


Abstract
6-Acetyl-8-cyclopentyl-5-methyl-2-([5-(piperazin-1-yl)pyridin-2-yl]amino)pyrido(2,3-d)pyrimidin-7(8H)-one [palbociclib (PD-0332991)] is a cyclin-dependent kinase 4/6 inhibitor approved for the treatment of metastatic breast cancer and is currently undergoing clinical trials for many solid tumors. Glioblastoma (GBM) is the most common primary brain tumor in adults and has limited treatment options. The cyclin-dependent kinase 4/6 pathway is commonly dysregulated in GBM and is a promising target in treating this devastating disease. The blood-brain barrier (BBB) limits the delivery of drugs to invasive regions of GBM, where the efflux transporters P-glycoprotein and breast cancer resistance protein can prevent treatments from reaching the tumor. The purpose of this study was to examine the mechanisms limiting the effectiveness of palbociclib therapy in an orthotopic xenograft model. The in vitro intracellular accumulation results demonstrated that palbociclib is a substrate for both P-glycoprotein and breast cancer resistance protein. In vivo studies in transgenic mice confirmed that efflux transport is responsible for the limited brain distribution of palbociclib. There was an ∼115-fold increase in brain exposure at steady state in the transporter deficient mice when compared with wild-type mice, and the efflux inhibitor elacridar significantly increased palbociclib brain distribution. Efficacy studies demonstrated that palbociclib is an effective therapy when GBM22 tumor cells are implanted in the flank, but ineffective in an orthotopic (intracranial) model. Moreover, doses designed to mimic brain exposure were ineffective in treating flank tumors. These results demonstrate that efflux transport in the BBB is involved in limiting the brain distribution of palbociclib and this has critical implications in determining effective dosing regimens of palbociclib therapy in the treatment of brain tumors.
Introduction
The cyclin-dependent kinase (CDK) 4/6 pathway is a major regulator of the G1 to S phase transition in the cell cycle (Peyressatre et al., 2015). The p16-CDK4–cyclin D–retinoblastoma (Rb) axis is commonly dysregulated in many cancers and this pathway is a promising target for cancer therapy. During normal cell cycle progression, CDK4 complexes with cyclin D and phosphorylates Rb (VanArsdale et al., 2015). This phosphorylation event leads to downstream signaling via the E2F family of transcription factors and is linked to G1/S phase cell cycle progression (Fry et al., 2004; Baughn et al., 2006; Barton et al., 2013). This pathway is hyperactive in many types of cancers, and inhibitors of this pathway, such as 6-acetyl-8-cyclopentyl-5-methyl-2-([5-(piperazin-1-yl)pyridin-2-yl]amino)pyrido(2,3-d)pyrimidin-7(8H)-one [palbociclib (PD-0332991)], have the potential to be widely used across many solid tumors (Finn et al., 2015). Tumor suppressor proteins, such as p16, regulate the cell cycle by preventing CDK4 from forming a complex with cyclin D. Amplification of CDK4, CDK6, or cyclin D as well as the deletion of CDKN2A (the gene that encodes for p16) is commonly observed in glioblastoma (GBM). Any one of these alterations leads to dysregulation of this critical pathway in cell cycle progression (Thangavel et al., 2013).
Palbociclib (PD-0332991) is a promising CDK4/6 inhibitor for malignancies with alterations in this pathway. Palbociclib was approved for the treatment of metastatic breast cancer in early 2015 for patients with estrogen receptor–positive, Her2-negative tumors (Turner et al., 2015). Although palbociclib is currently approved for breast cancer, the potential use of palbociclib in other indications is under investigation. This p16–cyclin D–CDK4/6-Rb pathway is commonly dysregulated in breast cancer (hormone receptor–positive), melanoma (90%), and GBM (78%) tumors, making it an attractive therapeutic target (Cancer Genome Atlas Research Network, 2008; Peyressatre et al., 2015; Turner et al., 2015). Previous studies have examined the effectiveness of palbociclib therapy against GBM xenograft cell lines (Michaud et al., 2010). Michaud et al. determined that of the 21 GBM xenografts they examined, 16 (76%) were sensitive to palbociclib treatment in vitro. The five tumor lines that were resistant to palbociclib therapy all had mutations in Rb, which is downstream of CDK4/6. These data indicate there is a clear rationale to consider palbociclib and other CDK4/6 inhibitors to treat brain tumors.
A critical factor in the use of palbociclib in the treatment of brain tumors is achieving effective drug delivery to all tumor cells, including those invasive cells that reside behind an intact blood-brain barrier (BBB) (Agarwal et al., 2011b). The BBB acts as both a physical and biochemical barrier, limiting the brain delivery of numerous treatments (Abbott, 2013). Tight junction proteins, such as occludin and claudin, prevent the paracellular transport of compounds from the blood into the brain, and efflux transporters actively prevent compounds from reaching the brain via the transcellular route (Abbott, 2013). P-glycoprotein (P-gp) and breast cancer resistant protein (BCRP) are two efflux transporters that are highly expressed at the BBB (Uchida et al., 2011) and can prevent potentially effective agents from reaching the brain. GBM is the most common primary brain tumor in adults and survival following diagnosis, even after aggressive treatment, is about 1 year (Stupp et al., 2005). Therefore, the purpose of this study was to determine the mechanisms that limit the delivery, and hence efficacy, of palbociclib therapy in an orthotopic xenograft model of patient-derived GBM.
