dithio-bis(2-nitrobenzoic acid/0.1 M sodium phosphate, pH 6.8/10 mM EDTA were added, and the contents were incubated at room temperature for 5 min. Controls substituted water for substrate. Bacterial lysates (diluted to 3 mg/ml in homogenization buffer) were assayed for O-acetylation of N-OH-ABP by HPLC separation and measurement of ABP-deoxyguanosine adduct as described previously (Hein et al., 2006).
For determinations of apparent Michaelis-Menten kinetic parameters Km (micromolar) and Vmax (nanomoles per minute per milligram), initial velocities were determined using a range of substrate concentrations. The data were fitted to a one-phase exponential association nonlinear regression curve using GraphPad Prism software, version 2.01 (GraphPad Software Inc., San Diego, CA). A specificity constant was calculated (Vmax/Km), which, assuming equal Nat1, Nat2, and Nat3 protein expression, could approximate the rate of substrate capture (Fersht, 1998).
Thermostability. Bacterial lysates diluted to 1 mg/ml in homogenization buffer were incubated at either 37 or 50°C for 5, 10, 20, 40, 60, or 80 min, transferred immediately to ice, and assayed for N-acetylation of AF. Results were normalized to activity of unincubated enzyme (time 0 = 100%) for the percentage of activity remaining. Heat inactivation half-life (t1/2) and heat deactivation constant (k) were determined by fitting the data to a first-order decay curve using GraphPad Prism software.
Results
Cloning and Sequencing Rat Nat3. The Nat3 ORF in inbred strains F344, WKY, and SPRD were identical (Gen-Bank accession nos. AY253757, AY253758, and AY253759). The Nat3 ORF in these rat strains differed from the predicted Nat3 ORF, XM_224762, from rat strain BN, at ORF nucleotide 358.
Recombinant Expression of Rat Nat3. Western blotting with rat Nat3 antiserum selectively detected the presence of recombinant Nat3 enzyme in bacterial lysates derived from E. coli transformed with rat Nat3. In contrast, an N-acetyltransferase band was not visible in bacterial lysates derived from E. coli transformed with rat Nat1, Nat2, or vector (Fig. 1). Ponceau S staining confirmed equal total protein loading in each lane. Due to differences in immunoreactivity of the antibodies used to detect each isozyme, and the indistinguishable size of the Nat band in total protein stain, the amount of recombinant protein in each lysate could not be compared.
Recombinant Rat N-Acetyltransferase Activity.N-Acetylation activity of recombinant rat Nat1, Nat2, and Nat3 for 13 substrates is summarized in Fig. 2. Recombinant rat Nat3 N-acetylated ABP, AF, MDA MOCA, 35DMA, 3EA, and 5AS but not INH, PA, PABA, SMZ, pABG, PAA, or 3EA. Recombinant rat Nat1 N-acetylated all substrates, with the exception of pABG. Recombinant rat Nat2 N-acetylated all substrates except INH. Recombinant rat Nat1, Nat2, and Nat3 each catalyzed the O-acetylation of N-OH-ABP (Table 2). No N-acetylation activity was detected toward OT, 26DMA, or 3A4MAA for any rat N-acetyltransferase. Background activity was extremely low or undetectable for all substrates tested.
Apparent Michaelis-Menten kinetics and specificity constant for recombinant rat Nat1, Nat2, and Nat3

Western blot analysis of bacterial lysates using anti-Nat3 antiserum. The rat Nat3 selective antiserum identified rat Nat3 protein (∼33.5 kDa) in lysates containing recombinantly expressed rat Nat3. No Nat band was visible in lanes of the negative control lysates (vector) or the lysates containing recombinantly expressed Nat1 or Nat2. Ponceau S red staining of the membrane was used as a loading control to verify equal protein loading in every lane.
Apparent Michaelis-Menten Kinetics. Recombinant rat Nat2 had the highest apparent Vmax and Km for the N-acetylation of ABP and AF, and for the O-acetylation of N-OH-ABP (Table 2). Although recombinant rat Nat2 had the lowest affinity for these substrates, it catalyzed their N-acetylation and O-acetylation at much higher rates than recombinant Nat1 or Nat3. For each of these substrates, recombinant rat Nat1 and Nat3 apparent Km values were comparable, indicating similar affinities for the substrates, although recombinant Nat3 had a lower apparent Vmax and therefore the slowest reaction velocity relative to recombinant rat Nat1 and Nat2. O-Acetylation activity was detected for all three recombinant isozymes at much lower rates than for N-acetylation (lower Vmax). In addition, recombinant rat Nat1, Nat2, and Nat3 affinity for the N-hydroxylated substrate N-OH-ABP was much lower (higher Km) in comparison with AF or ABP.

