


Abstract
(+)- And (−)-amphetamine and methamphetamine wereN-oxygenated by the cDNA expressed adult human flavin-containing monooxygenase form 3 (FMO3), their corresponding hydroxylamines. Two major polymorphic forms of human FMO3 were studied, and the results suggested preferentialN-oxygenation by only one of the two enzymes. Chemically synthesized (±)-amphetamine hydroxylamine was also a substrate for the human FMO3 and it was converted to phenylpropanone oxime with a stereoselectivity ratio of trans/cis of 5:1. Human FMO3 also N-oxygenated methamphetamine to produce methamphetamine hydroxylamine. Methamphetamine hydroxylamine was alsoN-oxygenated by human FMO3, and the ultimate product observed was phenylpropanone. For amphetamine hydroxylamine, studies of the biochemical mechanism of product formation were consistent with the production of an N,N-dioxygenated intermediate that lead to phenylpropanone oxime. This was supported by the observation that α-deutero (±)-amphetamine hydroxylamine gave an inverse kinetic isotope effect on product formation in the presence of human FMO3. For methamphetamine, the data were consistent with a mechanism of human FMO3-mediated N,N-dioxygenation but the immediate product, a nitrone, rapidly hydrolyzed to phenylpropanone. The pharmacological activity of amphetamine hydroxylamine, phenylpropanone oxime, and methamphetamine hydroxylamine were examined for effects at the human dopamine, serotonin, and norepinephrine transporters. Amphetamine hydroxylamine and methamphetamine hydroxylamine were apparent substrates for the human biogenic amine transporters but phenylpropanone oxime was not. Presumably, phenylpropanone oxime or nitrone formation from amphetamine and methamphetamine, respectively, represents a detoxication process. Because of the potential toxic nature of amphetamine hydroxylamine and methamphetamine hydroxylamine metabolites and the polymorphic nature of N-oxygenation, human FMO3-mediated metabolism of amphetamine or methamphetamine may have clinical consequences.
Methamphetamine (METH) and amphetamine (AMPH) overuse cause complex physiological effects including rapid heart rate, elevated blood pressure, increased body temperature and respiration rate, and pupillary dilation (Innes and Nickerson, 1975). There is a heightened sense of well being or euphoria, increased alertness and vigor, decreased sleep time, and reduced food intake. It is possible that the impending METH and congener epidemic could be hazardous to more individuals than the cocaine epidemic because of the neurotoxic properties of METH when smoked or administered i.v. and the increased incidence of violence and assaultive behaviors. The ready chemical synthesis of METH and AMPH may contribute to the widespread increase in procurement and abuse.
Repeated administration of METH and AMPH causes persistent serotonin and dopaminergic terminal degeneration in the frontal cortex, hippocampus, and amygdala in rats and nonhuman primates (Ricaurte et al., 1982). After administration, METH and AMPH enter the cell through the nerve terminal storage vesicles and 1) cause dopamine (DA) and norepinephrine (NE) to leak out into the synaptic cleft to increase DA levels; 2) inhibit DA degradation, thus increasing the cytoplasmic pool of DA; 3) increase DA loss from storage vesicles; and 4) inactivate a storage proton pump and increase DA concentration in the cytoplasm (Gibb et al., 1997; Wrona et al., 1997). Cytoplasmic accumulation of DA (or serotonin) and intraneuronal oxidation of the neurotransmitter may be linked to neuronal degeneration (Liang and Rutledge, 1982). One hypothesis is that Fenton-type chemistry produces hydroxyl radicals and DA and serotonin are rapidly attacked to provide neurotoxins that directly contribute to cytotoxicity (Wrona et al., 1997). Other mechanisms contributing to neurotoxicity employing endogenous substances may also contribute to the neurodegenerative effects of METH and AMPH (Gibb et al., 1997).
Another possible mechanism contributing to toxicity is metabolism of METH and AMPH to reactive metabolites that could participate in DA, serotonin, or NE autoxidation. Compared with structurally related biogenic amines, the N-oxidative metabolism of AMPHs is significantly decreased. Aromatic hydroxylation,N-dealkylation, and deamination dominate the metabolism of this class of amines, although considerable interspecies differences have been observed (Caldwell, 1976). N-oxidative products of AMPH have been observed in the urine of animals or in in vitro preparations, including hydroxylamine (Beckett and Al-Sarraj, 1973; Lindeke et al., 1973; Danielson et al., 1977; Florence et al., 1982), oxime (Beckett et al., 1971; Beckett and Jones, 1975; Wright et al., 1977; Matsumoto and Cho, 1982), and nitroso (Franklin, 1974; Jonsson and Lindeke, 1976) metabolites. In humans, (S)(+)-AMPH undergoes more extensive metabolism than (R)(−)-AMPH (Caldwell, 1976). Replacement of the α proton with deuterium retards N-dealkylation of the (S)(+)-isomer but no kinetic isotope effect is observed for (R)(−)-AMPH metabolism in some animal hepatic preparations (Parli and McMahan, 1973).
Flavin-containing monooxygenases (FMO) comprise a family of five monooxygenases that N- and S-oxygenate nucleophilic heteroatom-containing drugs, xenobiotics, and dietary materials (Cashman, 1995). In humans, the prominent hepatic form is FMO3 (Lomri et al., 1992), which consists of two enzymes that are polymorphic at codon 158 (Brunelle et al., 1997). We have examined theN-oxygenation of AMPH, METH, and hydroxylamine metabolites because 1) primary amines are excellent substrates for human FMO3 (Lin et al., 1996), 2) closely related primary phenethylamines (Lin and Cashman, 1997a) and tyramine (Lin and Cashman, 1997b) are excellent substrates for human FMO3, and 3) it is unlikely that cytochrome P-450 or monoamine oxidase catalyze metabolic bioactivation of the AMPH nitrogen atom. In addition, we examined METH as a substrate for human FMO3 to extend this analysis to secondary phenethylamines.
In this report, we describe the human FMO3-catalyzedN-oxygenation of (+) and (−)-AMPH and (±)-AMPH hydroxylamine.3The results show that in the presence of human FMO3, there is appreciable formation of AMPH hydroxylamine. AMPH hydroxylamine is also a substrate for human FMO3 and the trans oxime product is formed in marked preference over the cis oxime. METH and METH hydroxylamine are also substrates for human FMO3, although they are considerably less efficiently N-oxygenated than AMPH. Studies of the pharmacological activity of AMPH hydroxylamine and METH hydroxylamine and AMPH oxime suggest that hydroxylamine formation is a bioactivation process leading to potentially biologically active materials, whereas formation of oxime metabolites that are largely devoid of biological activity constitute a detoxication process.
