


Abstract
Doxorubicin (DOX) and daunorubicin (DAUN) are effective anticancer drugs; however, considerable interpatient variability exists in their pharmacokinetics. This may be caused by altered metabolism by nonsynonymous single-nucleotide polymorphisms (ns-SNPs) in genes encoding aldo-keto reductases (AKRs) and carbonyl reductases. This study examined the effect of 27 ns-SNPs, in eight human genes, on the in vitro metabolism of both drugs to their major metabolites, doxorubicinol and daunorubicinol. Kinetic assays measured metabolite levels by high-performance liquid chromatography separation with fluorescence detection using purified, histidine-tagged, human wild-type, and variant enzymes. Maximal rate of activity (Vmax), substrate affinity (Km), turnover rate (kcat), and catalytic efficiency (kcat/Km) were determined. With DAUN as substrate, variants for three genes exhibited significant differences in these parameters compared with their wild-type counterparts: the A106T, R170C, and P180S variants significantly reduced metabolism compared with the AKR1C3 wild-type (Vmax, 23–47% decrease; kcat, 22–47%; kcat/Km, 38–44%); the L311V variant of AKR1C4 significantly decreased Vmax (47% lower) and kcat and kcat/Km (both 43% lower); and the A142T variant of AKR7A2 significantly affected all kinetic parameters (Vmax and kcat, 61% decrease; Km, 156% increase; kcat/Km, 85% decrease). With DOX, the R170C and P180S variants of AKR1C3 showed significantly reduced Vmax (41–44% decrease), kcat (39–45%), and kcat/Km (52–69%), whereas the A142T variant significantly altered all kinetic parameters for AKR7A2 (Vmax, 41% decrease; kcat, 44% decrease; Km, 47% increase; kcat/Km, 60% decrease). These findings suggest that ns-SNPs in human AKR1C3, AKR1C4, and AKR7A2 significantly decrease the in vitro metabolism of DOX and DAUN.
Introduction
Aldo-keto reductases (AKRs) and carbonyl reductases (CBRs) are members of a highly divergent superfamily of NAD(P)H-dependent oxidoreductase enzymes that have been shown to metabolize a broad range of endogenous and exogenous carbonyl-containing compounds, including steroid hormones, aflatoxins, sugar/lipid aldehydes, and prostaglandins (Matsunaga et al., 2006; Hoffmann and Maser, 2007; Jin and Penning, 2007; Penning and Drury, 2007). AKRs and CBRs are categorized as phase I metabolizing enzymes responsible for converting compounds containing reactive aldehyde and ketone functional groups to their corresponding hydroxy metabolites. This metabolic conversion increases the water solubility of the metabolites and facilitates their elimination from the body directly or via phase II conjugation reactions.
In addition to the aforementioned compounds, AKRs and CBRs have been implicated in the metabolism of the anthracycline antibiotics, daunorubicin (DAUN) and doxorubicin (DOX) (Cummings et al., 1991; Jin and Penning, 2007). DAUN and DOX rank among the most effective antineoplastic agents ever developed in cancer therapy (Lakhman et al., 2005; Blanco et al., 2008). DAUN plays a vital role in the treatment of acute myeloid and lymphoblastic leukemias, whereas DOX is extensively used against numerous cancers, including breast cancer, childhood solid tumors, non-Hodgkin's lymphoma, and soft tissue carcinomas (Hunault-Berger et al., 2001; Fassas and Anagnostopoulos, 2005; Cortés-Funes and Coronado, 2007). Even though cancer treatments involving these drugs have resulted in longer life expectancy, both anthracyclines have been associated with variation in the onset of life-threatening adverse events, such as chronic cardiotoxicity (Barry et al., 2007; Deng and Wojnowski, 2007; Menna et al., 2007). Both the frequency of chronic cardiotoxicity and mortality rate are correlated with the lifetime cumulative dose of either drug. Moreover, the risk of chronic cardiotoxicity increases when DOX and DAUN are used in combination with other anticancer drugs, such as herceptin, paclitaxel, docetaxel, vincristine, and cyclophosphamide (Mordente et al., 2001; Danesi et al., 2002; Floyd et al., 2005; Gianni et al., 2007).
The cause of the interpatient variation in DAUN- or DOX-induced adverse events is unknown. However, nonsynonymous single-nucleotide polymorphisms (ns-SNPs) in the genes expressing enzymes that metabolize these anthracyclines may be one factor contributing to this interpatient variable toxicity. Previous in vitro studies demonstrated that ns-SNPs in human AKR1A1, CBR1, and CBR3 genes produce enzymes with significantly reduced metabolism of DAUN and/or DOX compared with their wild-type counterparts (Bains et al., 2008, 2009, 2010). To test this hypothesis, we examined whether variant enzymes generated by ns-SNPs in eight other human AKR and CBR genes (AKR1B1, AKR1B10, AKR1C1, AKR1C2, AKR1C3, AKR1C4, AKR7A2, and CBR4) significantly affect their ability to metabolize DAUN or DOX. The AKRs were selected because prior experiments have shown that the wild-type enzyme was able to metabolize DOX and/or DAUN (Ohara et al., 1995; O'Connor et al., 1999; Martin et al., 2006; Kassner et al., 2008). CBR4 was included in this study because this CBR isoform is the only one remaining for which there are no published data on its ability to metabolize DOX and DAUN. The CBR1, CBR3, and CBR4 genes encode the three distinct isoforms of the CBR enzymes that are found in humans (Hoffmann and Maser, 2007; Oppermann 2007; Endo et al., 2008).
The purpose of this study is to improve our understanding of the effect of genetic variation in the human AKR and CBR4 genes on the in vitro metabolism of DOX, DAUN, and standard test substrates. Presently, there are 28 documented ns-SNPs in these genes in the human genome, as listed in the National Centre for Biotechnology Information Database. The frequencies of these variant alleles among different populations range from 0.8 to 62.5% (Table 1). Using purified, bacterially expressed human histidine-tagged enzymes, we compared the metabolic capability of the wild type and variants by monitoring the formation of the corresponding carbon-13 alcohol metabolites, daunorubicinol (DAUNol) and doxorubicinol (DOXol). We demonstrated that certain variants of AKR1C3 (A106T, R170C, and P180S), AKR1C4 (L311V), and AKR7A2 (A142T) significantly reduced DAUN and DOX metabolism compared with the wild-type enzymes based on the following kinetic parameters: Vmax (DAUN, 23–61% decrease; DOX, 41–44% decrease), Km (156 and 47% increase with the A142T variant in the presence of DAUN and DOX, respectively), kcat (DAUN, 22–61% decrease; DOX, 39–45% decrease), and kcat/Km (DAUN, 38–85% decrease; DOX, 52–69% decrease). Furthermore, after comparing kcat/Km values between these two drugs among the wild-type and variant enzymes, we observed that DAUN is generally a better substrate for the AKRs, whereas DOX is a better substrate for CBR4.
Allele frequencies of the nonsynonymous single-nucleotide polymorphic variants of human AKR and CBR4 enzymes from different ethnic groups
Allele frequencies were obtained from the National Center for Biotechnology Information (NCBI) database (http://www.ncbi.nlm.nih.gov). The ethnic groups are designated as follows: A (African), C (white), Ch (Chinese), E (European), H (Hispanic), J (Japanese), PRH (Pacific Rim Heritage), and UFV (Utah, French and Venezuelan). X refers to a combined population from Michigan along with UFV. The chromosome sample counts (n) for each variant are also given.