Rat Nat1, Nat2, and Nat3 N-acetylation of various substrates. Activity is nanomoles of product per minute reaction time per milligram of total protein. Error bars indicate standard error (n = 3). Substrate abbreviations are defined under Materials and Methods. Activity was not detected for substrates marked ND. The scale of each graph differs depending on the activity of each isozyme. The rat Nat2 graph has a split y-axis to accommodate the wide range of activities.
Thermostability. Lysates were assayed at 37 and 50°C, as described under Materials and Methods; however, thermostability differences among the recombinant rat Nat isozymes were most readily observed at 50°C (Fig. 3). Recombinant Nat1 was the most stable with no reduction in activity after 120 min incubation at 50°C (data only shown to 80 min). Recombinant Nat3 lost most of its activity following 80 min of incubation, and it was less stable than recombinant Nat1 but more stable than recombinant Nat2. Recombinant Nat2 was relatively unstable, having lost most of its activity within 5 min of incubation at 50°C.
Discussion
Cloning and sequencing of rat Nat3 from rat genomic DNA and recombinant expression of rat Nat1, Nat2, and Nat3 in parallel provided an opportunity to characterize rat Nat3 in comparison with rat Nat1 and Nat2. This study identifies a rat Nat3 ORF encoding for a functional arylamine N-acetyltransferase enzyme with intermediate intrinsic stability relative to rat Nat1 and Nat2 and that catalyzes both N-acetylation and O-acetylation reactions. Multiple studies in bacterial and mammalian expression systems have reported very low to undetectable mouse Nat3 activity toward a limited number of substrates (Kelly and Sim, 1994; Fretland et al., 1997; Estrada-Rodgers et al., 1998). Recombinant rat Nat3 had low activity relative to recombinant Nat1 and Nat2, but future studies of Nat1, Nat2, and Nat3 mRNA and protein expression in rat tissues are necessary to determine the relative contribution of rat Nat3 to acetylator phenotype in the rat.
Seven arylamine substrates were N-acetylated by recombinant rat Nat3, including ABP, AF, 5AS, 35DMA, 3EA, MDA, and MOCA, although each of these substrates was also N-acetylated at higher rates by recombinant rat Nat1 and Nat2. In contrast, recombinant rat Nat1 selectively N-acetylated INH, MOCA, and PA, whereas recombinant rat Nat2 selectively N-acetylated PAA, PABA, pABG, ABP, AF, 5AS, 35DMA, 3EA, and MDA (Fig. 2). Substrates PABA and SMZ are classic human NAT1- and NAT2-selective substrates, respectively (Grant et al., 1991). Recombinant rat Nat2 PABA selectivity is analogous to human NAT1. Although acetylated at relatively low rates by recombinant Nat1 and Nat2, SMZ was not selective for either recombinant rat Nat1 or Nat2, which may be attributed to additional steric interference at the rat Nat1 active site from larger tyrosine residues at 125 and 129 instead of the smaller human NAT2 serine residues at 125 and 127 (Table 3).