Materials and Methods
Chemicals
Chemicals, reagents, buffers, and solvents used in this study were of the highest purity available from commercial sources. Phenylpropanone, hydroxylamine hydrochloride, sodium cyanoborohydride (NaCNBH3), sodium cyanoborodeuteride, and hydroxymethylamine were purchased from Aldrich Chemical Company (Milwaukee, WI). (+)- and (−)-AMPH and -METH were provided by the National Institute on Drug Abuse Drug Supply Program, National Institutes of Health (Rockville, MD). RTI-55 and 2β carbomethoxy-3β-(4-fluorophenyl)tropane were kind gifts from Dr. Ivy Carroll (RTI, Research Triangle Park, NC). Other buffers, reagents, and solvents were obtained from Fisher Chemical (San Jose, CA). Ether was dried by distillation from sodium/benzophenone. Tropolone, DA, pargyline, and all of the compounds of the NADPH-generating system were from Sigma Chemical (St. Louis, MO). Chromatography was done with silica gel 60, 230-400 mesh Acros Chemical (Pittsburgh, PA), and thin-layer chromatography used precoated silica gel F254 plates from E. Merck (Darmstadt, Germany). [3H]DA, [3H]5-hydroxytryptamine [3H]5HT, [3H]NE, and [125I]RTI-55 were purchased from Du Pont-New England Nuclear (Boston, MA). The human DA transporter (hDAT) used in these experiments is the cDNA pcDNA1-hDAT described previously (Eshleman et al., 1994, 1995a). The human serotonin transporter (hSERT) cDNA was generously supplied by Dr. Randy Blakely (Vanderbilt University, Nashville, TN) subcloned into pcDNA1 and transfected into human embryonic kidney (HEK)-293 cells (HEK-hSERT) (Ramamoorthy et al., 1993). Dr. Blakely also kindly supplied us with HEK cells transfected with pcDNA3-human NE transporter (hNET) (HEK-hNET).
Instrument Analysis
1H NMR spectra were recorded on a Varian spectrometer (Varian Analytical Instruments, Sunnyvale, CA) operating at a frequency of 300 MHz. The proton chemical shift values were listed in parts per million relative to tetramethylsilane. Fast atom bombardment (FAB) mass spectra were recorded on a VG 70SEQ instrument. Both instruments are housed at the Department of Medicinal Chemistry, University of Washington (Seattle, WA). Liquid chromatography-atmospheric pressure-chemical ionization-mass spectra were recorded with a VG Platform single quadrupole mass spectrometer at the National Center for Toxicological Research (Jefferson, AR).
Synthetic Procedures
Synthesis of cis and transPhenylpropanone Oxime.
Cis and transphenylpropanone oxime have been previously synthesized (Beckett and Jones, 1975; Florence et al., 1982). Phenylpropanone (compound1; 0.13 g, 1 mmol) was dissolved in 10 ml of tetrahydrofuran (THF). Hydroxylamine hydrochloride (0.14 g, 2 mmol, 2.0 equiv.) was placed in 5 ml of water and added to a stirred solution of phenylpropanone. Sodium carbonate (0.2 g, 2 mmol, 2 equiv.) in 5 ml of water was also added to the reaction mixture. After about 2 h, the reaction was complete as judged by thin-layer chromatography (TLC). The reaction mixture was alkalinized with ammonium hydroxide (to give a pH = 10) and phenylpropanone oxime, (compound 2),extracted from the aqueous reaction mixture with 15 ml ethyl acetate, washed with 10 ml brine, and the organic layer dried over Na2SO4 and concentrated to give a light yellow oil as a mixture of cis andtrans oximes (compounds 2a,b; Fig.1) that were separated by silica column chromatography (hexane/ethyl acetate, 20:80, v/v). For phenylpropanone oxime (compound 2), the yield was 78% Rf = 0.14 that afforded a 4:1 mixture oftrans/cis oximes. The 1H NMR (CDCl3) δ: 8.0 to 8.6 (w, 1H), 7.22 to 7.4 (m, 5H), 3.46 (s, 2H), 1.72 (s, 3H) showed that the desired compound was obtained.

Chemical synthesis of cis andtrans phenylpropanone oxime (compounds 2a,b), from phenylpropanone (compound 1), and AMPH hydroxylamine, (compound 3), or deuterated AMPH hydroxylamine (compound4).
Synthesis of AMPH Hydroxylamine.
For synthesis of AMPH hydroxylamine, phenylpropanone oxime (compound 2; 0.15 g, 1 mmol) was dissolved in MeOH with bromophenol blue (0.1 mg) as a pH indicator. The reaction was maintained at a pH below 3.0 with cold HCl (10%) so that over-reduction or dimerization was minimized. NaCNBH3 (0.67 ml, 1 M solution in THF, 0.67 equiv.) was added dropwise to the reaction mixture. The reaction was stirred at room temperature and was complete after about 3 h as judged by TLC. The reaction mixture was extracted with ether, washed with brine, dried over Na2SO4, and concentrated in vacuo. The product was purified by flash silica gel chromatography (ethyl acetate/hexane, 20:80, v/v) to give a solid in 90% yield, Rf = 0.17. Mass spectral data (FAB)m/z (percentage of abundance) 151 (M+, 17), 149 (28), 119 (98), 117 (75), 115 (50), 103 (32), 91 (38), 60 (100) and 1H-NMR (CDCl3) δ: 7.1 to 7.4 (m, 5H), 3.18 (m, 1H), 2.85 (m, 1H), 2.64 (m, 1H), 1.1 (d, J = 6.35 Hz, 3H) showed that the desired AMPH hydroxylamine (compound 3), was obtained and the spectral data was in accordance with a previous report (Beckett and Al-Sarraj, 1973).
Synthesis of Deuterated AMPH Hydroxylamine.
The synthesis of deuterated AMPH hydroxylamine (compound 4) by a different route has been described before (Parli and McMahan, 1973). Phenylpropanone oxime (compound 2; 0.15 g, 1 mmol) was dissolved in 5 ml of MeOH followed by the addition of bromophenol blue (0.1 mg). sodium cyanoborodeuteride (0.67 ml, 1 M solution in THF, 0.67 mol equiv.) and 2 N HCl-methanol was added dropwise with stirring at 0°C and then brought to room temperature for 3 h. After the reaction was judged complete by TLC, the reaction was stopped by evaporation of the mixture and the residue was taken up in ether and the organic layer was washed with brine and dried over Na2SO4 and concentrated in vacuo. The product arising from the deuteration of oxime compound2 was purified by flash silica gel column chromatography (ethyl acetate/hexane, 20:80, v/v), Rf = 0.17 and gave a solid, with a yield of 90% (Fig. 1). The deuterated AMPH hydroxylamine (compound 4) was fully characterized by1H NMR (CDCl3) δ: 7.1 to 7.4 (m, 5H), 3.19 (m, trace H), 2.85 (d, J = 13.2 Hz, 1H), 2.63 (d, J = 13.2 Hz, 1H), 1.1 (d,J = 6.35 Hz, 3H), and 13C NMR (CDCl3) 138.8, 129.5, 128.7, 126.5, 58.7, 40.1, 17.7, and mass spectral data (FAB) m/z: 153 (M+). The data was consistent with 0.73 deuterium incorporated into the AMPH hydroxylamine (compound 4).
Synthesis of N-Hydroxylamine.
The synthesis of METH hydroxylamine (compound 5) was done by a modification of the general method described above. Phenylpropanone was dissolved in MeOH and hydroxylamine hydrochloride was added. The reaction was kept at a pH = 4 for 5 h and then NaCNBH3 was added. The reaction was complete in 5 h as determined by TLC. To stop the reaction, HCl was added and then made basic before extracting into ethyl acetate. The organic fraction was washed with brine and the organic layer was dried over Na2SO4. After concentrating to an oil and separation by silica gel chromatography, METH hydroxylamine (compound 5) was obtained in 68% yield, Rf = 0.24 (Fig. 2).1H NMR (CDCl3): δ 7.34 (m, 5H), 3.63 (m, 2H), 3.08 (s, 3H), 2.62 (t, J = 12 Hz, 1H), 1.23 (d, J = 6 Hz, 3H), and13C NMR (acetone-d6): 140.3, 130.3, 129.4, 128.2, 66.9, 65.2, 45.3, 39.3 and mass spectral data (FAB) m/z 166 (M + 1), 95, 92, 91, 74, 65, 58, 56 were consistent with the assigned structure.