Materials and Methods
Chemicals and Enzymes.
Agarose, ampicillin (sodium salt), chloramphenicol, daunorubicin hydrochloride [(8S,10S)-8-acetyl-10-[(3-amino-2,3,6-trideoxy-a-l-lyxo-hexopyransoyl)oxy]-7,8,9,10-tetrahydro-6,8,11-trihydroxy-1-methoxy-5,12-naphthacenedione hydrochloride], dl-glyceraldehyde, doxorubicin hydrochloride [10-[(3-amino-2,3,6-trideoxy-a-l-lyxohexopyranosyl)oxy]- 7,8,9,10-tetrahydro-6,8,11-trihydroxy-8-(hydroxyacetyl)-5,12-naphthacenedione hydrochloride], DNaseI, idarubicin, kanamycin, (S)-1-indanol, lysozyme, menadione, methanol, potassium phosphate (KH2PO4), N,N,N′,N′ tetramethylethylenediamine, NADP+, NADPH, sodium phosphate (NaH2PO4), RNaseI, urea, 1-acenaphthenol, and 9,10-phenanthrenequinone were supplied by Sigma-Aldrich (St. Louis, MO). High-performance liquid chromatography (HPLC)-grade acetonitrile, agar, ammonium persulfate, formic acid, ethanol, glycine, glycerol, glacial acetic acid, imidazole, and Tris were purchased from Thermo Fisher Scientific (Waltham, MA). NaCl and yeast extract were ordered from Merck Biosciences (Darmstadt, Germany). Bacto tryptone and pooled human liver cytosol were obtained from BD Biosciences (San Jose, CA), and isopropyl β-d-1-thiogalactopyranoside (IPTG) was supplied by MBI Fermentas (Hanover, MD). Tween 20 was purchased from EMD Biosciences (San Diego, CA). Klenow fragment, T4 ligase, factor Xa (FXa), and restriction enzymes were purchased from New England Biolabs (Ipswich, MA). Doxorubicinol was obtained from Qventas Inc. (Branford, CT).
Molecular Cloning of Human AKR and CBR4 Genes and Preparation of Genetic Variants.
The human AKR1C3 wild-type coding region was excised from a pOTB7 recombinant plasmid (Invitrogen, Carlsbad, CA) by using XmnI and DdeI (blunt end with Klenow fragment) and subcloned into HindIII (blunt end)–XhoI (blunt end) sites of the pET28a prokaryotic expression vector (EMD Biosciences) with T4 ligase. This construct gave rise to a human AKR1C3 enzyme with an amino terminal 6×His tag separated from the enzyme by a 32-amino acid residue linker. A FXa cleavage site and methionine start site were inserted at the amino terminus between the linker and AKR1C3 gene using the QuikChange Site-Directed Mutaganesis Kit (Stratagene, La Jolla, CA) with the 5′-CTCCGTCGACAAGCTATAGAAGGAAGAATGGATTCCAAACACCAG-3′ (forward) and 5′- CTGGTGTTTGGAATCCATTCTTCCTTCTATAGCTTGTCGACGGAG-3′ (reverse) primers (the aforementioned sites are underlined). The QuikChange polymerase chain reaction (PCR) amplification protocol for site-directed mutagenesis was modified as follows: two separate 50-μl reactions (one for each primer) were subjected to 10 cycles of denaturation at 95°C for 1 min, annealing at 60°C for 1.5 min, and elongation at 68°C for 6.5 min. A 25-μl aliquot of each PCR amplification was combined with 0.75 μl of PfuTurbo DNA polymerase (Stratagene). This reaction was subjected to 18 cycles of the same PCR protocol above.
The human AKR1B10 and CBR4 wild-type coding regions were PCR-amplified from a pOTB7 (Invitrogen) and pCMV-SPORT6 (Invitrogen) recombinant plasmid, respectively, using the following primers, which contained an EcoRI adapter in the forward primers and a XhoI adapter in the reverse primers (adapters are underlined): 5′-GTACCGCTCGAATTCATGGCCACGTTTGTGG-3′ (forward) and 5′-GTCTGCTACCTCGAGTCAATATTCTGCATCG-3′ (reverse) for AKR1B10 and 5′-GTACCGCTCGAATTCATGGACAAAGTGTGTGCTG-3′ (forward) and 5′-GTCTGCTAACTCGAGGTGCCCTTGATGCTAATC-3′ (reverse) for CBR4. PCR was performed in a 50-μl reaction buffer containing 100 ng of template, 200 ng of each primer, 0.2 mM dNTPs, and 1.25 units of PfuTurbo DNA polymerase. The amplifying conditions for PCR involved an initial denaturation step at 95°C for 1 min, followed by 35 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 1 min, and extension at 68°C for 10 min. Subsequently, a final extension reaction was performed at 68°C for 10 min. The AKR1B10 and CBR4 PCR products were cut with EcoRI and XhoI, then subcloned into pET28a. Respectively, these constructs gave rise to a human AKR1B10 and CBR4 enzyme with an amino terminal 6×His tag and a 26-amino acid residue linker between the tag and enzyme. A FXa cleavage site was inserted at the amino terminus between the linker and gene via site-directed mutagenesis with the following primers: 5′-GGATCCGAATTCATAGAAGGAAGAATGGCCACGTTTGTG-3′ (forward) and 5′-CCACAAACGTGGCCATTCTTCCTTCTATGAATTCGGATCC-3′ (reverse) for AKR1B10 and 5′-CGGATCCGAATTCATAGAAGGAAGAATGGACAAAGTGTGTGC-3′ (forward) and 5′-GCACACACTTTGTCCATTCTTCCTTCTATGAATTCGGATCCG-3′ (reverse) for CBR4 (the FXa sites are underlined in the primer sequences).
The pET15b constructs containing the human AKR1B1, AKR1C1, AKR1C4, and AKR7A2 genes were generously provided by Dr. C. R. Wolf (Biomedical Research Centre, Ninewells Hospital and Medical School, University of Dundee, Dundee, Scotland). These constructs gave rise to their respective AKR genes with a 10-amino acid residue linker between the 6×His tag and gene. A FXa cleavage site was inserted at the amino terminus between the linker and gene by using site-directed mutagenesis with the following primers (the FXa site is underlined): AKR1B1, 5′-CGCGGCAGCCATATAGAAGGAAGAATGGCAAGCCGTCTCC-3′ (forward) and 5′-GGAGACGGCTTGCCATTCTTCCTTCTATATGGCTGCCGCG-3′ (reverse); AKR1C1, 5′-CGCGGCAGCCATATAGAAGGAAGAATGGATTCGAAATATC-3′ (forward) and 5′-GATATTTCGAATCCATTCTTCCTTCTATATGGCTGCCGCG-3′ (reverse); AKR1C4, 5′-GCGGCAGCCATATAGAAGGAAGAATGGATCCCAAATATC-3′ (forward) and 5′-GATATTTGGGATCCATTCTTCCTTCTATATGGCTGCCGC-3′ (reverse); AKR7A2,5′-GCGGCAGCCATATAGAAGGAAGAATGGATCCCAAATATC -3′ (forward) and 5′-GATATTTGGGATCCATTCTTCCTTCTATATGGCTGCCGC-3′ (reverse). In the case of the construct with the AKR1C4 gene, a ns-SNP resulting in a change from cysteine to tyrosine at position 170 was detected after sequencing. Therefore, we performed site-directed mutagenesis as described above with the following primers (ns-SNP is underlined) to generate the wild-type AKR1C4 gene: 5′-GTCAACTTCAACTGCAGGCAGCTGGAG-3′ (forward) and 5′-CTCCAGCTGCCTGCAGTTGAAGTTTGAC-3′ (reverse). A FXa cleavage site was inserted at the amino terminus between the linker and AKR1C4 gene by using site-directed mutagenesis with the 5′- GCGGCAGCCATATAGAAGGAAGAATGGATCCCAAATATC-3′ (forward) and 5′-GATATTTGGGATCCATTCTTCCTTCTATATGGCTGCCGC-3′ (reverse) primers.