Materials and Methods
Chemicals
Palbociclib (PD-0332991) was purchased from Chemietek (Indianapolis, IN). [3H]prazosin and [3H]vinblastine were purchased from Perkin Elmer Life and Analytical Sciences (Waltham, MA) and Moravek Biochemicals (La Brea, CA), respectively. (3S,6S,12aS)-1,2,3,4,6,7,12,12a-octahydro-9-methoxy-6-(2-methylpropyl)-1,4-dioxopyrazino(1′,2′:1,6)pyrido(3,4-b)indole-3-propanoic acid-1,1-dimethylethyl ester (Ko143) was purchased from Tocris Bioscience (Ellisville, MO), and (R)-4-[(1aR,6R,10bS)-1,2-difluoro-1,1a,6,10b-tetrahydrodibenzo-(a,e)cyclopropa(c)cycloheptan-6-yl]-α-[(5-quinoloyloxy)methyl]-1-piperazine ethanol, trihydrochloride [LY335979 (zosuquidar)] was kindly provided by Eli Lilly and Co. (Indianapolis, IN). Cell culture reagents were purchased from Invitrogen (Carlsbad, CA), and all other chemicals were from Sigma-Aldrich (St. Louis, MO).
In Vitro Studies
In vitro studies were conducted using Madin-Darby canine kidney II (MDCKII) cells. Vector control and gene encoding the murine breast cancer resistance protein (Bcrp1)-transfected MDCKII cells were gifts from Dr. Alfred Schinkel (The Netherlands Cancer Institute, Amsterdam, The Netherlands) and vector control and gene encoding the human p-glycoprotein (MDR1)-transfected (MDCKII-MDR1) cell lines were provided by Dr. Piet Borst (The Netherlands Cancer Institute). Cell lines were cultured in Dulbecco’s modified Eagle’s medium with 10% (v/v) fetal bovine serum and penicillin (100 U/ml), streptomycin (100 μg/ml), and amphotericin B (250 ng/ml). MDCKII-MDR1 cells were cultured in media with colchicine (80 ng/ml) to maintain positive selection pressure for P-gp expression. Cells were maintained in 25-ml tissue culture flasks at 37°C in a humidified incubator with 5% CO2.
Intracellular Accumulation Studies.
Palbociclib intracellular accumulation studies were performed as previously described (Mittapalli et al., 2012). Briefly, the cells were preincubated for 30 minutes with blank cell assay buffer or cell assay buffer containing 200 nM Ko143 or 1 μM zosuquidar. Following the preincubation, 2 µM palbociclib was added to each well for 60 minutes at 37°C. 1% Triton-X was used to lyse the cells, and the lysate was analyzed via liquid chromatography–tandem mass spectrometry (LC-MS/MS) for drug and protein concentration (BCA protein assay) to normalize accumulation.
In Vivo Studies
Animals.
Concentration-time profile studies were conducted in Friend leukemia virus strain B male and female wild-type, gene encoding the murine p-glycoprotein (Mdr1a/b−/−) (P-gp knockout), Bcrp1−/− (Bcrp knockout), and Mdr1a/b−/−Bcrp1−/− (triple knockout) mice (Taconic Farms, Germantown, NY). Animals were maintained in a 12-hour light/dark cycle with unlimited access to food and water and were 8–12 weeks old at the time of the experiment. In vivo studies were approved by the Institutional Animal Care and Use Committee at the University of Minnesota.
Brain Distribution of Palbociclib after a Single Oral Dose.
Wild-type, Mdr1a/b−/−, Bcrp1−/−, and Mdr1a/b−/−Bcrp1−/− mice received an oral dose (10 mg/kg) of palbociclib (vehicle: 1% carboxymethyl cellulose and 1% Tween 80). Following euthanasia in a CO2 chamber, brain and blood samples were collected at 0.5, 1, 2, 4, 8, 12, and 24 hours postdose (n = 4 per time point). Plasma was isolated from whole blood via centrifugation (3500 rpm for 15 minutes at 4°C), the whole brain was removed and washed with ice cold water, and superficial meninges were removed by blotting with tissue paper. Samples were stored at −80°C until analysis via LC-MS/MS.
Steady-State Brain Distribution of Palbociclib.