Chemical synthesis of METH hydroxylamine (compound5) from phenylpropanone (compound 1).
Enzyme Preparations
The two prominent human FMO3 enzymes, present at approximately 53% and 47% allele frequencies for the Caucasian populations examined (Akerman et al., 1997) (i.e., human FMO3 Glu-158 and Lys-158, respectively), have been cloned and expressed as maltose binding fusion proteins (MBP) in Escherichia coli. The details of the human FMO3 cDNA cloning, protein expression studies, and assay conditions of the FMO3-MBP have been described previously (Brunelle et al., 1997). For the present study, human FMO3-MBP Glu-158 and Lys-158 formed 41.9 and 14.1 nmol 10-[(N,N-dimethylamino)pentyl]-2-(trifluoromethyl)phenothiazineN-oxide (5-DPT N-oxide)/min/mg of protein, respectively.
Metabolic and Analytical Methods for Human FMO3-MBP Activity
A typical incubation contained 50 mM potassium phosphate buffer (pH = 8.4), 0.5 mM NADP+, 0.5 mM glucose phosphate, 1.0 IU of glucose 6-phosphate dehydrogenase, 10 to 50 μg of cDNA-expressed human FMO3-MBP, and 1.2 mM diethylenetriaminepentaacetic acid (final volume 0.25 ml). At various time intervals, the incubations were stopped by the addition of 8 volumes of cold ethyl acetate. After addition of 20 mg of sodium carbonate and a brief centrifugation, the organic layer was separated from the aqueous phase and the organic fraction was evaporated to dryness. The aqueous layer was re-extracted with 0.5 ml of acetonitrile and centrifuged, and the organic portion was combined with the previous organic extract and evaporated to dryness. The combined organic extracts were placed in acetonitrile and then immediately analyzed for products by the HPLC method procedure described below.
The profile of AMPH metabolites were determined by HPLC analysis of the combined organic extracts of the incubation mixture. The metabolic products were separated and quantified with a Hitachi L-6200A HPLC interfaced to a Hitachi D-2500 Chromato-Integrator with a Hitachi L-4000H UV detector set at 220 nm (Hitachi Scientific Instruments, Mountain View, CA) . The system was fitted with a C-18 analytical column (25 × 0.4 cm) from Rainin Corp. (Emeryville, CA). The mobile phase for analysis of AMPH and AMPH hydroxylamine consisted of an isocratic system set at 70% A and 30% B where A was water with 1% THF and B was acetonitrile containing 0.1% HClO4at a flow rate of 1.5 ml/min. This system efficiently separated AMPH and AMPH hydroxylamine that had retention times of 2.85 and 3.29 min, respectively. The mobile phase for the separation of AMPH hydroxylamine and cis and trans oxime consisted of 75% A and 25% B where the A and B solvents were as described above. This system separated AMPH hydroxylamine and trans and cisphenylpropanone oxime with retention times of 3.3, 7.1, and 7.9 min, respectively. The HPLC procedure was as above except the mobile phase for the separation of METH, METH hydroxylamine, and phenylpropanone consisted of 70% A and 30% B where A was water and B was acetonitrile containing 0.1% HClO4 at a flow rate of 1.5 ml/min. This system separated METH, METH hydroxylamine, and phenylpropanone with retention times of 3.8, 5.2, and 9.8 min, respectively. For analysis of METH and METH hydroxylamine, the incubations were stopped by the addition of acetonitrile, centrifuged at 10,000g, and directly injected onto the HPLC column for quantification. On the basis of the UV absorption of each material, metabolites were quantified by comparing the metabolite and substrate peak areas of the chromatogram and calculating the percentage of conversion as described previously (Lin and Cashman, 1997).
AMPH hydroxylamine (compound 3) was also tested as an inhibitor of human FMO3-MBP mediated 10-[(N,N-dimethylamino)pentyl]-2-(trifluoromethyl)phenothiazine (5-DPT) N-oxygenation by a general procedure described previously (Brunelle et al., 1997). Thus, Lineweaver-Burke analysis of 5-DPT N-oxygenation in the presence or absence of various concentrations of AMPH hydroxylamine was done to assess whether synthetic hydroxylamine (compound 3) was a competitive inhibitor of human FMO3-MBP.
Receptor Binding Assays
[125I]RTI-55 Binding.
HEK-hDAT, -SERT, or -hNET cells were grown until confluent on 150-mm diameter tissue culture dishes and incubated in DMEM supplemented with 5% fetal calf serum, 5% bovine calf serum, and penicillin/streptomycin at 37°C in a humidified 10% CO2 environment. Medium was removed from plates, plates were washed with 10 ml PBS, lysis buffer (10 ml, 2 mM HEPES and 1 mM EDTA) was added, and plates were placed on ice for 10 min. Cells were scraped from plates and centrifuged for 20 min at 30,000g and the pellet was resuspended in 6 to 24 ml of 0.32 M sucrose, (a resuspension volume that results in binding ≤10% of the total radioactivity) with a Polytron at setting 7 for 5 s. Assays contained 50 μl of membrane preparation (approximately 12–30 μg protein, depending on the cell line), 25 μl of drug, and 25 μl of [125I]RTI-55 (40–80 pM final concentration) in a final volume of 250 μl Krebs HEPES buffer (25 mM HEPES, 122 mM NaCl, 5 mM KCl, 1.2 mM MgSO4, 2.5 mM CaCl2, 1 μM pargyline, 100 μM tropolone, 2 mg glucose/ml, and 0.2 mg ascorbic acid/ml, pH 7.4) was used for all assays. Specific binding was defined as the difference in binding observed in the presence and absence of 5 μM mazindol (HEK-hDAT and -NET) or 5 μM imipramine (HEK-hSERT). Membranes were preincubated with drugs for 10 min before addition of [125I]RTI-55 unless indicated otherwise. The reaction was incubated for 90 min at room temperature in the dark and was terminated by filtration onto Whatman GF/C filters (Whatman Inc., Clifton, NJ) using a 96-well Tomtech cell harvester (Tomtech, Orange, CT). The incubation time was designed to be within the equilibrium time of [125I]RTI-55 binding and to allow slowly associating compounds to bind to the transporter. Scintillation fluid (50 μl) was added to each filtered spot and radioactivity remaining on the filter was determined using a Wallace β-plate reader (Wallace Labs., Cranbury, NJ). Competition experiments were conducted with duplicate determinations.
Inhibition of Substrate Uptake by HEK-hDAT, -hSERT, and -hNET.