The construct with the AKR7A2 gene included a ns-SNP at position 142, resulting in a change from alanine to threonine, which turned out to be one of the variants that we used in this study. Therefore, we performed site-directed mutagenesis as described above with the following primers to generate the wild-type gene: 5′-CTTCTACCTACACGCACCTGACCACG-3′ (forward) and 5′-CGTGGTCAGGTGCGTGTAGGTAGAAG-3′ (reverse). This construct gave rise to a wild-type AKR7A2 enzyme with a 10-amino acid residue linker between the 6×His tag and gene.
The pET28a- and pET15b-variant constructs were prepared by site-directed mutagenesis using the QIAGEN (Mississauga, ON, Canada) protocol with the primers listed in Supplemental Table 1. All constructs were verified by dideoxy sequencing at the University of British Columbia Nucleic Acid Protein Service unit. The truncated variant for AKR1C3 (E36Term) was not prepared because the translated protein is devoid of the important amino acid residues required for catalysis (Asp50, Tyr55, Lys84, and His117) (Schlegel et al., 1998; Di Luccio et al., 2006; Jin and Penning, 2007). Therefore, it was assumed that the protein would be inactive.
Expression and Purification of Recombinant Human AKR and CBR4 Wild-Type and Variant Enzymes.
The pET constructs of the AKR and CBR4 wild-type and variants were heat shock-transformed into Escherichia coli BL21 (DE3) pLysS competent cells and expressed under the control of an IPTG-inducible T7 polymerase. Cells were plated on Luria-Bertani broth agar (1% bacto-tryptone, 0.5% yeast extract, 0.5% NaCl) supplemented with antibiotics (25 μg/ml chloramphenicol and 50 μg/ml kanamycin sulfate for the pET28a constructs or 100 μg/ml ampicillin for the pET15b constructs). Colonies were randomly picked and cultured overnight at 37°C in 3 ml of Luria-Bertani broth with kanamycin and chloramphenicol at the same concentrations stated previously. Cultures were expanded to 800 ml and grown at 37°C until an OD600 of 0.5 was reached. IPTG was added to a final concentration of 1 mM, and cells were allowed to grow for an additional 5 h, after which the cultures were harvested by centrifugation (4000g for 20 min at 4°C), followed by removal of the supernatant.
The cells containing the recombinant 6×His-tagged AKR enzymes were resuspended at 5 ml per gram wet weight with buffer A (300 mM NaCl, 50 mM NaH2PO4, pH 8.0). The cell suspension was lysed with lysozyme (final concentration of 1 mg/ml, incubated on ice for 30 min), and subsequently disrupted using six 10-s bursts (with a 10-s cooling period between each burst) from a sonic dismembranator with a microtip set at 200 to 300 W. This was followed by incubation with DNaseI and RNaseI (5 and 10 μg/ml, respectively) for 15 min on ice and then centrifugation (10,000g, 20 min, 4°C). The cell lysate was subjected to nickel-nitrilotriacetic acid affinity (Ni-NTA) chromatography to isolate the recombinant proteins (Bains et al., 2008). Ultimately, the AKRs were eluted by using multiple fractions of buffer A with increasing concentrations of imidazole (30, 50, 100, and 250 mM).
Cells containing the recombinant 6×His-tagged CBR4 enzymes were resuspended in buffer B (100 mM NaH2PO4, 10 mM Tris-Cl, pH 8.0) with 8 M urea for lysis. The extracted CBR4 proteins were subjected to purification by Ni-NTA chromatography under denaturing conditions according to the QIAGEN protocol. In the end, CBR4 was eluted by using multiple fractions of buffer B with decreasing pH levels (6.3, 5.9, and 4.5). The eluted fractions were then dialyzed at 4°C to gradually remove the urea (buffer B with 6, 4, 2, and 1 M urea for 2 h each followed by 0 M urea overnight).
Protein purity was assessed by running elution fractions on 18% SDS-polyacrylamide gels, which were stained with SYPRO Ruby (Invitrogen Canada, Inc., Burlington, ON, Canada) overnight (16 h). After staining, the protein was detected by using a Storm 840 Molecular Dynamics Imager (GE Healthcare, Little Chalfont, Buckinghamshire, UK) at excitation and emission wavelengths of 450 and 520 nm, respectively.
Western blot analyses of the purified fractions were conducted according to the procedure described by Odyssey (LI-COR Biosciences, Lincoln, NE). After 18% SDS-polyacrylamide gel electrophoresis, proteins were transferred at 20 V in Towbin's buffer (25 mM Tris, 192 mM glycine, and 20% v/v methanol) overnight (at 4°C) to a Hybond-C Extra nitrocellulose membrane (GE Healthcare). The membranes were blocked in Odyssey blocking buffer, and the enzyme was detected by using either a MaxPab polyclonal mouse antihuman AKR1B1, AKR1B10, AKR1C1, AKR1C2, AKR1C3, AKR1C4, AKR7A2, or CBR4 antibody (Abnova Corporation, Taipei City, Taiwan) (diluted 1:2500) as the primary antibodies and IRDye 800CW goat anti-mouse IgG as the secondary antibody (diluted 1:5000) (LI-COR Biosciences). Both primary and secondary antibodies were in blocking buffer containing 0.1% Tween 20. The bound secondary antibody was detected by using the Odyssey Infrared Imaging system (LI-COR Biosciences).
Enzymatic Activity of AKR and CBR4 Enzymes in the Presence of Test Substrates.