Comparisons of amino acid residues known to control substrate specificity
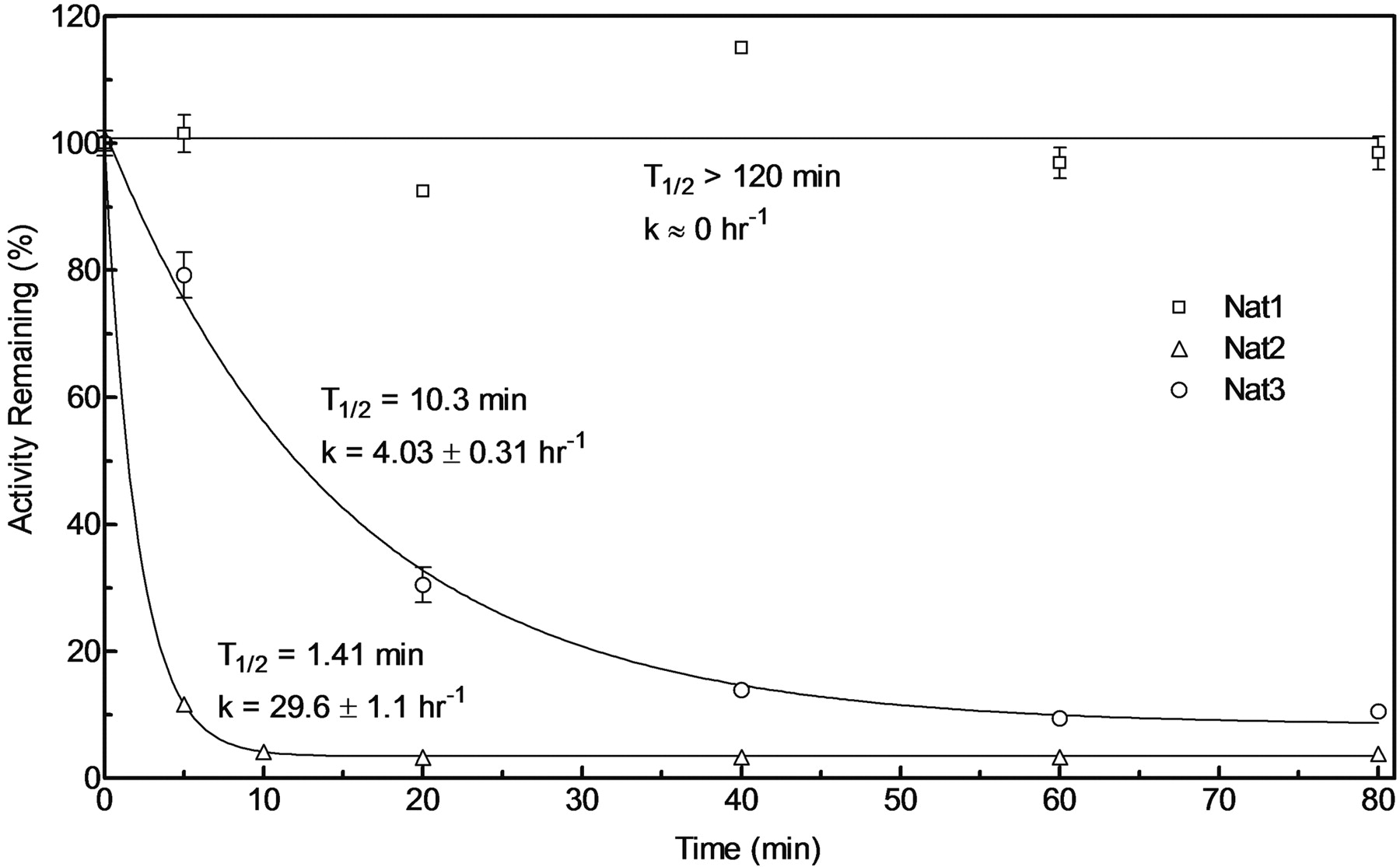
Thermostability of rat N-acetyltransferases. Percentage of 2-aminofluorene N-acetyltransferase activity remaining plotted on the ordinate versus incubation time at 50°C on the abscissa. Heat deactivation rate (k) and heat inactivation half-life (t1/2) are indicated for each Nat isozyme.
Kawamura et al. (2005) describes a similar analysis of the substrate selectivities of purified human NAT1 and NAT2, and hamster Nat2, highlighting the similarities between hamster Nat2 and human NAT1. The results of that study of purified N-acetyltransferase enzymes agree with this study of unpurified, recombinantly expressed rat N-acetyltransferases, thereby confirming both the result and expression system used in this study. Both studies measure activities for PABA, pABG, AF, INH, and PA, and they found relative substrate selectivities that support the homologous relationship of human NAT1 to rat Nat2 and of human NAT2 to rat Nat1. The Kawamura et al. (2005) study likewise noted the similar substrate selectivity between human NAT1 and the rodent, in that case hamster, Nat2.
Recombinant rat Nat1, Nat2, and Nat3 differed in N-acetylation and O-acetylation reaction rates. Recombinant rat Nat2 catalyzed N-acetylation and O-acetylation at much higher rates than Nat1 or Nat3, regardless of substrate; however, these results were obtained by recombinant expression in a bacterial system. Because rats do not use the same degradation mechanisms as bacteria, expression in a mammalian system will be necessary to understand the influence of eukaryotic cell degradation mechanisms on the expression of rat Nat enzymes. Investigations into the expression of these arylamine N-acetyltransferases in mammalian cell culture and in rat tissues are in progress.
Rat Nat3 consisted of a single open reading frame of 870 base pairs encoding a 290-amino acid protein, analogous to the previously identified human and rat N-acetyltransferase genes. Rat Nat3 nucleotide sequence was 77.2 and 75.9% identical to human NAT1 and NAT2, respectively. Rat Nat3 amino acid sequence was 68.6 and 67.2% identical to human NAT1 and NAT2, respectively (Fig. 4). Whereas the predicted Nat3 ORF from the BN rat strain has a guanine (G) at nucleotide 358, the inbred rat strains F344, WKY, and SPRD rats had an adenine (A) at 358. The change A358>G results in a valine-to-isoleucine change at residue 120. However, mammalian N-acetyltransferase genes from human, rhesus monkey (Macaca mulatta), chimpanzee (Pan troglodytes), cattle (Bos taurus), cat (Felis catus), rabbit (Oryctolagus cuniculus), Chinese hamster (Cricetulus griseus), golden hamster (Mesocricetus auratus), mouse (Mus musculus), and chicken (Gallus gallus) have an adenine at nucleotide position 358.

