


Abstract
The rat isolated perfused kidney was used to investigate the linearity of the renal disposition of morphine and its potential oxidative and glucuronidative metabolism by the kidney. In a set of single-dose experiments, morphine was administered to recirculating perfusion medium to achieve initial concentrations of 0.2, 2 and 20 μM (n = 4 at each concentration). In a set of multiple-dose experiments, morphine was administered to perfusate as sequential bolus doses to achieve concentrations of 0.2, 2, 20 and 200 μM (n = 6). HPLC was used to determine the concentration of morphine in perfusate and urine. Normorphine, morphine-3-glucuronide and morphine-6-glucuronide could not be detected in perfusate or urine, a result that suggests an absence of oxidative and glucuronidative metabolism of morphine by the rat kidney. The volume of distribution of morphine within the kidney was high (31 ± 3 ml/g at 0.2 μM), which indicates extensive accumulation, and remained constant with increasing perfusate concentration. The ratio of unbound renal excretory clearance to glomerular filtration rate was always greater than unity for all kidneys, which indicates that the renal excretion of morphine involves net tubular secretion. This ratio was constant (P > .05) over the 100-fold concentration range of the single-dose study. In the multiple-dose study, the ratio was marginally but significantly (P < .05) higher at concentrations of 2, 20 and 200 μM than at 0.2 μM, a difference that cannot be explained by saturation of tubular secretion. The results suggest that the tubular secretion of morphine is not saturated over a wide range of concentrations (0.2–200 μM).
In humans and experimental animals, morphine is extensively metabolizedvia conjugative and oxidative pathways to metabolites that exhibit pharmacological activity (Milne et al., 1996). Although only 10% of a dose of morphine is excreted unchanged in urine, the kidney plays a major role in the excretion of M3G, M6G (Milne et al., 1992; Somogyi et al., 1993) and possibly normorphine (Milne et al., 1996).
The renal clearance of morphine, which exists predominantly as a cation at physiological pH, involves filtration at the glomerulus, tubular secretion and possibly reabsorption. A significant degree of tubular secretion has been observed in humans (Milne et al., 1992;Somogyi et al., 1993), chickens (May et al., 1967; Watrous et al., 1970; Hakim and Fujimoto, 1971) and sheep (Milne et al., 1995). These findings are supported byin vitro studies involving the stop-flow microperfusion of the renal proximal tubule from the rat (Ullrich and Rumrich, 1995), cortical slices from mice (Teller et al., 1976) and proximal tubular segments from the rabbit (Schäli and Roch-Ramel, 1982). Furthermore, secretion has been observed in the IPK of the rat (Ratcliffe et al., 1985; van Crugten et al., 1991). In the chicken (May et al., 1967; Watrous et al., 1970) and rabbit (Schäli and Roch-Ramel, 1982), secretion of morphine involves a cation transport system, as evidenced by competition between morphine and other organic cations, such as cyanine 863 (May et al., 1967; Watrous et al., 1970), mepiperphenidol (Watrous et al., 1970; Schäli and Roch-Ramel, 1982) and quinine (Schäli and Roch-Ramel, 1982). Studies in humans (Somogyi et al., 1993) and the rat IPK (van Crugten et al., 1991) suggest that the renal clearance of morphine also involves a component of reabsorption, and Ullrich and Rumrich (1995) recently suggested that the renal disposition of morphine involves carrier-mediated bidirectional tubular transport.
Drug metabolizing enzymes, such as UDP-glucuronosyltransferase, sulfotransferase and cytochromes P-450, are known to exist in the mammalian kidney (Jones et al., 1979; Anders, 1980; Hjelleet al., 1986), and morphine is one of many drugs that has been suggested to undergo renal metabolism. Indeed, evidence exists for the glucuronidation of morphine by microsomes prepared from fetal (Pacifici and Rane, 1982) and adult (Yue et al., 1988;Cappiello et al., 1991) human kidneys, and by proximal tubules isolated from the rabbit (Schäli and Roch-Ramel, 1982). In addition, studies conducted in vivo in sheep (Milneet al., 1993; Milne et al., 1995) and dogs (Jacqzet al., 1986) have provided evidence for glucuronidation of morphine by the kidney. In contrast, glucuronidation of morphine was not observed in rat kidney microsomes (Rush et al., 1983;Rush and Hook, 1984) or the rat IPK (Ratcliffe et al., 1985;van Crugten et al., 1991). However, after studying the disposition of morphine in intact rats and rats in which the bile duct was cannulated and renal pedicles were ligated, Horton and Pollack (1991) hypothesized that the kidney is involved in the metabolic clearance of morphine. Hence some controversy remains about the role of the rat kidney in the metabolism of morphine. It is possible that the apparent conflict between the studies conducted in rats is due to the formation by the kidney of other metabolites, such as normorphine.