The enzyme activities of the purified 6×His-tagged AKR and CBR4 wild-type and variant enzymes were measured by using a Fluoroskan Ascent FL (Thermo Fisher Scientific) by following the initial rate of either NADPH oxidation or NADP+ reduction (depending on the test substrate) at excitation and emission wavelengths of 355 and 460 nm, respectively. The assays were conducted using 1-acenaphthenol (for AKR1C isoforms), dl-glyceraldehyde (for AKR1B1), (S)-1-indanol (for AKR1B10), 9,10-phenanthrenequinone (for AKR7A2), and menadione (for CBR4) as test substrates (Burczynski et al., 1998; O'Connor et al., 1999; Martin et al., 2006; Byrns et al., 2008; Endo et al., 2008; Takahashi et al., 2008). In brief, 3 μg of purified protein was incubated with 2.3 mM NADP+ or 0.2 mM NADPH and test substrate [1 mM acenapthenol and dl-glyceraldehyde, 0.5 mM (S)-1-indanol, 0.05 mM 9,10-phenanthrenquinone, and 5–500 μM menadione] in a reaction mixture of 150 μl of 100 mM KH2PO4, pH 7.4. Assays involving AKR1B1, AKR1C1, AKR1C2, AKR1C3, AKR7A2, and CBR4 were performed at 25°C, whereas assays with the remaining enzymes were conducted at 37°C (these were the temperatures at which the assays were performed in the aforementioned published literature with the test substrates). In these assays, the concentration of dimethyl sulfoxide-methanol (4:1 mixture), which was required to dissolve the substrates (except for dl-glyceraldehyde for which water was used) was kept below 4% (v/v) in the final reaction mixture. Readings were collected at 20-s intervals for 1.5 h with shaking between each reading. Maximal rates were calculated from the Ascent program (version 2.6) (Thermo Fisher Scientific) using a 5-min interval (15 total readings) with the steepest slope. The enzymatic activity was calculated from the maximal rates using a standard curve constructed from the fluorescence measurements of solutions of known NADPH concentrations. Enzyme activity in the presence of 1-acenapthenol and (S)-1-indanol was measured as nanomoles of NADP+ reduced per minute per milligram of purified protein, whereas activity for the other substrates was measured as nanomoles of NADPH oxidized per minute per milligram of purified protein. Activities were ascertained and compared with published rates to determine whether the purified enzymes were functional.
Kinetic Activity of AKR and CBR4 Enzymes in the Presence of Anthracyclines.
Activity measurements for the reduction of the anthracyclines were performed by incubating either DOX or DAUN (50–700 μM; in some cases up to 1000 μM) with 3 μg of purified AKR or CBR4 enzyme in a total volume of 150 μl containing 100 mM potassium phosphate, pH 7.4, and 1 mM NADPH at 37°C. Protein concentrations were determined by the Bradford protein assay using bovine serum albumin as a standard. The reaction was stopped by adding 300 μl of ice-cold acetonitrile, which contained idarubicin as an internal standard, followed by vortex mixing and centrifugation at 10,000g for 10 min at 4°C to remove protein. The supernatant was removed for HPLC analysis. HPLC separation was performed by using a Waters Alliance 2695 system (Waters, Milford, MA) with an analytical column (Waters Symmetry C18, 75 × 4.6 mm i.d., 3 μm) and a guard column (Phenomenex SecurityGuard C18, 40 × 4.6 mm i.d.; Phenomenex, Torrance, CA). HPLC conditions were as follows: mobile phase A, 0.1% formic acid and B, acetonitrile; gradient elution, 0 to 1 min, 15% B; 1 to 8 min, 15% B to 35% B; 8 to 10 min, 35% B; 10 to 10.1 min, 35% B to 15% B; column re-equilibration for 2 min at a constant flow rate of 1 ml/min. The column was heated to 30°C, and the autosampler temperature was set to 10°C with 5 μl of sample injected onto the column. Fluorescence detection of DOX and DAUN and their carbon-13 hydroxy metabolites, DOXol and DAUNol, was performed with excitation and emission wavelengths of 460 and 550 nm, respectively (Waters 2475 Multi λ Detector). Quantitation of DOXol and DAUNol was performed on the basis of a weighted (1/x2) linear regression determined from solutions of known concentrations of DOXol. Because a chemical standard for DAUNol could not be obtained, its concentrations were calculated as DOXol equivalents using a response ratio of 1.0. All HPLC data processes, including chromatogram integration, calibration, and quantitative calculations, were performed with Waters Empower software (version 2.0).
The kinetic constants of Vmax and Km were determined by fitting rate measurement data using nonlinear least-squares fitting of a Michaelis-Menten hyperbola (Prism version 4.0; GraphPad Software Inc., San Diego, CA). Values for kcat were calculated from Vmax values using the apparent molecular weight for the 6×His-tagged AKRs and CBR4 (wild type and variants). The kcat/Km values for the wild-type and variant enzymes were also calculated. After Michaelis-Menten data analysis, Eadie-Hofstee plots were generated to check for deviation from linearity with changing substrate concentrations.
Statistical Analysis.
Statistical analyses were performed by using Instat (version 3.6; GraphPad Software Inc.). Results were expressed as means ± S.D. Enzyme activities were compared by using a one-way analysis of variance followed by Tukey-Kramer multiple comparisons tests. Differences were considered significant at p < 0.05.
Results
Expression and Purification of Human AKR and CBR4 Wild-Type Enzymes and Variants.
The expression of the 6×His-tagged recombinant human AKR and the CBR wild-type enzymes were confirmed by Western blot analyses, which showed a band with mobility corresponding to the calculated molecular mass of the tagged AKR or CBR protein (38.5 kDa for AKR1B1, 40.3 kDa for AKR1B10, 39.4 kDa for AKR1C1, 40.2 kDa for AKR1C2, 41.9 kDa for AKR1C3, 39.7 kDa for AKR1C4, 39.3 kDa for AKR7A2, and 29.5 kDa for CBR4) (Supplemental Fig. 1). Total protein staining of the SDS-polyacrylamide gel electrophoresis gel demonstrated that the wild-type fraction was purified from its transformed bacterial lysate, because no other proteins were detected (Supplemental Fig. 1). For the AKR wild-type and variant enzymes, the majority of pure enzyme was recovered in the 250 mM imidazole elution fractions; no further protein was eluted with higher imidazole concentrations. For the CBR4 wild-type and variant enzymes, the majority of pure enzyme was recovered in the pH 4.5 elution fractions after protein purification under denaturing conditions with 8 M urea. The results for the expression and purification of the variant forms of each enzyme paralleled that of the corresponding wild-type enzyme (data not shown).
Enzymatic Rates of AKR and CBR4 Wild-Type and Variants with Test Substrates.
Enzymatic rates for the AKR1C isoforms were determined using 1-acenaphthenol as a standard test substrate, whereas 9,10-phenanthrenequinone, dl-glyceraldehyde, (S)-1-indanol, and menadione were used to calculate AKR7A2, AKR1B1, AKR1B10, and CBR4 activity, respectively (Table 2). A total of six 6×His-tagged variants exhibited significantly reduced activity rates compared with their corresponding wild-type enzymes. In the presence of 1-acenaphthenol, the 6×His-tagged wild-type AKR1C isoforms were found to have rates ranging from 1660 ± 190 to 3370 ± 160 nmol NADP+ reduced/min · mg purified protein, which was in accordance with published values (listed in Table 2). Three of the five variants for AKR1C3 (A106T, R170C, and P180S) were found to have significantly lower rates compared with the wild type (29–39% lower). In addition, the F46Y and L311V variants had significantly lowered enzymatic rates compared with their respective AKR1C2 and AKR1C4 wild types (30–42% lower). For the 6×His-tagged AKR7A2 wild-type enzyme, the rate was found to be 2553 ± 229 nmol NADPH oxidized/min · mg purified protein, which parallels the published rate of 3100 ± 107 nmol · mg using 9,10-phenanthrenequinone. Only one variant (A142T) was found to have a significantly reduced enzymatic rate compared with the wild-type AKR7A2 (35% lower). The wild-type AKR1B isoforms were found to be active with rates (AKR1B1, 354 ± 48 nmol/min · mg; AKR1B10, 980 ± 80 nmol/min · mg) similar to published values [AKR1B1, 441 ± 14 nmol/min · mg with dl-glyceraldehyde; AKR1B10, 1000 nmol/min · mg with (S)-1-indanol]; none of the variant forms of these enzymes had significantly altered activity compared with their respective wild-type enzymes. In the case of the CBR4 wild-type enzyme, the kinetic parameters (Vmax, 543 ± 41 nmol/min · mg; Km, 47 ± 9 μM; kcat, 16.1 ± 1.2 min−1; kcat/Km, 0.34 ± 0.07 min−1 · μM−1) closely reflected the published values with menadione (calculated Vmax, 508 nmol/min · mg; Km, 36 μM; kcat, 12.7 min−1; kcat/Km, 0.35 min−1 · μM−1). The kinetic parameters for the L70M variant were similar to the wild type; hence, the leucine to methionine substitution at position 70 in this protein does not seem to alter the in vitro enzyme capability of the CBR4 enzyme, at least with menadione as the test substrate.