Alzet osmotic mini pumps (1003D; Durect Corporation, Cupertino, CA) were implanted in the peritoneal cavity of wild-type, Mdr1a/b−/−, Bcrp1−/−, and Mdr1a/b−/−Bcrp1−/− mice to deliver 1 μl/h as a constant infusion to determine the steady-state brain and plasma concentrations of palbociclib (n = 4). Palbociclib (10 mg/ml in dimethylsulfoxide) was loaded into the pumps and primed at 37°C overnight in sterile saline. Pumps were implanted into the peritoneal cavity as described previously (Agarwal et al., 2010). Briefly, isoflurane was used to anesthetize mice, and the hair was removed from the abdominal cavity. A small incision was made in the lower right abdominal wall and the peritoneal membrane was exposed, and then a small incision was made in the peritoneal membrane and the primed pump was inserted into the cavity. The peritoneal membrane was sutured, and the skin incision was closed with surgical clips. The surgical procedure was conducted on a heating pad until the animals were fully recovered. Forty-eight hours following pump implantation, the mice were sacrificed and blood and brain samples were isolated. Samples were stored at −80°C until analysis via LC-MS/MS.
Palbociclib Efficacy in GBM22 Xenograft.
Tumor-bearing studies were conducted in female athymic nude mice (Harlan Sprague-Dawley athymic nude-Foxn1nu mice) as described in detail previously (Carlson et al., 2011; Pokorny et al., 2015). The patient-derived xenografts (PDXs) were derived from individual primary human GBM at the Mayo Clinic (Rochester, MN) and maintained through serial passages in the flank (Carlson et al., 2011). Short-term explant cultures were maintained exclusively in the Sarkaria Laboratory in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum and 1% penicillin/streptomycin media prior to intracranial or flank implantation. Mice were anesthetized using ketamine (100 mg/kg) and xylazine (10 mg/kg), and intracranial tumors were implanted 1 mm anterior and 2 mm lateral from the bregma. GBM22 is a PDX that has homozygous deletion of CDKN2A/B, hemizygous deletion of CDK4, gain of CDK6, and loss of CDKN2C, CCND1, CCND2, and RB1 (Cen et al., 2012). GBM22 tumor cells were implanted either in the flank or intracranially. Eleven days following intracranial tumor implantation, palbociclib was dosed at 150 mg/kg until mice became moribund (n = 10). Fourteen days following flank tumor implantation, palbociclib was dosed at either 150 or 10 mg/kg until tumors reached 1500 mm3 (n = 8–10). Mice were euthanized by CO2 inhalation.
In Vivo Pharmacological Inhibition of Efflux Transporters.
A microemulsion formulation of elacridar, a dual inhibitor of P-gp and BCRP, was prepared as described previously (Sane et al., 2013). Cremophor EL, Carbitol, and Captex 355 were formulated in a 6:3:1 ratio. On the day of the experiment, elacridar was added to this mixture and sonicated to form a 3 mg/ml solution that was then diluted with water to form a 1 mg/ml microemulsion for injection. Wild-type mice received blank microemulsion or 10 mg/kg elacridar via intraperitoneal injection and a single oral dose of palbociclib (10 mg/kg). Two hours following the administration of elacridar and palbociclib, blood and brain samples were collected and analyzed via LC-MS/MS.
LC-MS/MS Analysis of Palbociclib.
The concentration of palbociclib in cell lysate, plasma, tumor, and brain homogenate samples was determined using a sensitive LC-MS/MS assay. Brain and tumor homogenate samples were prepared by adding three volumes of 5% bovine serum albumin before homogenizing using a tissue homogenizer (PowerGen 125; Thermo Fisher Scientific, Waltham, MA). Samples were spiked with 25 ng dasatinib as the internal standard and extracted by the addition of one to two volumes of pH 11 buffer and 5–10 volumes of ethyl acetate, followed by vigorous shaking for 5 minutes and then 5 minutes of centrifugation at 7500 rpm. The organic layer was transferred to microcentrifuge tubes and dried under nitrogen. Samples were reconstituted in 100 µl of mobile phase and transferred to high-performance liquid chromatography vials for analysis. An AQUITY ultra performance liquid chromatography system (Waters, Milford, MA) was used with a Phenomenex Synergi Polar 4 µ polar-RP 80A column (75 × 2 m; Torrance, CA). The ionization was conducted in the positive mode, and the m/z transitions were 448.34–379.95 and 488.21–400.88 for palbociclib and dasatinib, respectively. The retention time was 2.8 minutes for palbociclib and 4.8 minutes for dasatinib. The mobile phase (73:27::1 mM ammonium formate with 0.1% formic acid to acetonitrile) was delivered at a constant flow rate of 0.25 ml/min.
Pharmacokinetic Calculations.
Parameters from the concentration-time profiles in plasma and brain samples were obtained by noncompartmental analysis performed using Phoenix WinNonlin 6.2 (Pharsight, Mountain View, CA). The area under the curve (AUC) for plasma (AUCplasma) and the brain (AUCbrain) were calculated using the log-linear trapezoidal approximation (AUC0-tlast). The standard error around the mean of the AUCs was estimated using the sparse sampling module in WinNonlin. Relative brain exposure comparisons between wild-type, Mdr1a/b−/−, Bcrp1−/−, and Mdr1a/b−/−Bcrp1−/− mice were made using the drug targeting index [(AUCbrain/AUCplasma)knockout/(AUCbrain/AUCplasma)wild type].