Human, mouse, and rat N-acetyltransferase nucleotide (top) and amino acid (bottom) sequence comparisons (percentage identical). Sequence comparisons were determined using EMBOSS pairwise alignment algorithms (Rice et al., 2000).
The Cys68, His107, Asp122 catalytic triad is present in rat Nat3, analogous to other mammalian Nats. The three residues (125, 127, 129) known to influence N-acetyltransferase substrate selectivity (Goodfellow et al., 2000) are unique to rat Nat3 among the human NATs and rat Nats, but they are identical to those found in the mouse Nat3 sequence (Table 3), suggesting that Nat3 arylamine substrate selectivity is similar to mouse Nat3. The rat Nat3 C-terminal undecapeptide is 81.8% identical to mouse Nat3, 63.6% identical to human NAT1 and both rat Nat1 and Nat2, and 54.5% identical to human NAT2 (Table 3). Rat Nat3 nucleotide sequence is 75.7% identical to rat Nat1 and 78.0% identical to rat Nat2; rat Nat3 amino acid sequence is 71.0% identical to rat Nat1 and 71.7% identical to rat Nat2 (Fig. 4). Rat Nat3 amino acid and nucleotide sequences are most identical to those of mouse Nat3, at 87.9 and 91.0%, respectively. The rat Nat3 catalytic core (63-131) (Rodrigues-Lima et al., 2001) is 72.5% identical to rat Nat1 and Nat2 catalytic cores, 73.9% identical to mouse Nat1 and Nat2 catalytic cores, and 89.9% identical to mouse Nat3 catalytic core. The rat Nat3 nucleotide sequence is 70.3% identical to the sequence of the human NAT pseudogene (NATP), which is lower than the sequence identity of NATP with human NAT1 and NAT2, and rat Nat1 and Nat2. This low sequence identity combined with the difference in chromosomal orientation to the other two Nats (NATP is located between NAT1 and NAT2) strongly suggests that human NATP and rat Nat3 are not homologous.
In summary, recombinant rat Nat3 was functional and catalyzed the N-acetylation of several arylamine substrates, including 3-ethylaniline, 3,5-dimethylaniline, 5-aminosalicylic acid, 4-aminobiphenyl, 4,4′-methylenedianiline, 4,4′-methylenebis(2-chloroaniline), and 2-aminofluorene, and the O-acetylation of N-hydroxy-4-aminobiphenyl. The relative affinities of arylamine carcinogens such as 4-aminobiphenyl, N-hydroxy-4-aminobiphenyl, and 2-aminofluorene for recombinant rat Nat3 were comparable to recombinant rat Nat1 and higher than for recombinant rat Nat2. This study is the first to report a third arylamine N-acetyltransferase isozyme with significant functional capacity. Future studies will involve characterization of recombinant rat N-acetyltransferases in mammalian cell systems, and use of the rat as an animal model for investigation of N-acetyltransferase activity, expression, and regulation in vivo.
Footnotes
-
This work was partially supported by United States Public Health Service Grant CA34627 from the National Cancer Institute and Training Grant ES011564 from the National Institute of Environmental Health Sciences. A preliminary report of this work was presented at the 95th Annual Meeting of the American Association for Cancer Research; 2004 Mar 27-31; Orlando, FL. American Association for Cancer Research, Philadelphia, PA.
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.106.108399.
-
ABBREVIATIONS: NAT1, human N-acetyltransferase 1; NAT2, human N-acetyltransferase 2; Nat1, rat or mouse N-acetyltransferase 1; Nat2, rat or mouse N-acetyltransferase 2; Nat3, rat or mouse N-acetyltransferase 3; NATP, human N-acetyltransferase pseudogene; ORF, open reading frame; kb, kilobase(s); BN, Brown Norway; F344, Fisher 344; SPRD, Sprague-Dawley; WKY, Wistar Kyoto; PCR, polymerase chain reaction; 3EA, 3-ethylaniline; 35DMA, 3,5-dimethylaniline; OT, o-toluidine; 26DMA, 2,6-dimethylaniline; 5AS, 5-aminosalicylic acid; PABA, p-aminobenzoic acid; PAA, p-aminoacetanilide; 3A4MAA, 3-amino-4-methoxyacetanilide; pABG, N-(p-aminobenzoyl)glutamate; PA, procainamide; ABP, 4-aminobiphenyl; MDA, 4,4′-methylenedianiline; MOCA, 4,4′-methylenebis(2-chloroaniline); AF, 2-aminofluorene; INH, isoniazid; SMZ, sulfamethazine; N-OH-ABP, N-hydroxy-4-aminobiphenyl; HPLC, high-performance liquid chromatography.
-
↵1 This work constitutes partial fulfillment for the Ph.D. program in Pharmacology and Toxicology at the University of Louisville School of Medicine.
- Received May 24, 2006.
- Accepted July 6, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}