To date, the effect of alterations in concentration on morphine renal transport has not been investigated. In view of the significant degree of renal tubular secretion of morphine, it is conceivable that the relative contribution of the three renal clearance mechanisms (i.e., filtration, secretion and reabsorption) is concentration-dependent. Hence the present studies were designed to investigate the effect of concentration of morphine on its renal disposition in the rat IPK. An additional aim was to resolve the uncertainty surrounding the role of the rat kidney in the metabolism of morphine via conjugative and oxidative routes.
Materials and Methods
Chemicals
The following drugs and chemicals were used in this study: morphine hydrochloride (MacFarlane Smith Ltd., Edinburgh, UK), normorphine hydrochloride (Makor Chemicals, Jerusalem, Israel), [14C]-carboxy-inulin (3 mCi/g; Du Pont, Dreiech, West Germany), fraction V BSA (Miles Diagnostics Inc., Kankakee, IL),l-cysteine, glycine, l-glutamic acid andd-mannitol (Sigma Chemical Co. (St. Louis, MO),d-glucose and n-butyl alcohol of HPLC grade (E. Merck, Darmstadt, Germany) and acetonitrile of “far UV” grade and methanol and chloroform of HPLC grade (BDH Laboratory Supplies, Poole, England). All other chemicals were of analytical grade or equivalent.
Rat IPK
The study was approved by the Institute of Medical and Veterinary Science Animal Ethics Committee (Adelaide, South Australia). Male Sprague-Dawley rats (345–505 g) from the Gilles Plains Animal Resource Centre (Adelaide, Australia) were maintained at approximately 21°C on a 12-hr light/dark cycle, with free access to food and water. The IPK preparation was based on methods described previously (Elliset al., 1990; Mancinelli et al., 1995). Rats were anesthetised with an i.p. injection of sodium pentobarbital (60 mg/kg Nembutal; Boehringer Ingelheim, Artarmon, NSW, Australia) before surgery. The abdominal cavity was opened to expose the right renal artery, and the surgical procedure of Mancinelli et al., (1995) was used thereafter.
The perfusate medium (150 ml) consisted of erythrocyte-free Krebs-Henseleit bicarbonate buffer (pH 7.4) containing 65 g/l of BSA,d-glucose (5 mM), l-cysteine (0.5 mM), glycine (2.3 mM) and l-glutamic acid (0.5 mM). Before use, BSA was dissolved in Krebs-Henseleit buffer (130 g/l) and purified by dialyzing against three exchanges of protein-free buffer over 3 days at 4°C. Dialyzed albumin solutions were stored frozen at −20°C. On the day of a perfusion experiment, the concentration of BSA in the perfusion medium was adjusted to 65 g/l, and the three amino acids and glucose were added. Perfusate was then filtered successively through 1.2-μm and 0.45-μm filters (Millipore, Bedford, MA). Perfusate was recirculated through the perfusion system, before cannulation of the kidney, for at least 30 min, which enabled the perfusate to become adequately oxygenated (>0.6 mM). [14C]-inulin (0.35 μCi) was added to the perfusate reservoir 15 to 20 min after the kidney was cannulated, and morphine was then added to the perfusate in accordance with the protocols described below. Urine was collected into preweighed tubes, and the volume was determined gravimetrically. Perfusate was collected from the reservoir at the midpoint of each urine collection interval or as specified. Urine and perfusate samples were stored at −20°C until analysis. Upon completion of a perfusion experiment, the kidney was weighed.