Statistical Analysis.
GraphPad Prism 6.04 (San Diego, CA) software was used for statistical analysis. The sample sizes used were based on previous work and were determined based on approximately 80% power to detect differences greater than 10-fold in distribution studies or 2-fold in efficacy studies. Data in all experiments are represented as mean ± S.D. Comparisons between the two groups were made using an unpaired t test. Multiple comparisons were made using one-way analysis of variance followed by Bonferonni’s multiple comparisons test. A significance level of P < 0.05 was used for all studies.
Results
Intracellular Accumulation of Palbociclib.
MDCKII vector control and P-gp– or Bcrp-transfected cell lines were used to study the intracellular accumulation of the CDK4/6 inhibitor palbociclib. Functionality of efflux in the cell lines was validated using [3H]vinblastine as a positive control for P-gp–mediated transport and [3H]prazosin as a positive control for Bcrp-mediated transport. The accumulation of [3H]vinblastine was 90% lower in the P-gp–overexpressing cell line than vector control cells (Fig. 1A) [vector control (normalized): 100 ± 6.7%; P-gp: 9.4 ± 0.8%; P < 0.0001]. This effect was abolished when P-gp was inhibited by 1 μM LY335979. Intracellular accumulation of palbociclib was 25% lower in the P-gp–overexpressing cells than in the vector-control cells (Fig. 1A) (vector control: 100 ± 12.3%; P-gp: 74.8 ± 6.0%; P = 0.0333) and the efflux was again reversed by LY335979. In the Bcrp-overexpressing cells, the accumulation of [3H]prazosin was 73% lower than in the vector control cells (Fig. 1B) (vector control: 100 ± 6.1%; Bcrp: 27.0 ± 2.4%; P < 0.0001) and reversed by Ko143. Intracellular accumulation of palbociclib was significantly diminished in the Bcrp-overexpressing cells versus the vector control cells (Fig. 1B) (vector control: 100 ± 7.9%; Bcrp: 23.7 ± 2.8%; P < 0.0001). The reduced intracellular accumulation of palbociclib was reversed in the presence of the P-gp–specific inhibitor LY335979 (P-gp: 77.5 ± 6.0%; P-gp with LY3359779: 143.6 ± 13.8%; P < 0.0001). Similarly, the difference in intracellular accumulation of palbociclib was abolished in the presence of the Bcrp-specific inhibitor Ko143 (Bcrp: 23.7 ± 2.8%; Bcrp with Ko143: 85.6 ± 1.3%; P < 0.0001). Taken together, these in vitro data demonstrate that palbociclib is a substrate of both P-gp and Bcrp, and the active efflux of palbociclib by each transporter can be inhibited by transporter-specific inhibitors.

Intracellular accumulation of palbociclib in vector-controlled and MDR1- or Bcrp1-overexpressing MDCKII cells. (A) The intracellular accumulation of vinblastine (positive control) and 2 µM palbociclib in MDCKII-vector controlled and MDR1 cells. (B) Intracellular accumulation of prazosin (positive control) and 2 µM palbociclib in MDCKII-vector controlled and MDR1 cells. *P < 0.05 and ***P < 0.001 when compared with wild-type control; #P < 0.001 when compared with transfected cells without an inhibitor (n = 3–6).
Concentration-Time Profiles in Four Genotypes.
The brain and plasma concentration time profiles were determined in Friend leukemia virus strain B wild-type, Bcrp1−/−, Mdr1a/b−/−, and Mdr1a/b−/−Bcrp1−/− mice following a single oral dose (10 mg/kg) of palbociclib. The plasma concentrations are elevated in the Mdr1a/b−/− and Mdr1a/b−/−Bcrp1−/− mice, and the brain concentrations vary widely among the four genotypes, with wild-type mice having the lowest brain exposure, followed by Bcrp1−/−, Mdr1a/b−/−, and Mdr1a/b−/−Bcrp1−/− mice (Fig. 2). The brain-to-plasma AUC ratios in the wild-type, Bcrp1−/−, Mdr1a/b−/−, and Mdr1a/b−/−Bcrp1−/− mice were 0.064, 0.14, 1.42, and 7.36, respectively. The Mdr1a/b−/−Bcrp1−/− mice have a 115-fold increase in the brain exposure of palbociclib when compared with the wild-type mice [i.e., drug targeting index, (AUCbrain/AUCplasma)knockout/(AUCbrain/AUCplasma)wild type (Table 1)].

Concentration-time profiles in Friend leukemia virus strain B wild-type, Bcrp1−/−, Mdr1a/b−/−, and Mdr1a/b−/−Bcrp1−/− mice. Brain and plasma concentration-time profile in (A) wild-type mice; (B) Bcrp1−/− mice; (C) Mdr1a/b−/− mice; and (D) Mdr1a/b−/−Bcrp1−/− mice (n = 4 per time point).