HEK-hDAT, -hSERT, or -hNET cells were grown on 150 mm diameter tissue culture dishes as described above. Media was removed and plates were washed twice with PBS. PBS (2.5 ml) was then added to each plate, plates were placed in a 25°C water bath for 5 min, and cells were gently scraped from plates and triturated with a pipette. Aliquots (50 μl) of the suspended cells were added to 96-well plates containing drugs and Krebs-HEPES buffer in a final assay volume of 0.5 ml. After a 10-min preincubation in a 25°C water bath,3H-labeled neurotransmitter (50 μl, 20 nM final concentration: [3H]DA, [3H]5-HT, and [3H]NE, 56, 26.9, and 60 Ci/mmol, respectively) was added and the assay was incubated for 10 min. The reaction was terminated by filtration onto GF/C filters presoaked in 0.05% polyethylenimine using a Tomtech cell harvester. Scintillation fluid (50 μl) was added to each filtered spot and radioactivity remaining on the filters was determined as described above. Specific uptake was defined as the difference in uptake in the absence and presence of 5 μM mazindol (hDAT and hNET) or 5 μM imipramine (hSERT).
Transporter-Mediated [3H]1-Methyl-4-Phenylpyridinium (MPP+) Release. Stable Expression of Transporters in Mammalian Cells.
Stable transfection of C6 glioma cells for use in transporter-mediated release assays was carried out as previously described (Eshleman et al., 1995) using pcDNA1-hDAT or pcDNA1-hSERT (14 μg) and pBabePuro (2 μg; gift from Dr. Rachel Neve, Harvard University, Belmont, MA), which confers resistance to puromycin. Puromycin-resistant cells were isolated by selection at a concentration of 2 μg/ml, a concentration that kills control C6 cells within 1 week (A. Janowsky, unpublished observation). Release assays were conducted as previously described (Johnson et al., 1998). Briefly, when C6 glioma cells transfected with hDAT (C6-hDAT) or -hSERT cells were confluent in 24-well plates, [3H]MPP+ (2 nM), in Krebs-HEPES buffer (25 mM HEPES, 120 mM NaCl, 5 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 1 μM pargyline, 0.2% glucose, and 0.02% ascorbic acid, pH 7.4) was added to each well in a final volume of 0.5 ml. Loading of the cells with3H-labeled substrate was conducted for 20 or 60 min at room temperature for C6-hDAT cells or C6 glioma cells transfected with hSERT cells, respectively. Nonspecific uptake was defined as the difference in uptake observed in the presence and absence of 5 μM mazindol. The loading buffer was removed and cells were quickly washed with room-temperature release buffer (2 × 0.3 ml), which was identical to the loading buffer except that release buffer lacked Ca2+. Release buffer (0.5 ml final volume, room temperature) was added to the plates and, after a 10- min preincubation period in a 37°C water bath, drugs were added to the wells. Trichloroacetic acid (3%) was added, and radioactivity remaining in the cells was determined by liquid scintillation spectrometry. All experiments were conducted with triplicate determinations.
Data Analysis
GraphPad Prism (GraphPad Software, San Diego, CA) was used to determine all kinetic, saturation and competition binding data. IC50 values were converted toKi values using the Cheng-Prusoff equation.
Results
Chemical Synthesis and Chemical Stability.
As a prelude to the enzymatic and pharmacological studies, the chemical stability of AMPH and AMPH hydroxylamine (compound 3) and phenylpropanone oximes (compounds 2a and 2b) to auto-oxidation and hydrolysis was determined. The phenylpropanone oximes (compound2) were examined for their ability to isomerize during the experimental conditions used. As determined by HPLC analysis over the time course of about a month, AMPH and the cis andtrans phenylpropanone oximes (compounds 2aand b) were completely stable in water under the reaction and work-up conditions that were used. At elevated pH values, thetrans oxime did racemize to the cis oxime to a small extent. This is in agreement with what has been described before (Caldwell, 1976). However, the amount of racemization observed did not influence the determination of the stereoselectivity of the product formation reactions described below. AMPH hydroxylamine (compound3) was also placed in potassium phosphate buffer (pH = 8.4) and periodically, aliquots were withdrawn and directly analyzed by the HPLC method described above. Under these conditions, AMPH hydroxylamine (compound 3) was not indefinitely stable and was slowly converted to oxime (compound 2) with a half-life of approximately 3.5 h. In the presence of acetonitrile, AMPH hydroxylamine (compound 3) also slowly decomposed to oxime (compound 2) with a half-life of 78 days. Surprisingly, in addition to oxime formation, AMPH hydroxylamine (compound 3) formed an acetonitrile adduct that was reasonably stable to the chromatographic conditions used. The acetonitrile adduct (i.e.,m/z 193, presumably from the MH+) andcis and trans oximes (i.e., m/z 150 as well as the fragment ion due to the loss of 16 amu) were characterized using on-line liquid chromatography-atmospheric pressure-chemical ionization-mass spectrometry.
The chemical stability of METH, METH hydroxylamine, and phenylpropanone to decomposition was also studied. As determined by direct HPLC analysis of aqueous potassium phosphate buffer (i.e., pH = 8.4) or acetonitrile solutions of METH and phenylpropanone, no detectable degradation was observed for solutions of these materials stored at room temperature for over a month. In the presence of acetonitrile, no detectable formation of the acetonitrile adduct of METH hydroxylamine was observed over the time course of approximately 1 month. It was notable that in the presence of aqueous phosphate buffer (i.e., pH = 8.4) the half-life of METH hydroxylamine (compound 5; i.e., approximately 13 h) was significantly longer than that for AMPH hydroxylamine (compound 3).
On the basis of control experiments and stability studies, it was judged that for either series of amine compounds (i.e., AMPH or METH) as well their metabolites (i.e., hydroxylamines, oximes, or phenylpropanone) attentive work-up of the enzymatic samples and rapid bioanalysis precluded confounding issues caused by nonenzymatic processes. As a precaution for any spurious nonenzymatic process, for each enzymatic reaction conducted, parallel experiments were done without enzyme present to serve as a control. In practice, generally, very little nonenzymatic product was observed for the studies done (data not shown), but the minor amount of product was subtracted from the enzyme rate to yield the true enzyme rate.
N-Oxygenation of AMPH and AMPH Hydroxylamine by Human FMO3-MBP.