Enzymatic activities for reported test substrates by recombinant 6×His-tagged AKR and CBR wild-type and variant allele enzymes
Values correspond to mean ± S.D. obtained from three experiments performed with three independent enzyme preparations (n = 9) for each isoform. Reported activities for the wild-type enzymes are also given for comparison purposes.
After the studies with the test substrates, we tested for a significant difference in activity between the tagged and untagged enzymes. The untagged enzymes were generated by incubating the tagged wild-type AKR proteins with FXa for 6 h at 23°C. The reaction was mixed with Ni-NTA resin and run through a column to separate the 6×His tag and linker from the native enzyme. Western blot analysis demonstrated this treatment completely removed the tag and linker from the wild-type enzymes (Supplemental Fig. 2). There were no significant differences in enzymatic activity between the tagged and native AKR1C using 1-acenaphthenol as substrate (rates of the native enzymes were 1800 ± 130, 2710 ± 140, 3060 ± 210, and 1500 ± 210 nmol/min · mg for AKR1C1, AKR1C2, AKR1C3, and AKR1C4, respectively), suggesting that the amino acid linker and 6×His tag engineered on the amino terminus of the wild-type gene products have no effect on enzyme activity. Likewise, we found similar results between the tagged and native AKR1B isoforms, AKR7A2, and CBR4 enzymes [rates of the native enzyme were 300 ± 50 (AKR1B1), 910 ± 40 (AKR1B10), and 2550 ± 250 (AKR7A2) nmol/min · mg; kinetic parameter values for the native CBR4 were: Vmax, 508 ± 32 nmol/min · mg; Km, 50 ± 10 μM; kcat, 15.0 ± 0.9 min−1; kcat/Km, 0.30 ± 0.07 min−1 · μM−1]. Although we did not cleave off the tag and linker for the variants, we assumed that there would be no significant difference between the rates of the tagged and untagged variant enzymes. Keeping these results and assumptions in mind, the uncleaved wild-type and variant enzymes were used for subsequent activity assays using the anthracyclines as substrates.
Kinetic Characterization of AKR and CBR4 Wild-Type and Variant Enzymatic Activities with DOX and DAUN as Substrates.
We measured the in vitro formation of the major alcohol metabolites to evaluate the impact of the single amino acid substitutions in the human AKR and CBR4 enzymes on the reduction of anthracycline drugs. A 50-min incubation period was found to be in the linear range of enzymatic activity for each of the concentrations of DOX and DAUN used to conduct the enzymatic assays (3 μg of purified protein). In addition, the concentration of cofactor (1 mM NADPH) was sufficient for maximal enzymatic activity during this 50-min incubation period. Higher concentrations of cofactor were also examined (1.5 and 2 mM); however, the enzymatic rates associated with these concentrations did not differ from that of 1 mM (data not shown).
Full chromatographic resolution of DAUNol, DOXol, DAUN, DOX, and idarubicin (internal standard) was achieved for all chemical standards and in vitro samples (Bains et al., 2008). DOXol, DOX, DAUNol, DAUN, and idarubicin were observed to elute at 4.6, 5.5, 6.1, 6.9, and 7.4 min, respectively. Incubation of the 6×His-tagged AKR and CBR4 wild-type and variant enzymes with DOX generated a single new chromatographic peak that was identified as DOXol. Likewise, incubating the AKR and CBR4 wild-type and variant enzymes with DAUN generated a single new chromatographic peak that was identified as DAUNol. The identification of the metabolite peaks was confirmed by incubation of DOX and DAUN with human liver cytosol and the generation of compounds that had identical chromatographic behaviors. There were no detectable peaks at the DAUNol or DOXol retention time in the absence of the AKR and CBR4 proteins. In addition to the enzymatic assays performed in this study, we incubated enzyme only as well as enzyme and substrate without the addition of cofactor as controls. The production of DOXol and DAUNol from these controls was below the limit of detection (25 nM) using HPLC fluorescence. Therefore, we conclude the production of the alcohol metabolites is caused by enzymatic processes.
Michaelis-Menten kinetic curves were constructed for the AKR wild type (Fig. 1) and each of the variant enzymes in the presence of differing concentrations of DAUN, and the kinetic parameter values were determined (Table 3). A total of five allelic variants of AKR1C3, AKR1C4, and AKR7A2 exhibited significant reductions in enzymatic activity with DAUN compared with their respective wild-type enzymes: a 38 to 44% reduction in kcat/Km for the A106T, R170C, and P180S variants of AKR1C3 [this reduction is caused by corresponding decreases in Vmax (23–47%) and kcat (22–47%)]; a 43% reduction in kcat/Km for the L311V variant of AKR1C4 [this reduction is caused by corresponding decreases in Vmax (47% lower) and kcat and kcat/Km (43% lower)]; and a 85% reduction in kcat/Km for the A142T variant of AKR7A2 [this reduction is caused by corresponding decreases in Vmax and kcat (both 61%) and an increase in Km (156%)].

In vitro enzymatic activities for the purified 6×His-tagged AKR1B1 (A), AKR1B10 (B), AKR1C1 (C), AKR1C2 (D), AKR1C3 (E), AKR1C4 (F), AKR7A2 (G), and CBR4 (H) wild-type enzymes with daunorubicin. Activities were measured by following the rate of daunorubicinol production. Three independent batches of each enzyme were purified, and assays were performed in triplicate with each batch. Enzymatic activities are reported as mean ± S.D. (n = 9). Dotted lines refer to variants that have significant differences in kcat/Km parameter values compared with the wild type.
Kinetic constants for daunorubicin metabolism by recombinant 6×His-tagged AKR and CBR4 wild-type and variant enzymes
Values correspond to mean ± S.D. obtained from three experiments performed with three independent enzyme preparations (n = 9) for each isoform.
With DOX as a substrate (Fig. 2; Table 4), three allelic variants of AKR1C3 and one for AKR7A2 demonstrated significant reductions in enzymatic activity with respect to the wild-type enzymes: a 52 and 69% reduction in kcat/Km for both the R170C and P180S variants of AKR1C3, respectively [this reduction is caused by corresponding decreases in Vmax (41 and 44%) and kcat (39 and 45%)]; and a 60% reduction in kcat/Km for the A142T variant of AR7A2 [this reduction is caused by corresponding decreases in Vmax (41%) and kcat (44%) and an increase in Km (47%)].