Summary parameters from concentration-time profiles following 10 mg/kg oral dose
DTI = (AUCbrain/AUCplasma)knockout/(AUCbrain/AUCplasma)wild type.
Brain Distribution of Palbociclib at Steady State.
The brain distribution of palbociclib at steady state was determined after a constant rate infusion into the intraperitoneal cavity using Alzet osmotic pumps for 48 hours at 10 µg/h. The steady-state brain-to-plasma ratios were 0.2 ± 0.07, 0.4 ± 0.1, 2.5 ± 0.1, and 27.8 ± 5.9 for wild-type, Bcrp1−/−, Mdr1a/b−/−, and Mdr1a/b−/−Bcrp1−/− mice, respectively (Fig. 3; Table 2). The brain-to-plasma ratio was ∼140-fold higher in the Mdr1a/b−/−Bcrp1−/− mice when compared with the wild-type mice.

Steady-state distribution of palbociclib in Friend leukemia virus strain B wild-type, Bcrp1−/−, Mdr1a/b−/−, and Mdr1a/b−/−Bcrp1−/− mice. (A) Brain and plasma steady-state concentrations. (B) Brain-to-plasma ratio. **P < 0.01; ***P < 0.001 (n = 4).
Summary of steady-state concentrations
DTI = (Brain-to-plasma ratio)knockout/(Brain-to-plasma ratio)wild type.
Flank versus Intracranial Survival Studies.
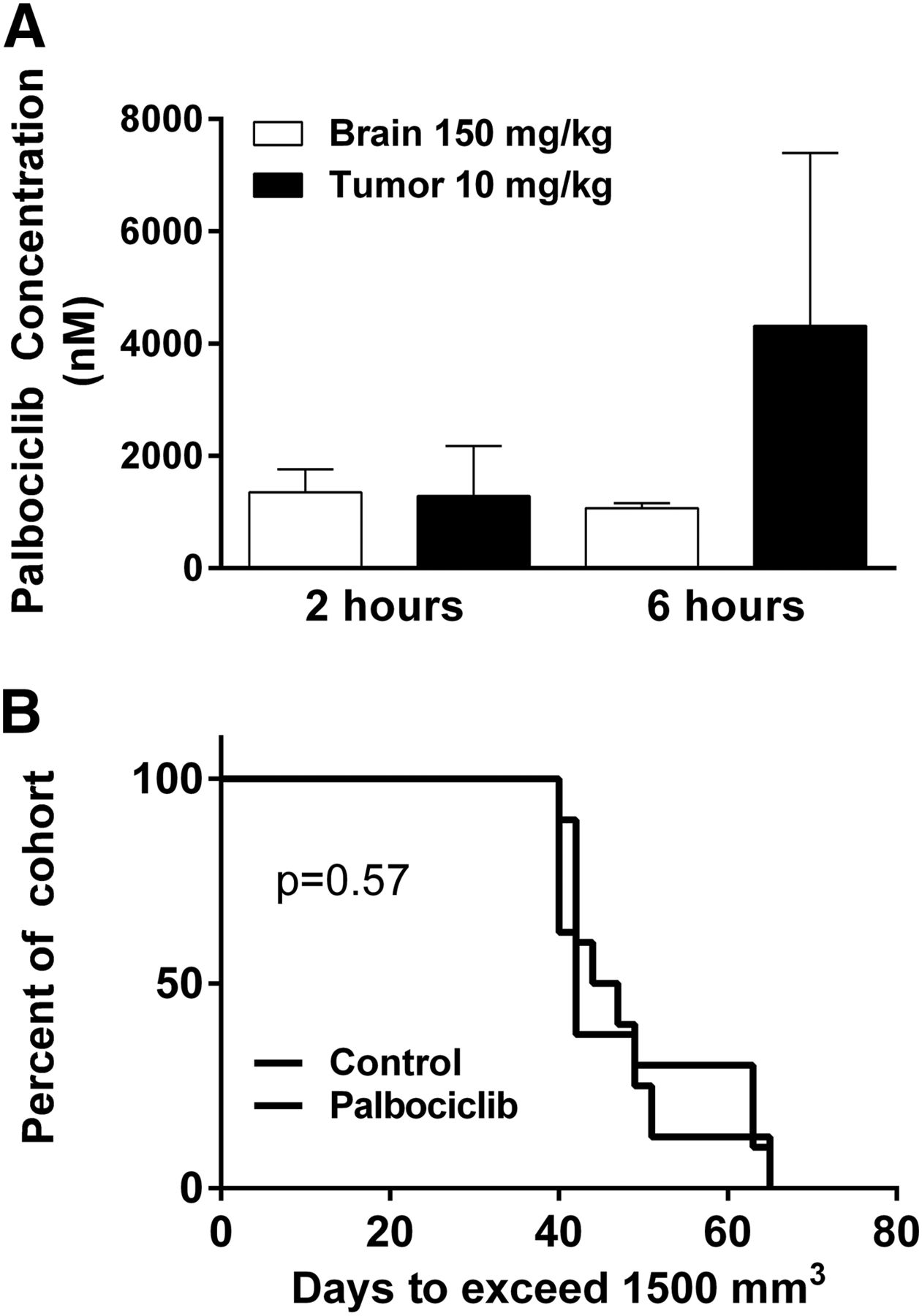
GBM22 tumor cells were implanted in either the flank or intracranially in athymic nude mice to assess the effect of the BBB on the efficacy of palbociclib treatment. Mice with established subcutaneous tumors (flank) were randomized to therapy (150 mg/kg per day) or vehicle control, and the time to exceed 1500 mm3 was measured. Palbociclib therapy provided a significant prolongation in the time to reach 1500 mm3 (P < 0.0001; Fig. 4; Fig. 5A). In stark contrast to what was seen in the subcutaneous model, the time to reach moribund in the treatment group was no different than that of the vehicle-treated group in the intracranial tumor model (P = 0.55; Fig. 5B).