Perfusate was delivered to the kidney via a rotary pump (Masterflex model 7521-35, Cole Palmer, Chicago, IL), an 8-μm in-line filter (Millipore), a silastic tubing oxygenator, a glass bubble-trap, a flow meter and finally a glass cannula. Venous outflow drained directly into the perfusate reservoir. Renal arterial pressure was maintained at 100 ± 15 mm Hg by adjustment of the perfusate flow rate (35–50 ml/min). The concentration of oxygen in perfusate flowing into the kidney was determined routinely, using an Orion Model 820 Dissolved Oxygen Meter (Boston, MA), to be in excess of 0.6 mM.
Functional viability and performance of the kidney were assessed by measurement of the GFR, determined as the renal clearance of [14C]-inulin), UFR, perfusion pressure, perfusate pH, urine pH and the %TR of water, glucose and sodium.
Experimental Design
A: Single-dose IPK studies.
Approximately 5 min after the addition of [14C]-inulin, morphine was administered as a bolus dose into the perfusate reservoir, to achieve an initial concentration of 0.2 (low, n = 4), 2 (medium,n = 4) or 20 μM (high, n = 4), respectively. Urine samples were collected over 10-min intervals for 90 min, and perfusate samples (1.4 ml) were collected immediately after dosing and thereafter at 0.5, 1, 2.5 and 5 min and at the midpoint of each urine collection interval. We determined fu at concentrations of 0.1, 1 and 10 μM at 37°C by ultrafiltration of four replicates at each concentration through YMT membranes using the MPS-1 Micropartition system (Amicon Corp, Danvers, MA).
B: Multiple-dose IPK studies.
Multiple-dose IPK studies (n = 6) consisted of four periods (0–15, 15–30, 30–45 and 45–60 min) in which morphine was administered to the perfusate reservoir as a series of bolus doses to achieve initial morphine concentrations of 0.2 μM (low), 2 μM (medium), 20 μM (high) and 200 μM (extra-high), respectively. Before the addition of the initial morphine bolus dose, [14C]-inulin was added to the perfusate reservoir. For the first 5 min after each bolus dose, morphine was allowed to equilibrate. To make possible calculation of CLR M after each bolus dose, urine was collected over the final 10 min of each of the four periods, and perfusate was sampled every 5 min. The binding of morphine to perfusate protein was studied at concentrations of 0.2, 2, 20 and 200 μM, as described above.
Analytical Methods
The concentration of morphine in perfusate was determined using a validated reverse-phase HPLC method that utilized normorphine as the internal standard. Before analysis, perfusate samples (0.5 ml) collected during the medium, high and extra-high periods were diluted 1 in 2, 1 in 20 and 1 in 200, respectively, in blank perfusate. Each sample was supplemented with internal standard (200 μl of 1 μg/ml normorphine) and 0.2 M bicarbonate buffer pH 9 (0.5 ml). Extraction solvent (6 ml of chloroform containing 20% n-butyl alcohol) was added to the buffered mixture, and the tube contents were rotary-mixed at 33 rpm for 10 min and then centrifuged at 1800 × g for 10 min. After removal of the upper aqueous phase by aspiration, 5 ml of the organic phase was transferred to a 10-ml tube containing 0.05% sulfuric acid (200 μl), and this mixture was rotary-mixed (33 rpm, 10 min) and then centrifuged (1800 × g, 10 min). A 150-μl aliquot of the acid phase was transferred to a HPLC automatic injector vial, and 100 μl was injected onto the HPLC column.
The HPLC system consisted of a LC-10AT pump, SIL-10A automatic injector, SCL-10A system controller, SPD-6A UV spectrophotometric detector (210 nm) and C-R6A Chromatopac integrator, all from Shimadzu (Tokyo, Japan), and a Nova-pak C18, 4-μm Radial-Pak cartridge and Nova-Pak C18 Guard-Pak insert, both from Waters (Milford, MA). Mobile phase (70 mM potassium dihydrogen orthophosphate buffer, pH 3, containing 1.5% acetonitrile and 1% methanol) was delivered in single-pass mode to the column at a flow rate of 1 ml/min. Typical retention times for normorphine and morphine were 4.9 and 6.2 min, respectively. The chromatographic run time for each sample was 35 min because of the presence of late-eluting endogenous compounds. Calibration curves, ranging from 0.013 to 1.33 μM, were linear (r 2 > 0.998), and repeat analysis of quality-control samples containing morphine at concentrations of 0.027 μM, 0.27 μM and 1.06 μM indicated that the interday and intraday accuracy and precision of the assay were within 11%.