In vitro enzymatic activities for the purified 6×His-tagged AKR1B1 (A), AKR1B10 (B), AKR1C1 (C), AKR1C2 (D), AKR1C3 (E), AKR1C4 (F), AKR7A2 (G), and CBR4 (H) wild-type enzymes with doxorubicin. Activities were measured by following the rate of doxorubicinol production. Three independent batches of each enzyme were purified, and assays were performed in triplicate with each batch. Enzymatic activities are reported as mean ± S.D. (n = 9). Dotted lines refer to variants that have significant differences in kcat/Km parameter values compared with the wild type.
Kinetic constants for doxorubicin metabolism by recombinant 6×His-tagged AKR and CBR4 wild-type and variant enzymes
Values correspond to mean ± S.D. obtained from three experiments performed with three independent enzyme preparations (n = 9) for each isoform.
Eadie-Hofstee plots for the AKR and CBR4 wild-type and variant enzymes verified linearity at the same concentrations of DAUN and DOX used to conduct the assays (r2 > 0.88 for all plots). Furthermore, by comparing kcat/Km values, we observed that DAUN is a better substrate for the AKR wild-type and variant enzymes compared with DOX (1.9- to 3-fold higher for AKR1B1, 129.1- to 256.5-fold higher for AKR1B10, 4.9- to 6.7-fold higher for AKR1C1, 2.4- to 8.2-fold higher for AKR1C3, 2.5- to -4.5 fold higher for AKR1C4, and 6.0- to 17.1-fold higher for AKR7A2). Only the AKR1C2 enzyme (wild type and variant) exhibited no significant difference in kcat/Km values between DAUN and DOX, suggesting that both drugs are equally good substrates for this enzyme. With the CBR4 wild-type and variant enzymes, DOX was demonstrated to be a superior substrate as seen with a 31.9- to 40-fold increase in kcat/Km over DAUN.
Discussion
The focus of this study was to examine the effect of ns-SNPs on the enzymatic activity of human AKR and CBR4 using test substrates, as well as DAUN and DOX. The wild-type and variant alleles of these genes were cloned into an E. coli expression vector with a 6×His tag added to the amino terminus of the expressed protein. The respective wild-type and variant proteins for each gene were purified, and purity was assessed by electrophoretic and Western blot analyses. To determine whether the 6×His tag influenced enzyme activity, the tag was removed by incubation with FXa and the enzyme was repurified. Using the test substrates, the activity of the tagged enzyme did not significantly differ from its native counterpart. Therefore, we were able to assess the impact of the single amino acid substitutions on the activity of each enzyme, without cleaving the tag in the subsequent assays involving DAUN and DOX.
DOXol and DAUNol, which are the carbon-13 alcohol metabolites of DOX and DAUN, respectively, were quantified in these assays, because previous published studies demonstrated that they are the major metabolites in cancer patients receiving treatment with either of these anthracycline anticancer drugs (Lipp and Bokemeyer, 1999; Plebuch et al., 2007). Our assays show that both drugs are converted to their respective major metabolites by the wild-type and variant enzymes for the reductases examined in this study.
Of the 27 allelic variants that were studied (excluding the E36Term truncated variant), five were found to significantly decrease metabolism of DAUN compared with their respective wild-type enzymes: the A106T, R170C, and P180S variant forms of AKR1C3; the L311V variant of AKR1C4; and the A142T variant of AKR7A2. In the presence of DOX as a substrate, three of the five aforementioned variants significantly reduced activity compared with their respective wild-type enzymes: R170C, P180S, and A142T. To our knowledge, this study is the first demonstration of the effect of these variant enzymes on DAUN and DOX metabolism.
We used three-dimensional models of human AKR1C3, AKR1C4, and AKR7A2 enzymes (Fig. 3) to examine the location of the five mutations relative to the active and cofactor binding sites. Reduction of the carbonyl group by AKR enzymes is thought to involve the cooperation of four amino acids (tyrosine, lysine, aspartic acid, and histidine) forming a catalytic tetrad, which are positionally conserved in the individual reductases within this superfamily (Jez et al., 1997; Penning 2003; Barski et al., 2008). The locations of these conserved residues for the three aforementioned AKR enzymes are as follows: AKR1C3 and AKR1C4, Asp50, Tyr55, Lys84, and His117; AKR7A2, Asp72, Tyr77, Lys105, and His141. The roles that these residues play in the reduction of carbonyl-containing substrates to their alcohol products are shown in Fig. 4 (Schlegel et al., 1998; Di Luccio et al., 2006; Jin and Penning, 2007; Barski et al., 2008; Di Costanzo et al., 2009).
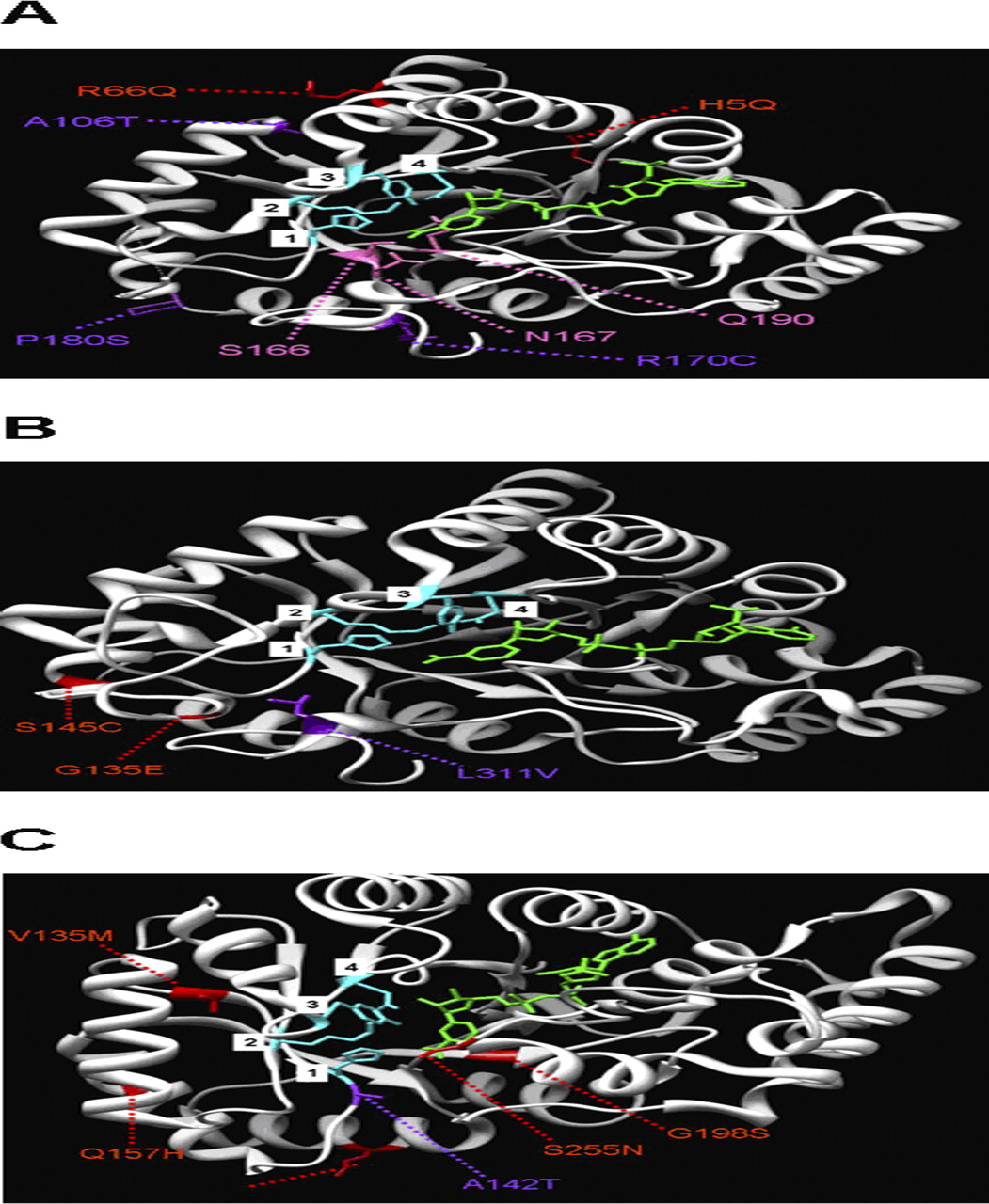
Three-dimensional molecular structure of human AKR1C3 (A), AKR1C4 (B), and AKR7A2 (C) wild-type enzymes complexed with the cofactor NADP+ (green) [Protein Data Bank ID: 2FGB (AKR1C3), 2FVL (AKR1C4), and 2BP1 (AKR7A2)]. The mutations that significantly altered enzyme activity (purple) and those that did not alter enzyme activity (red) are shown. In addition, the residues comprising the catalytic tetrad are illustrated (blue): AKR1C3 and AKR1C4, 1) His117, 2) Lys84, 3) Tyr55, and 4) Asp50; AKR7A2, 1) His141, 2) Lys105, 3) Tyr77, and 4) Asp72. In addition, provided are three residues of AKR1C3 that participate in cofactor binding: Ser166, Asp167, and Glu190 (pink). The molecular graphic images of the AKRs were produced by using the University of California, San Francisco Chimera program (Resource for Biocomputing, Visualization, and Informatics, University of California, San Francisco, CA).