Xenograft (GBM22) tumor volume in subcutaneous tumor-bearing mice following continuous treatment with 150 mg/kg per day of palbociclib or vehicle (n = 8 to 9).

Efficacy of palbociclib therapy (150 mg/kg per day) in patient-derived xenograft (GBM22). (A) Efficacy in subcutaneous GBM22 tumor bearing mice (n = 8 to 9) based on time to exceed tumor size of 1500 mm3. (B) Survival in GBM22 xenograft intracranial tumor model (n = 10).
The concentration of palbociclib in the brain following a 150 mg/kg dose was not significantly different than the concentration in the subcutaneous tumor following a 10 mg/kg dose (Fig. 6A). Following this study, mice with subcutaneous tumors were randomized to two groups: palbociclib (10 mg/kg per day) or vehicle control. There was no effect on subcutaneous tumor growth in the 10 mg/kg palbociclib-treated group compared with vehicle control (P = 0.57; Fig. 6B). This informative study demonstrates that the concentrations reaching invasive tumor cells at the growing edge of a brain tumor were subtherapeutic. These results show the relationship between site-specific drug delivery and efficacy.

Flank tumor exposure to mimic brain delivery. (A) Brain and flank tumor concentrations following either 10 (subcutaneous tumor) or 150 (brain) mg/kg oral dose (n = 4). (B) Efficacy in GBM22 xenograft subcutaneous tumor with daily dosing of 10 mg/kg (n = 8–10).
Pharmacological Inhibition of Efflux Transporters Improves Palbociclib Brain Delivery.
The effect of pharmacological inhibition of the efflux transporters on the brain exposure of palbociclib was studied to determine the feasibility of this strategy to enhance the brain delivery of palbociclib and possibly improve efficacy. Elacridar, a dual inhibitor of P-gp and Bcrp, was administered in a microemulsion formulation simultaneously with an oral dose of palbociclib. Two hours following dosing, plasma concentrations were no different in the wild-type plus microemulsion vehicle treated and wild-type plus elacridar treatment groups (vehicle: 867.7 ± 1187 nM; treatment: 363.1 ± 261.9 nM; Fig. 7A). Elacridar vehicle treatment provided no improved delivery when compared with the wild-type mice; however, elacridar treatment significantly (P = 0.0062) improved the brain delivery of palbociclib (wild-type + vehicle: 0.123 ± 0.072; WT + elacridar: 2.73 ± 1.56; Fig. 7B). These data indicate that pharmacological inhibition of both BCRP and P-gp may be useful in enhancing the brain delivery and hence the treatment efficacy of palbociclib in brain tumors.

Effect of coadministration of elacridar, a dual inhibitor of P-gp and Bcrp, on the brain distribution of palbociclib. (A) Brain and plasma concentrations 2 hours following administrations of elacridar (10 mg/kg i.p.) and palbociclib (10 mg/kg PO). (B) Brain-to-plasma ratio of palbociclib 2 hours postdose. **P < 0.01 (n = 4 to 5).
Discussion
GBM remains a lethal disease, and there is a serious unmet need for better therapeutic options for these patients. Alterations in the p16-CDK4–cyclin D–Rb pathway are commonly found (∼78%) in GBM, and therefore CDK4/6 inhibitors, such as palbociclib, provide a promising targeted therapy in the treatment of GBM. To use palbociclib effectively in the treatment of brain tumors, it is critical to understand the mechanisms that may limit the brain distribution of palbociclib and the relationship between delivery and efficacy.
Efflux transport in the BBB restricts the brain delivery of numerous therapeutic agents (Agarwal et al., 2010, 2011d; Wang et al., 2012; Parrish et al., 2015). P-gp and BCRP actively transport substrate drugs back into systemic circulation and can prevent potentially effective drugs from reaching invasive tumor cells that reside behind an intact BBB. In vitro intracellular accumulation in MDCKII-transfected cells indicate that palbociclib is a dual substrate of both P-gp and BCRP. Consistent with this substrate status, in vivo studies characterizing the brain exposure of palbociclib in wild-type, Mdr1a/b−/−, Bcrp1−/−, and Mdr1a/b−/−Bcrp1−/− mice demonstrate that both transporters are involved in limiting the brain delivery of palbociclib. Both P-gp and BCRP are also expressed in the intestine, and therefore the slight increase in systemic exposure after an oral dose in the knockout mice may be related to a decrease in efflux at the intestinal level leading to an increase in the drug absorbed.