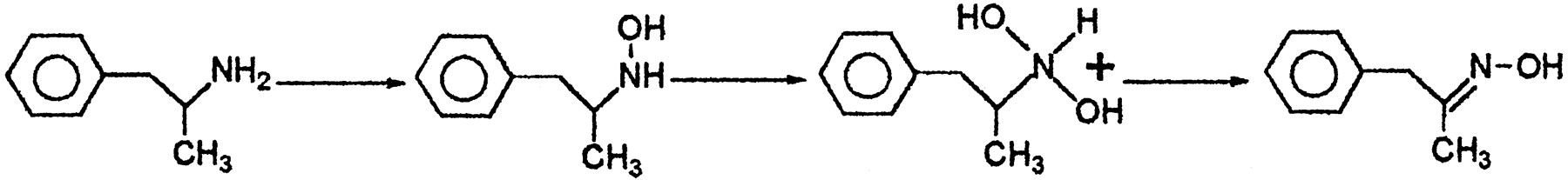
The N-oxygenation of AMPH and AMPH hydroxylamine in the presence of the two major human FMO3-MBP enzymes (i.e., forms with amino acids Lys-158 and Glu-158) was done to identify the products formed and to compare the metabolism with phenethylamine and tyramine. In contrast to phenethylamine or tyramine, where it was observed that human FMO3 forms exclusively trans oxime through a multistep process, for AMPH as substrate, AMPH hydroxylamine was observed as a metabolite, but hydroxylamine formation was detected at a lower rate. Undoubtedly, this was a consequence of the α-methyl group of the AMPH molecule exerting a steric effect on the enzyme. Presumably, this was due to a very slow second reaction in the A → B → C sequence that leads to oxime formation (Fig.3). To investigate this point, we examined the kinetics of AMPH N-oxygenation. In the presence of human FMO3-MBP Glu-158, N-oxygenation of (+)- and (−)-AMPH to AMPH hydroxylamine (compound 3) was linearly dependent on the presence of active enzyme (i.e., 0–200 μg of protein) and linearly dependent upon the time of the reaction (i.e., 0–20 min). N-oxygenation of (+)- and (−)-AMPH to AMPH hydroxylamine (compound 3) in the presence of human FMO3-MBP Lys-158 was linearly dependent on the presence of active enzyme (i.e., 0–250 μg of protein) and linearly dependent on the time of the reaction (i.e., 0–20 min). For both enzymes, the formation of hydroxylamine showed typical Michaelis-Menton kinetics. Thus, plots of the reciprocal of the substrate concentration versus the reciprocal of the velocity of AMPH hydroxylamine (compound 3) formation were linear. The calculatedKmapp andVmax values for human FMO3-MBP Glu-158-mediated conversion (−)-AMPH to AMPH hydroxylamine were 44.2 mM and 6.5 nmol/min/mg of protein and for (+)-AMPH, 11 mM and 1.06 nmol/min/mg of protein, respectively (Table1). For human FMO3-MBP Lys-158, the calculated Kmapp andVmax values for conversion of (−)-AMPH to AMPH hydroxylamine were 12.4 mM and 0.5 nmol/min/mg of protein and for (+)-AMPH the calculatedKmapp andVmax values for formation of AMPH hydroxylamine were 18.1 mM and 4.5 nmol/min/mg of protein, respectively (Table 1).

Proposed enzymatic N-oxygenation of AMPH by human FMO3. AMPH is N-oxygenated to produce hydroxylamine. Hydroxylamine isN,N-dihydroxylated to produce an unstable intermediate that dehydrates to produce mainly transoxime.
N -oxygenation of AMPH by human FMO3-MBP
Because a previous report (Rauckman et al., 1979) showed that hydroxylamines could have deleterious effects on FMO, the influence of AMPH hydroxylamine (compound 3) as an inhibitor of 5-DPTN-oxygenation by human FMO3-MBP Lys-158 and Glu-158 was examined. Plots of the reciprocal of the substrate concentration versus the reciprocal of the velocity of 5-DPT N-oxide formation were linear. For human FMO3-MBP Lys-158 the calculatedKmapp values for 5-DPTN-oxide formation in the presence or absence of 50 μM AMPH hydroxylamine (compound 3) were essentially the same (i.e., 108 μM and 72 μM, respectively). Likewise, for human FMO3-MBP Glu-158, the calculated Kmapp values for 5-DPT N-oxide formation in the presence or absence of 50 μM AMPH hydroxylamine were virtually the same (i.e., 11 μM and 32 μM, respectively). The results suggested that concentrations of AMPH hydroxylamine (compound 3) that far exceeded the levels expected to be present in enzyme incubations had virtually no effect on the ability of human FMO3-MBP to N-oxygenate 5-DPT and that compound 3 did not uncouple human FMO3.
To examine a role of human FMO3-MBP in the N-oxygenation of AMPH hydroxylamine, we analyzed the formation of phenylpropanone oxime. Formation of trans oxime was highly stereoselective. Thus, the ratio for formation of trans to cis AMPH oxime was observed to occur with a stereopreference of 5:1. In the presence of human FMO3-MBP Lys-158 or Glu-158, N-oxygenation of AMPH hydroxylamine (compound 3) to phenylpropanone oxime was linearly dependent on the presence of protein (i.e., 0–50 μg of protein) and linearly dependent upon the incubation time of the reaction (0–30 min). For both human FMO3-MBP Glu-158 and Lys-158, formation of phenylpropanone oxime (compound 2) showed typical Michaelis-Menton kinetics. Plots of the reciprocal of the substrate concentration versus the reciprocal of the velocity of the oxime (compound 2) formation were linear. The calculatedKmapp andVmax for human FMO3-MBP Glu-158 and Lys-158 were determined and are listed in Table2.
N -oxygenation of AMPH hydroxylamine compound 3 by human FMO3-MBP
In addition, the Kmapp andVmax for oxime formation for α-deuterated AMPH hydroxylamine was determined for each human FMO3-MBP and the isotope effect was calculated from eq. 1 (Northrop, 1971).
In addition to the forward reaction that produced oxime (compound2) from AMPH hydroxylamine (compound 3), the conversion of AMPH hydroxylamine to the retroreduction product AMPH by human FMO3-MBP was also examined. It was notable that in contrast to a previous report examining the retroreduction of aliphatic hydroxylamines by pig FMO1 (Poulsen et al., 1986), no evidence for human FMO3-MBP-dependent retroreduction of AMPH hydroxylamine (compound3) was observed.
N-Oxygenation of METH and METH Hydroxylamine by Human FMO3-MBP.
The study of the N-oxygenation of α-substituted phenethylamines was extended to the secondary amine METH and the secondary hydroxylamine METH hydroxylamine (compound5; Fig. 4). In the presence of human FMO3-MBP Lys-158 or Glu-158, formation of METH hydroxylamine (compound 5) from METH was linearly dependent on time and protein concentration. It was notable that the formation of compound 5 from METH was significantly less than the formation of AMPH hydroxylamine (compound 3) from AMPH. Thus, formation of METH hydroxylamine (compound 5) in the presence of human FMO3-MBP Lys-158 or Glu-158 was 1.6 and 0.9 nmol of METH hydroxylamine formed/min/mg of protein, respectively. The low level of N-oxygenase activity observed precluded attempts to characterize the Kmapp andVmax for this reaction. Likewise, theN-oxygenation of METH hydroxylamine by human FMO3-MBP Lys-158 and Glu-158 was also dependent on protein concentration and time. The product observed formed from METH hydroxylamine was phenylpropanone, apparently arising from the hydrolysis of the precursor nitrone (see Fig. 4). In the presence of human FMO3-MBP Lys-158 or Glu-158, the formation of phenylpropanone from METH hydroxylamine was modest (i.e., 3.6 or 1.4 nmol of phenylpropanone formed/min/mg of protein). Once again, the low level of conversion of hydroxylamine (compound 5) to phenylpropanone (compound 1) and the difficulty of quantifying the product formed precluded a complete kinetic characterization of the reaction. The results suggested, however, that METH hydroxylamine (compound5) was N-oxygenated to produce aN,N-dihydroxy intermediate that was converted to the nitrone after loss of the elements of water. Unconjugated aliphatic nitrones are unstable and the instability to hydrolysis affords the final observed product, phenylpropanone (Fig. 4).

Proposed enzymatic N-oxygenation of METH. METH is N-oxygenated to produce hydroxylamine. Hydroxylamine is N,N-dihydroxylated to produce an unstable species that results in a nitrone after spontaneous elimination of the elements of water. Nitrone (compound 6) is not indefinitely stable and is hydrolyzed to phenylpropanone.