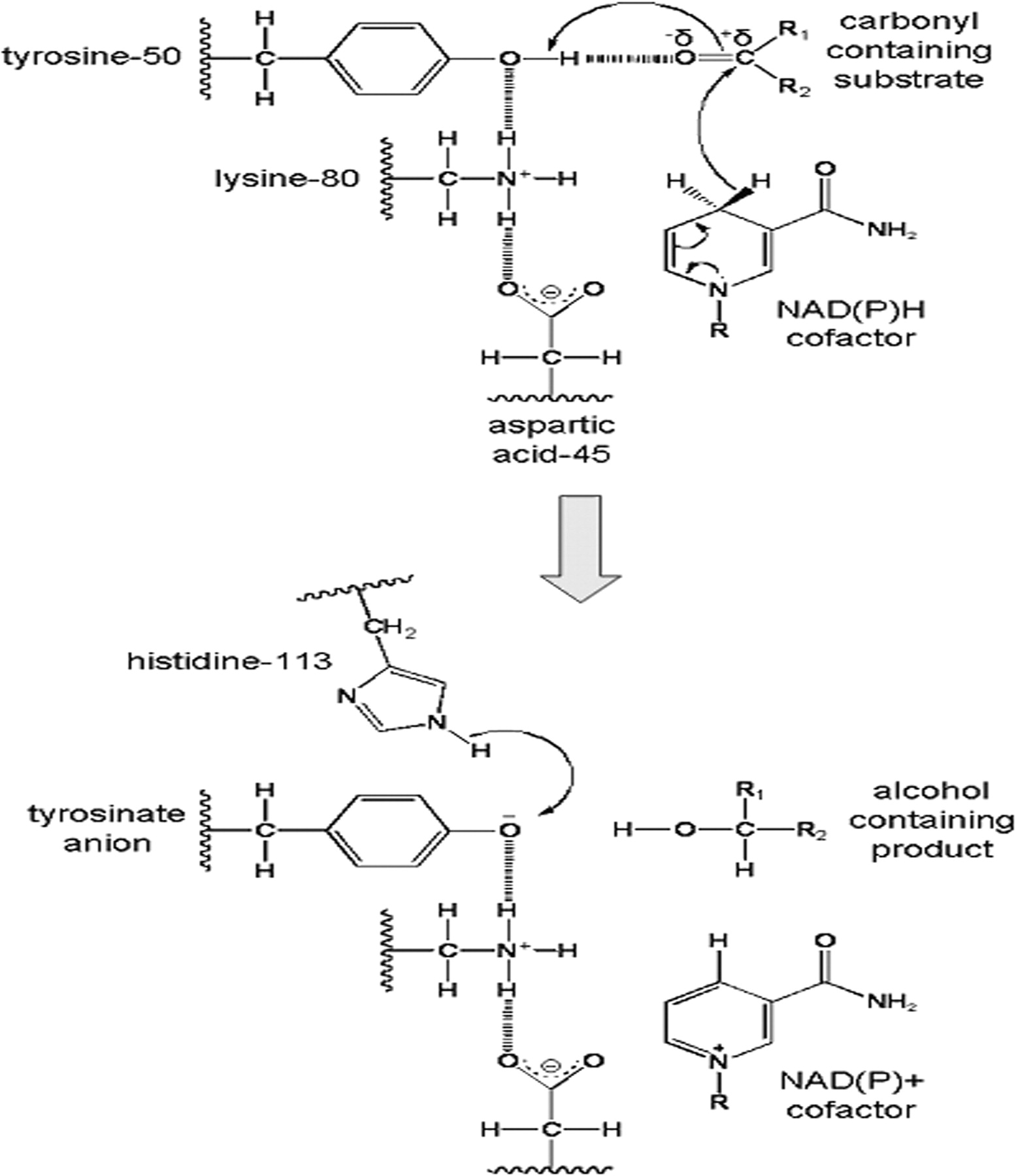
Proposed catalytic mechanism for AKR-mediated reduction reaction [modified based on the scheme provided by Barski et al. (2008)]. The numbering of the four catalytic residues is illustrated for AKR1A1. Top, in the reduction of the carbonyl-containing substrate, the Tyr50 residue is shown to be a proton donor for the carbonyl substrate. It also forms a hydrogen bond with the substrate, resulting in carbonyl polarization, which accelerates the hydride transfer of the pro-R hydrogen from NADPH to the carbonyl carbon. The hydrogen-bonding network provided by Lys80 and Asp45 serves to lower the pKa of tyrosine, making the proton transfer from tyrosine to the substrate easier. Bottom, the reduced carbonyl (i.e., the alcohol-containing substrate) then dissociates from the active site, and a net charge on the tyrosinate anion is stabilized by the hydrogen bonding network. His113 also participates in the catalytic mechanism by donating protons to the tyrosinate anion to restore the conserved tyrosine residue.
The R170C and P180S mutations in AKR1C3 are located close to amino acid residues in the cofactor binding site that participate in hydrogen bonding to the nicotinamide ring of NADPH (Ser166 in β-sheet 5, Asp167 in loop 5, and Glu190 in β-sheet 6), and therefore help anchor the cofactor to the AKR1C3 enzyme (Komoto et al., 2004). It is possible that these mutations affect the binding of the cofactor, which ultimately reduces the enzymatic conversion of DAUN and DOX to DAUNol and DOXol, respectively. It is, however, possible that the P180S mutation could alter the active site because its effect on metabolism seems greater for DOX (∼70% decrease in activity versus the AKR1C3 wild type) than DAUN (∼30% decrease). Currently, there are no data to indicate why or how the A106T polymorphism affects cofactor or substrate binding. We speculate that the chemical structural differences between the side-chain groups of these amino acids [alanine, -CH3; threonine, -CH(OH)(CH3)] are sufficient enough to alter the conformation of the AKR1C3 protein, thereby changing the structure of the active and/or cofactor binding sites. Furthermore, because a significant difference in enzymatic activity was detected between this variant and the wild type in the presence of DAUN, and not DOX, we speculate that the active site, rather than the cofactor binding site, is more severely affected by this amino acid substitution.