At steady state, no significant difference was observed in the brain distribution between the wild-type and Bcrp1−/− mice, likely because of P-gp–mediated compensation for the lack of BCRP efflux (Agarwal et al., 2011a); however, in Mdr1a/b−/− mice, there is an approximately 10-fold increase in brain exposure. The largest increase in brain exposure was observed in the Mdr1a/b−/−Bcrp1−/− mice, with an ∼115-fold increase in brain distribution, again showing that both transporters are critical in limiting delivery. Consistent with recent reports (de Gooijer et al., 2015; Raub et al., 2015), we have shown that the efflux transporters P-gp and BCRP play a significant role in the brain delivery of palbociclib. Furthermore, our data in the current study indicate that the limited brain delivery of palbociclib may be the reason behind the lack of efficacy of palbociclib in the orthotopic GBM22 model.
The subcutaneous GBM22 model, which has homozygous deletion of CDKN2A/B as well as other alterations in the p16-CDK4-Rb pathway, demonstrated that palbociclib therapy is highly effective in prolonging the time to reach 1500 mm3; however, using the same GBM22 cell line, there was no survival benefit in an orthotopic (intracranial) mouse model. Using these two different models to study the efficacy of therapy on the same patient-derived GBM cells, we observed two radically different outcomes based on tumor location. In the subcutaneous model, where there is no efflux barrier to impede delivery, palbociclib therapy significantly hindered tumor growth. However, these same GBM22 cells were insensitive to palbociclib therapy in the intracranial model, where efflux in the BBB is limiting delivery. The disconnect between the observed flank and intracranial efficacy demonstrates the essential role of the BBB in the treatment of brain tumors. There is a need for new treatment options for patients with primary and metastatic brain tumors, and caution is needed when examining efficacy without recognizing the issue of delivery to invasive tumor cells behind an intact BBB.
We determined the dose that would expose a subcutaneous tumor to “brain-like” concentrations of palbociclib to understand the relationship between delivery and efficacy. In this study, a low dose of palbociclib (10 mg/kg per day) replicated concentrations in the subcutaneous tumor that were observed in the brain following a high dose (150 mg/kg per day) of palbociclib. This low dose of palbociclib, which mimicked the brain exposure of palbociclib, provided no therapeutic benefit in the flank tumor model of GBM. This demonstrates that the concentrations of a possibly effective drug, palbociclib, which reach the invasive edge of the tumor protected by an intact BBB, are subtherapeutic and therefore fail to provide a therapeutic benefit.
The experimental paradigm of examining both delivery and efficacy in the heterotopic and orthotopic tumors results in powerful insights as to why a drug may fail in treating a tumor in the brain. On the one hand, if a drug at a maximally tolerated dose is ineffective in the flank tumor, then it is possible that the drug should no longer be considered as a treatment option for that tumor type. However, if a drug at the maximally tolerated dose is ineffective in treating a tumor in the brain but is effective in the flank, it is then important to recognize that the lack of efficacy in the brain may be due to inadequate delivery. These insights should guide the preclinical screening of compounds for GBM.
There may also be some inherent differences between brain tumors and subcutaneous tumors in addition to the BBB preventing adequate drug delivery to brain tumors. The possible changes in the microenvironment between a subcutaneous tumor and an intracranial tumor have not yet been determined. These differences have not been explored in this study, but are important to understand and may provide yet another reason to be cautious when using subcutaneous tumors for preclinical screening of compounds for GBM and other brain tumors. In this preclinical model, we show that inadequate brain drug delivery due to efflux at the BBB could be solely responsible for the failure of palbociclib therapy.
Michaud et al. demonstrated the efficacy of palbociclib in both in vivo and in vitro models (Michaud et al., 2010). They proposed that since palbociclib was effective in their in vivo U87MG tumor model and GBM39 xenograft, the BBB was not a barrier for this molecule. In the U87MG model, drug delivery to the tumor core was significantly greater than delivery to the other regions of the brain (including the brain around the tumor and opposite hemisphere). It should be noted that the U87MG model is a less invasive and more circumscribed tumor than many PDX and genetically engineered models and as such serves as a poor model to study the invasive nature of glioma (de Vries et al., 2009). It is important to recognize that GBM is a disease of the whole brain, with invasive cells infiltrating from the original tumor site to other areas of the brain (Agarwal et al., 2011b). Previous studies have demonstrated the importance of the BBB by matrix-assisted laser desorption/ionization mass spectrometry imaging, where a drug that is a substrate for BBB efflux transporters is homogeneously distributed throughout the tumor for the U87 model, but that same drug is not distributed homogeneously in a GS2 model, a model that shares the invasive characteristics observed in human GBM (Salphati et al., 2014). For this reason, it is crucial to deliver therapies to the whole brain, not just the tumor core. This will require therapies that cross an intact BBB and overcome the liability of efflux transport.