Effect of AMPH Hydroxylamine and METH Hydroxylamine and AMPH Oxime on Biogenic Amine Transporters.
As shown in Tables 3 to 5, the binding and uptake inhibition for DA, serotonin, and NE (or their surrogates) in the presence of compounds 2, 3, and 5 was examined. In addition, the percentage of stimulated release of MPP+ from hDAT cells was also studied. For the hDAT, compounds 2, 3, and 5 had no detectable binding affinity (i.e.,Ki >10 μM) (Table3). Inhibition of [3H]DA uptake for AMPH hydroxylamine (compound2) was significantly greater than for METH hydroxylamine (compound 5) and AMPH oxime (compound 3) was devoid of measurable uptake inhibition activity. The IC50 value for release of [3H]MPP+ from C6hDAT cells for AMPH and AMPH oxime was comparable, although compound3 showed a much lower maximum percentage of stimulated release. AMPH hydroxylamine was significantly more potent at stimulating [3H]MPP+release than was METH hydroxylamine (Table 3).
Binding, uptake, and release in hDAT cells
Binding and uptake in HEK-hNET cells
In the presence of the hSERT, compounds 2, 3, and 5 showed no detectable competitive inhibition of binding (i.e., Ki > 10 μM) and no detectable uptake inhibition of [3H]serotonin (Ki > 10 μM) was observed (Table4).
Binding, uptake, and release in hSERT cells
Compounds 2, 3, and 5 showed no detectable binding affinity to the hNET (i.e.,Ki > 10 μM) (Table5). For AMPH hydroxylamine (compound2), the IC50 value for uptake inhibition of [3H]NE was similar to that of AMPH and METH. METH hydroxylamine was significantly less potent than METH at inhibiting uptake of [3H]NE. In the presence of the hNET, AMPH oxime (compound 3), did not possess any measurable uptake inhibition activity (Table 5).
Discussion
There are two prominent common polymorphic forms of human FMO3, differing at a single amino acid at codon 158 (i.e., human FMO3 Lys-158 and Glu-158), present in approximately equal allelic frequency in the Caucasian populations examined (Akerman et al., 1997; Brunelle et al., 1997). In individuals deficient in human FMO3 (i.e., those suffering from trimethylaminuria, the inability to metabolize trimethylamine) a relatively high incidence of psychosocial diseases are apparent, including low self-esteem, frustration, anxiety, clinical depression, paranoia, suicidal personality, and addiction to drugs. Many of the manifestations of trimethylaminuria may be explained in terms of excessive central and sympathetic neural stimulation. It is possible that the symptomology can be explained in terms of abnormal biogenic amine metabolism, because we have observed that trimethylaminuria patients also have abnormal catecholamine metabolism (Cashman et al., 1997).
We previously hypothesized that oxime formation represented a detoxication process (Lin and Cashman, 1997a,b). Phenethylamine and tyramine follow an A → B → C reaction profile in which human FMO3 does not allow the hydroxylamine to desorb from the surface of the enzyme. Such “catalytic facilitation” has been observed before (Du et al., 1995; Lin and Cashman, 1997a,b) and it allows formation of a nontoxic metabolite (i.e., oxime) through the intermediacy of a potentially dangerous metabolite (i.e., hydroxylamine). In the presence of phenethylamine or tyramine, the only human FMO3 metabolite observed is the corresponding trans oxime. If for steric or other reasons, primary amines produce significant quantities of hydroxylamine metabolites, this may pose a significant toxicological threat. Although preliminary studies indicate that hydroxylamines of AMPH and METH are toxic to brain cells (J. Miller and J. Cashman, unpublished observations), the present study was undertaken to establish the metabolic basis for AMPH and METH N-oxygenation.
In contrast to phenethylamine or tyramine where no detectable amount of hydroxylamine was observed formed in the presence of human FMO3-MBP, AMPH was converted to AMPH hydroxylamine (compound 3). The α-methyl group of AMPH causes a dramatic change in the way human FMO3-MBP N-oxygenates phenethylamines and precludes catalytic facilitation. In good agreement with the previous study of the N-oxygenation of phenethylamine, human FMO3-MBP Glu-158N-oxygenates AMPH more efficiently than FMO3-MBP Lys-158. TheVmax/Kmappvalue for phenethylamine N-oxygenation in the presence of human FMO3-MBP Glu-158 and Lys-158 is on average 2500-fold and 670-fold greater, respectively, than theVmax/Kmappvalue for AMPH N-oxygenation. Although stereochemical aspects of a molecule may influence kinetic values, the large rate enhancement for phenethylamine N-oxygenation suggests that human FMO3-MBP is highly sensitive to substrate steric features.
Human FMO3-MBP shows considerable substrate stereoselectivity in theN-oxygenation of AMPH. For human FMO3-MBP Glu-158, theVmax/Kmappvalues suggests that the (−)-AMPH isomer is more efficientlyN-oxygenated than the (+)-AMPH isomer. In contrast, in the presence of human FMO3-MBP Lys-158, theVmax/Kmappvalues suggests that the (−)-isomer of AMPH is not very efficientlyN-oxygenated. Because the (+) isomer of AMPH is three to four times more potent than the (−) isomer in eliciting central nervous system excitatory effects in humans (Innes and Nickerson, 1975), stereoselective N-oxygenation of AMPH by human FMO3 may have clinical consequences. It is possible that individuals administered (+)-AMPH may form more hydroxylamine if they are homozygous for the Lys -158 allele. On the other hand, individuals homozygous for the Glu-158 allele may metabolize (+)-AMPH less efficiently and possibly be spared to a certain degree from the toxic consequences of AMPH hydroxylamine formation. Such individuals would be anticipated to detoxicate AMPH hydroxylamine to the oxime much more efficiently than humans homozygous for the Lys-158 humanFMO3 gene. Of course, any such correlation is also presumably dependent on a large number of other parameters including metabolic, genetic, and environmental factors.
Some investigators have suggested that oxime (compound 2) formation from AMPH was due to the spontaneous oxidation (i.e., nonenzymatic) of AMPH hydroxylamine (compound 3) (Beckett and Jones, 1975). Others have disputed this assertion (Parli and McMahan, 1973). Because the human FMO3 product is largely thetrans oxime and because the trans/cis oxime ratio is 84:16 and greater than the spontaneous trans/cis oxime formation ratio of 80:20, oxime formation requires human FMO3. Part of the explanation may be that a full understanding of the participation of animal and human FMO in the metabolism of AMPH was not appreciated at the time of the earlier studies. It is notable that dehydration of the N,N-dihydroxy intermediate is a nonenzymatic process simply involving the thermodynamically favored loss of water (Fig. 3). In contrast to phenethylamine and tyramine, the lack of exclusive formation of the trans oxime indicates that catalytic facilitation is not fully operative for the AMPH class of substrate. This suggests that the region on the FMO3 enzyme surface that provides a protein template for the spontaneous dehydration is sensitive to steric features of the substrate.