Previous studies have shown the importance of Leu311 in substrate binding to human AKR1C4. For example, a study by Matsuura et al., (1998), which used chimeric enzymes produced by switching the C-terminal loop in AKR1C4 with that of AKR1C1, demonstrated that the binding of substrates, inhibitors, and activators of AKR1C4 requires the amino acid residues located in the C-terminal loop of the wild-type AKR1C4 enzyme, such as Leu311. Furthermore, Matsuura et al. found that a leucine to valine mutation in amino acid 311 (L311V) decreased the catalytic activity of AKR1C4 for its substrates, but did not affect the enzyme's sensitivity to an inhibitor or affect the enzyme's response to an activator. Hence, their study suggested that Leu311 is important in substrate binding to the active site of the AKR1C4 enzyme. This finding was further substantiated in a study by Kume et al., (1999), which used purified recombinant human AKR1C4 enzymes to show that the L311V polymorphism resulted in a 3- to 5-fold decrease in enzyme activity compared with the wild type for a variety of xenobiotic and steroidal substrates. In addition to the effect of L311V, this study revealed that the S145C ns-SNP (serine to cysteine mutation at amino acid position 145) had no significant effect on enzyme activity compared with the wild type for a variety of xenobiotic and steroidal substrates. Our study demonstrates that the L311V mutation dramatically alters the ability of the enzyme to metabolize DAUN and DOX and, like the previous studies on xenobiotic and steroidal substrates, indicates that the S145C mutation has little affect on the ability of the enzyme to use DAUN and DOX as substrates.
The alanine to threonine mutation at amino acid position 142 in human AKR7A2 (the A142T ns-SNP) is adjacent to the His141 residue, which is one of the four amino acids that forms a catalytic tetrad for reduction of the substrate (the others being Tyr77, Lys105, and Asp72). Because of this proximity, the A142T polymorphism may hinder the ability of His141 to participate in the catalysis of DOX and DAUN, leading to reduced enzymatic activity compared with the wild-type AKR7A2 enzyme.
In addition to looking at the differences in enzyme activity between the wild-type and variant enzymes with both anthracyclines, our studies demonstrated that DAUN was generally a better substrate for all wild-type and variant forms of the human AKRs (except AKR1C2), as shown by 1.9- to 257-fold increases in kcat/Km over DOX. On the other hand, CBR4 and its variant metabolize DOX far more efficiently than DAUN, as seen by increases of 31.9- to 40-fold in kcat/Km for DOX over DAUN. Catalytic efficiencies for both DAUN and DOX were also compared between the wild-type AKRs and CBRs (Fig. 5). For comparison purposes, kcat/Km values for human recombinant 6×His-tagged CBR1 and CBR3 (Bains et al., 2010) were incorporated, along with human 6×His-tagged AKR1A1 (same as in Bains et al., 2008), for which we expressed, purified, and performed extensive Michaelis-Menten analysis with DAUN and DOX by using the same protocols described in this study (DAUN, Vmax = 4042 ± 233 nmol/min · mg, Km = 737 ± 79 μM, kcat = 2.76 ± 0.16 s−1 · M−1, kcat/Km = 3750 ± 564; DOX, Vmax = 75 ± 10 nmol/min · mg, Km = 540 ± 64 μM, kcat = 0.0051 ± 0.005, kcat/Km = 95 ± 14 s−1 · M−1). Human CBR1 was found to metabolize DAUN far better than the other AKRs and CBRs, whereas AKR1C3 was the better enzyme in DOX metabolism.

Catalytic efficiencies of human recombinant 6×His-tagged AKRs and CBRs in the presence of daunorubicin (A) and doxorubicin (B). In addition, reported are catalytic efficiencies of human recombinant 6×His-tagged AKR1A1, CBR1, and CBR3, which were added for comparison purposes with the AKRs and CBR4 from this study. Efficiency values are reported as mean ± S.D.
In conclusion, this study, along with our previous in vitro studies involving AKRs and CBRs, demonstrates that these enzymes are able to metabolize both DAUN and DOX, with the AKRs and CBR1 generally having higher specificity for DAUN and CBR3 and CBR4 generally having higher specificity for DOX. Significant reductions in enzyme activity were discovered in 12 variants for all of the AKRs and CBRs studied: five variants of AKR1C3, AKR1C4, and AKR7A2 in this study, along with seven variants of AKR1A1 (N52S and E55D), CBR1 (V88I and P131S), and CBR3 (C4Y, V93I, and V244M) in other studies (Bains et al., 2008, 2009, 2010). Mutations in genes encoding these enzymes may contribute to the interpatient variability seen with the development of serious cardiac side effects in DAUN- and DOX-treated patients. If AKRs and CBRs play a major role in anthracycline-induced cardiotoxicity, we propose that this condition is largely the result of accumulation of the parent drug. This may be true because the AKR and CBR isoforms, with variants exhibiting significantly reduced activity toward DAUN and/or DOX, are expressed in human heart tissue (Table 5; Supplemental Fig. 3). The data collected in this study beg the question of whether individuals bearing one or more of the previously mentioned AKR and CBR polymorphisms are at higher risk for developing cardiac side effects after treatment with either of these anthracyclines. This issue can be addressed with clinical association studies, which are currently underway in our laboratories to determine whether there is a correlation with cardiotoxicity.
Relative abundance ratio values of the cytosolic AKR and CBR isoforms in human heart lysate (single donor, 38-year-old female; ProSci Incorporated, Poway, CA) and pooled human liver lysate (50 donors; BD Biosciences)
Relative abundance values are reported as mean in relation to β-tubulin ± S.D. obtained from three experiments performed (n = 3).
Acknowledgments
We thank Dr. J. M. Lubieniecka, Dr. R. H. Takahashi, A. Szeitz, Dr. R. C. Mottus, Dr. T. A. Pfeifer, Dr. S. A. Raithatha, and Dr. J. W. Hodgson (University of British Columbia) for advice and technical expertise.
Footnotes
These studies were supported by the Canadian Institutes of Health Research [Grant MOP-68896]. O.S.B. was supported by a Canadian Institutes of Health Research Doctoral Research Award.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.110.173179.
↵
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- AKR
- aldo-keto reductase
- CBR
- carbonyl reductase
- DOX
- doxorubicin
- DAUN
- daunorubicin
- DOXol
- doxorubicinol
- DAUNol
- daunorubicinol
- HPLC
- high-performance liquid chromatography
- ns-SNP
- nonsynonymous single-nucleotide polymorphism
- Km
- substrate affinity
- kcat
- turnover rate
- kcat/Km
- catalytic efficiency
- Ni-NTA
- nickel-nitrilotriacetic acid affinity
- PCR
- polymerase chain reaction
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- FXa
- factor Xa.

- Received July 20, 2010.
- Accepted September 3, 2010.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}