Coadministration of elacridar with palbociclib significantly improves the brain delivery of palbociclib and provides a potential therapeutic strategy to overcome efflux transport in the BBB and achieve adequate brain delivery. These findings have important implications in the use of palbociclib in the treatment of GBM, as it may be necessary to employ adjuvant therapies that improve delivery to the invasive tumor.
In addition to primary brain tumors, such as GBM, metastatic brain tumors are associated with a poor prognosis, with limited treatment options (Maher et al., 2009). Lung, breast, and melanoma are the three cancers with the highest propensity to metastasize to the brain and often have mutations in the CDK4/6 pathway (Musgrove et al., 2011; Gállego Pérez-Larraya and Hildebrand, 2014). The clinical utility of CDK4/6 inhibitors in the treatment of cancer has been most extensively studied in the treatment of breast cancers (Turner et al., 2015). Since breast cancer is the second most common cause of brain metastasis, it is paramount that treatment modalities are able to penetrate the BBB. In melanoma, 38% of patients have genetic deletion of p16INK4a, leading to unrestricted tumor growth via this pathway. Since well over half of melanoma patients are found to have brain tumors at autopsy (Fife et al., 2004), effectively delivering treatments to tumor cells behind an intact BBB, even at the stage of micrometastases, is critical. Furthermore, there have been studies that indicate that the incidence of brain metastasis can increase when treating cancers that have a high propensity to metastasize to the brain with agents that are unable to penetrate a BBB (O’Sullivan and Smith, 2014; Peuvrel et al., 2014). Understanding the delivery issues associated with using palbociclib therapy in the treatment of brain metastases will be essential in effectively treating this patient population.
Taken together, these results clearly demonstrate that the BBB plays a major role in limiting the brain delivery of palbociclib and limits the delivery of palbociclib to invasive tumor cells residing behind an intact BBB. Future work remains to determine the effect of palbociclib in combination with BBB efflux inhibition in tumor-bearing mice. Brain distribution studies comparing palbociclib with other CDK4/6 inhibitors [such as ribociclib (LEE-011) and abemaciclib (LY2835219)] are also of significant importance.
Acknowledgments
We thank Jim Fisher, Clinical Pharmacology Analytical Services Laboratory, University of Minnesota, for support in the development of the palbociclib LC-MS/MS assay.
Authorship Contributions
Participated in research design: Parrish, Mittapalli, Sarkaria, Elmquist.
Conducted experiments: Parrish, Mittapalli, Bakken, Pokorny.
Performed data analysis: Parrish, Mittapalli, Sarkaria, Elmquist.
Wrote or contributed to the writing of the manuscript: Parrish, Sarkaria, Elmquist.
Footnotes
- Received July 30, 2015.
- Accepted September 8, 2015.
Funding for this work was supported by the National Institutes of Health [Grants RO1 CA138437, RO1 NS077921, and P50 CA108961]. K.E.P. was supported by the Ronald J. Sawchuk, Edward G. Rippie, Rowell, American Foundation for Pharmaceutical Education Pre-Doctoral, and University of Minnesota Doctoral Dissertation Fellowships.
Part of this work was previously presented: Parrish KE, Pokorny JL, Mittapalli RK, Bakken K, Sarkaria JN, Elmquist WF (2013) BBB efflux pump activity limits brain penetration of palbociclib (PD0332991) in glioblastoma. Mol Cancer Ther 12 (11 Supplement):C81.
Abbreviations
- AUC
- area under the curve
- BBB
- blood-brain-barrier
- BCRP
- breast cancer resistance protein
- Bcrp1
- gene encoding the murine breast cancer resistance protein
- CDK
- cyclin-dependent kinase
- GBM
- glioblastoma
- Ko143
- (3S,6S,12aS)-1,2,3,4,6,7,12,12a-octahydro-9-methoxy-6-(2-methylpropyl)-1,4-dioxopyrazino(1′,2′:1,6)pyrido(3,4-b)indole-3-propanoic acid-1,1-dimethylethyl ester
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- LY335979 (zosuquidar)
- (R)-4-[(1aR,6R,10bS)-1,2-difluoro-1,1a,6,10b-tetrahydrodibenzo-(a,e)cyclopropa(c)cycloheptan-6-yl]-α-[(5-quinoloyloxy)methyl]-1-piperazine ethanol trihydrochloride
- MDCKII
- Madin-Darby canine kidney II
- MDR1
- gene encoding the human p-glycoprotein
- Mdr1
- gene encoding the murine p-glycoprotein
- palbociclib (PD-0332991)
- 6-acetyl-8-cyclopentyl-5-methyl-2-([5-(piperazin-1-yl)pyridin-2-yl]amino)pyrido(2,3-d)pyrimidin-7(8H)-one
- PDX
- patient-derived xenograft
- P-gp
- p-glycoprotein
- Rb
- retinoblastoma
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}