Kinetic values indicated that AMPH hydroxylamine (compound3) was considerably better than either (+)- or (−)-AMPH as a substrate for human FMO3-MBP. That oxime (compound 2) was not directly formed from either (+)- or (−)-AMPH suggests that the α-effect was insufficient to overcome the steric constraints of the substrate. Even though the hydroxylamine was a better substrate than the primary amine, this was not sufficient to allow direct formation of oxime and accumulation of AMPH hydroxylamine (compound 3) was observed. Human FMO3-MBP Glu-158 was superior at forming oxime (compound 2) from AMPH hydroxylamine (compound 3) than was human FMO3-MBP Lys-158. This may have clinical consequences. Because oxime formation represents a detoxication process, individuals that cannot efficiently detoxicate AMPH hydroxylamine may be at an increased risk to the toxic properties of AMPH hydroxylamine. Thus, humans that are homozygous for human FMO3 Lys-158 may be less able to detoxicate AMPH. Presumably, increased residence time of the hydroxylamine could provide for enzyme and nonenzymic oxidation of the hydroxylamine to reactive metabolites. For example, it has been shown that biogenic hydroxylamines can be converted to nitroso metabolites that can inhibit various biomacromolecules including hemoproteins and alkylate thiol-containing proteins.
A previous study (Parli and McMahan, 1973) showed that there was no primary deuterium isotope effect on AMPH hydroxylamine formation but that oxime formation was slowed somewhat by a deuterium atom at the α carbon position. In the previous study, formation of AMPH hydroxylamine and oxime was highly dependent on the tissue of the species used and this suggested that the nature and amount of enzyme present may play a significant role in determining AMPH metabolite product ratios. For human FMO3, an inverse isotope effect was observed. The apparent kinetic isotope effect on oxime formation is probably not a measure of the true intrinsic isotope effect (i.e., the full effect originating from the single isotopically sensitive step in catalysis) because the second step involving dehydration of theN,N-dihydroxy intermediate is thought to be spontaneous. However, because the proposed two-step process leading to oxime (compound 2) is essentially irreversible, the forward “commitment to catalysis” is great and the apparent kinetic isotope effect is probably not suppressed. Steric isotope effects can be significant and inverse (Northrop, 1971). Introduction of a deuterium into the α position accelerates N,N-dihydroxylation of AMPH hydroxylamine (compound 4) by a secondary β-deuterium isotope effect. Such effects arise from the lower steric bulk of deuterium or electronic effects leading to possible changes in hyperconjugation. The data is most consistent with bulky N,N-dihydroxy intermediate formation taking place faster with a smaller α deuterium present. That similar large, inverse kinetic isotope effects were observed for both human FMO3 enzymes studied suggested that C-D bond breaking was not present in the critical transition state and this is in accord with a mechanism whereby C-D bond breakage occurs as spontaneous dehydration after the rate limiting step.
Human FMO3-MBP Lys-158 and Glu-158 also N-oxygenates METH. Like AMPH, the incipient METH hydroxylamine (compound 5) is not apparently efficiently N-oxygenated and can be detected in enzyme incubations. That METH hydroxylamine (compound 5) was formed shows that secondary α-substituted phenethylamines can beN-oxygenated by human FMO3 but this process is not nearly as efficient as the N-oxygenation of primary or α-substituted primary phenethylamines. The biological residence time of METH hydroxylamine may be considerable because steric factors that limitN-oxygenation may also manifest themselves in enzymatic retroreduction of secondary hydroxylamines to the parent amine. Although this was not expressly examined, it is possible that both AMPH hydroxylamine and METH hydroxylamine participate in microsomal retroreduction and “futile cycling” to the parent amines.
METH hydroxylamine (compound 5) is alsoN-oxygenated but forms a nitrone after elimination of the elements of water. Nitrones such as compound 6 are not indefinitely stable in aqueous solution and rapidly decompose to phenylpropanone. Nitrone (compound 6) has been observed formed in the presence of liver homogenates from rats and rabbit after derivatization and mass spectral analysis (Couts et al., 1978). Pig FMO1 has been shown to convert secondary hydroxylamines to stable nitrones (Cashman et al., 1990). The nitrone metabolite (compound6) is the same metabolite observed formed from METH via non-FMO-dependent processes (Couts et al., 1978). Thus,N-demethylation and N-deamination can lead to the same phenylpropanone metabolite as that arising from FMO-dependent nitrone formation and decomposition. It is possible that the contribution of FMO to the metabolism of biogenic amines and congeners has been underestimated in the past because products stemming from other enzymes give the same products.
Limited studies suggest that METH N-oxygenation by human FMO3-MBP Lys-158 was more efficient than FMO3-MBP Glu-158. Because METH hydroxylamine has been shown to be more cytotoxic to rat brain cells than METH itself (J. Miller and J. Cashman, unpublished observations), it is possible that individuals that consume METH and are homozygous for the human FMO3 Lys-158 allele could form more METH hydroxylamine and possibly predispose themselves to the toxic effects of such metabolites.
AMPH and METH are substrates for DA, 5-HT, or NE transporters and increase the release of monoamines from neuronal preparations via exchange through the transporters as well as competing for the binding site for monoamines on the transporters (Eshleman et al., 1994; Gibb et al., 1997; Wrona et al., 1997). Compared with AMPH, AMPH hydroxylamine was relatively potent at blocking uptake but had no potency at blocking binding (Table 3). For release of [3H]MPP+ from HEK hDAT cells, AMPH hydroxylamine potency was somewhat higher and the efficacy was the same. AMPH hydroxylamine may function as a substrate for the hDAT but not compete for the same binding site on the hDAT as RTI-55 or AMPH or METH. In contrast, oxime (compound 2) was devoid of binding affinity and effects on uptake. It is possible that oxime (compound 2) is a very poor substrate, but in agreement with the markedly reduced pharmacological activity of phenethylamine oxime and tyramine oxime (Lin and Cashman, 1997a,b), oxime formation constitutes a detoxication process. METH hydroxylamine is considerably weaker at blocking DA radioligand binding and at inhibiting uptake at the hDAT than METH itself. METH hydroxylamine is about equi-potent with METH at causing release of [3H]MPP+ in hDAT cells, but the efficacy is only half that of METH. Introduction of aN-hydroxyl moiety into AMPH and METH causes a large alteration in drug interaction with recognition sites associated with the uptake process, but has much less effect on [3H]MPP+ release.
AMPH hydroxylamine had no apparent affinity for the RTI-55 binding site on the hSERT and had little effect on 5-HT uptake. The potency of AMPH hydroxylamine was less than that of AMPH in causing [3H]MPP+ release (data not shown), but the efficacy was unchanged. It is possible that AMPH hydroxylamine is a substrate for the hSERT. AMPH oxime (compound2) had little effect on [3H]5-HT uptake and essentially no affinity for the radioligand binding site on the hSERT. The results of AMPH oxime suggest that oxime formation represents a detoxication process.
AMPH hydroxylamine and METH hydroxylamine had no detectable affinity for inhibition of radioligand binding to the hNET. AMPH hydroxylamine was nearly as potent a [3H]NE uptake inhibitor as was AMPH. METH hydroxylamine was significantly less potent at inhibiting [3H]NE uptake than was METH. AMPH oxime was devoid of uptake inhibition activity. Thus, AMPH hydroxylamine was markedly more efficacious than METH hydroxylamine at blocking NE uptake. That AMPH oxime was without activity suggests that it does not interact with the hNET. The selectivity of AMPH and METH hydroxylamines and AMPH oxime is notable. AMPH hydroxylamine selectivity follows the rank order hNET > hDAT ≫ hSERT. Likewise, the transporter selectivity for METH hydroxylamine follows the rank order hNET > hDAT ≫ hSERT.
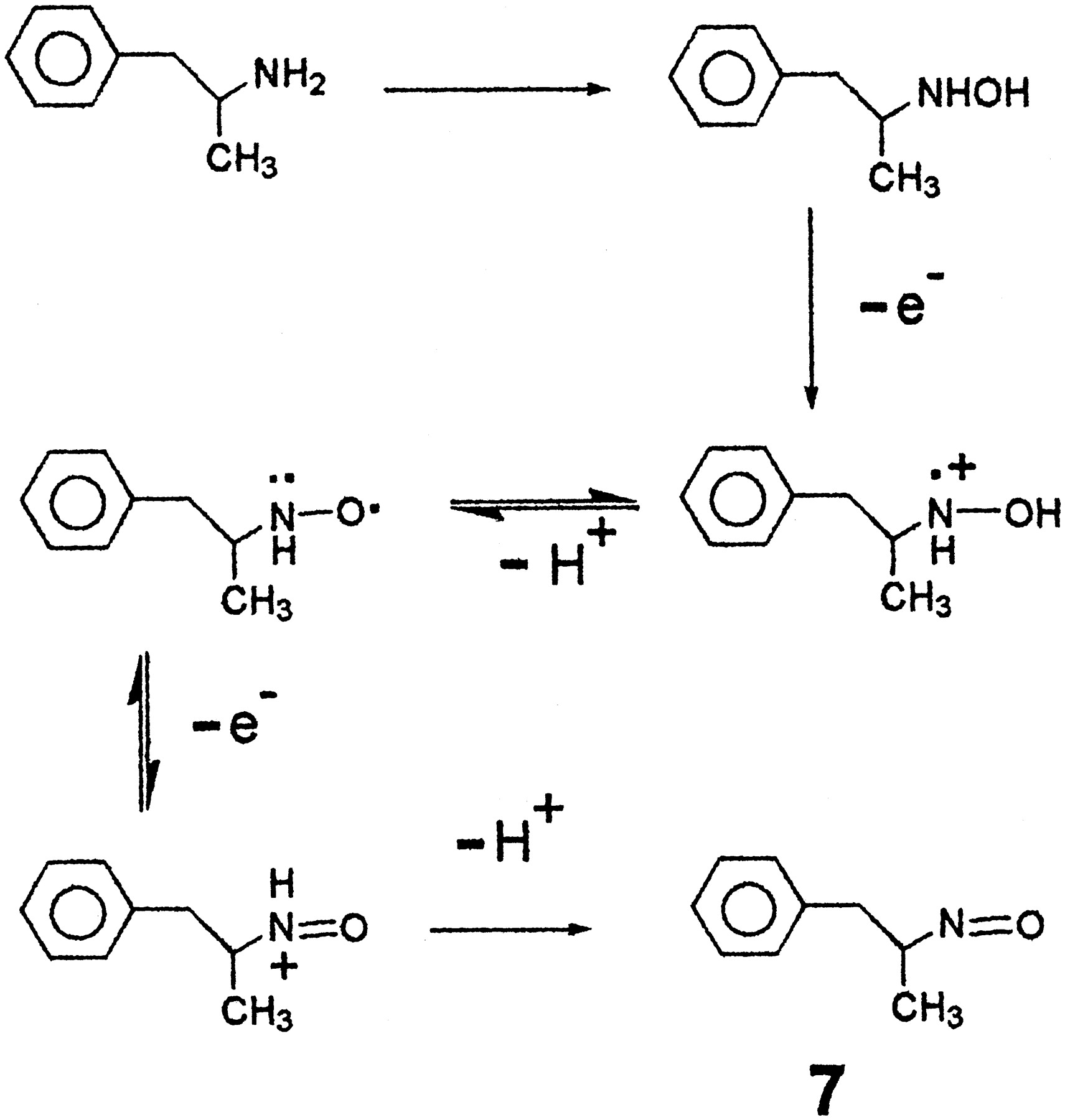
FMO has been detected in rat brain (Bhamre and Ravindranath, 1991) and it is likely that FMO3 is present in human brain. If the effective concentration of FMO3 is quite great in biogenic amine-containing cells, this may have pharmacological consequences for even highKm biogenic amine substrates. In view of the cytotoxicity of these metabolites and the fact that they are apparent substrates for human biogenic amine transporters, we conclude that N-oxygenation is a metabolic activation event. Further metabolism of the hydroxylamine to an oxime is a detoxication process. Among the possible cytotoxic events that could occur include direct action of the hydroxylamine metabolites or further sequential one electron metabolism to additional electrophilic metabolites (Fig.5). Hydroxylamines may directly interact with important biomacromolecules to impair cellular function or undergo conversion to nitroso metabolites that could also participate in alkylation of thiol-containing biomolecules that are essential for neuronal function.

.. Proposed sequential one electron oxidation of AMPH hydroxylamine to ultimately produce nitroso metabolite (compound7).
Acknowledgments
We acknowledge the generous gifts of (+)- and (−)-AMPH and METH from the National Institute on Drug Abuse Rare Chemical Collection. We also thank Dr. Daniel Doerge for supplying mass spectral data and to Dr. Greg Bruce for expression of human FMO3-MBP enzymes.
Footnotes
-
Send reprint requests to: John R. Cashman, Human BioMolecular Research Institute, 5310 Eastgate Mall, San Diego, CA. E-mail: ledcash{at}aol.com
-
↵1 This work was financially supported by National Institutes of Health Grants GM 36426, DA 11547 and DA 00269 (to J.R.C.) and Veterans Administration Merit Review and Career Scientist Program and NIH/NIDA Contract Number N01DA7-8071 (to A.J.).
-
↵2 Present address: Portland Veterans Administration Medical Center and Department of Psychiatry and Physiology, Pharmacology and Behavioral Neuroscience, Oregon Health Sciences University, 3181 S.W. Sam Jackson Park Rd. Portland, OR 97201.
-
↵3 Throughout the text, the synthetic AMPH hydroxylamine and METH hydroxylamine described is racemic.
- Abbreviations:
- AMPH
- amphetamine
- METH
- methamphetamine
- FMO3
- flavin-containing monooxygenase form 3
- NE
- norepinephrine
- DA
- dopamine
- MBP
- maltose binding protein
- NaCNBH3
- sodium cyanoborohydride
- FAB
- fast atom bombardment
- THF
- tetrahydrofuran
- TLC
- thin-layer chromatography
- 5-DPT
- 10-[(N,N-dimethylamino)pentyl]-2-(trifluoromethyl)phenothiazine
- 5-DPT N-oxide
- 10-[(N,N-dimethylamino)pentyl]-2-(trifluoromethyl)phenothiazineN-oxide
- hDAT
- human DA transporter
- hSERT
- human serotonin transporter
- hNET
- human NE transporter
- C6-hDAT cells
- C6 glioma cells transfected with hDAT
- DAT
- DA transporter
- 5-HT
- 5-hydroxytryptamine or serotonin
- MPP+
- 1-methyl-4-phenylpyridinium
- Received June 29, 1998.
- Accepted September 29, 1998.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}