The concentration of morphine in urine was determined by direct injection of 100 μl of diluted urine (1:10–1:5000) onto the HPLC system described above. Calibration curves containing morphine (0.026–1.33 μM) in diluted urine (1:50) were linear (r 2 > 0.996), and repeat analysis of quality-control samples containing morphine at concentrations of 0.135 μM, 0.54 μM and 1.06 μM indicated that the interday and intraday accuracy and precision of the assay were within 7%.
The potential role of the kidney in the formation of normorphine, the internal standard, was assessed by testing for the presence of normorphine in urine and perfusate samples that contained no internal standard. The possible presence of M3G and M6G in perfusate and urine samples was assessed by using a validated HPLC assay with UV detection involving solid-phase extraction of perfusate samples and direct injection of diluted urine samples according to a modification of a previously reported method (Evans and Shanahan, 1995a).
Glucose concentrations in perfusate and urine were determined by the glucose oxidase method, using a commercially available kit (Glucose Kit, Sigma Diagnostics, Sigma Chemical Co., St. Louis, MO). Sodium concentrations in perfusate and urine samples were determined by atomic absorbance spectrophotometry (Varian Techtron Atomic Absorbance Spectrophotometer, model no. AA6, Melbourne, Victoria, Australia).
Data Analysis
For the single-dose and multiple-dose studies, the CLR
M was determined as the rate of excretion into urine divided by the midpoint perfusate concentration, calculated according to equation 1.
For the single-dose studies, the time-averaged CLR(0–90)
M was calculated according to equation 4.
Data are presented as mean ± S.D. Analysis of variance was used to test for differences that were considered significant at the level of .05. Factorial design ANOVA was used to assess the concentration-dependence of protein binding for the single- and multiple-dose studies and to assess whether there were significant differences between the three groups of single-dose IPKs in terms of kidney function and the pharmacokinetic parameters for morphine. In addition, repeated-measures ANOVA was performed to determine whether there were any significant changes in these parameters with time. For the multiple-dose IPK studies, repeated-measures ANOVA was used to test for changes in kidney function parameters and morphine renal clearance measurements, across the four perfusion periods. Upon detection of a significant difference, pairwise comparisons were performed using the Fisher test. Simple bivariate regression analyses were performed to determine the relationship between morphine renal clearance parameters and functional parameters such as UFR, GFR and %TR of water.
Results
The functional viability of each IPK was assessed using GFR and %TR of water, glucose and sodium. A kidney was considered viable if the GFR and the %TR of water, glucose and sodium were greater than 0.4 ml/min and greater than 80%, 90% and 85%, respectively, over the experimental period. For the single- and multiple-dose IPK studies, each of these parameters was shown to remain relatively constant with time, which is in keeping with the results of previous studies from our laboratory (Mancinelli et al., 1995). No significant differences were observed among the three groups of single-dose IPKs for each of the functional parameters (P > .05). Therefore, morphine had no effect on the functional viability of the IPK.
There was no evidence for the presence of the oxidative metabolite normorphine or of the conjugative metabolites M3G and M6G in perfusate and urine samples, as determined by two individual HPLC methods. With the sensitivity limits of the respective assays for normorphine and M3G and M6G, it would have been possible to detect 0.1% conversion of morphine to normorphine over a 90-min perfusion and a 0.35% conversion of morphine to either M3G or M6G. Because normorphine was not formed by the kidney, its use as an internal standard in the analytical method was not compromised.
Binding of morphine to protein in perfusate was low and independent of morphine concentration over a 1000-fold range, P > .05, (table1). The average fu values used for the calculation of CLR M/fu · GFR ratios were 0.744 and 0.739 for the single- and multiple-dose IPK studies, respectively.
Fraction of morphine unbound in IPK perfusate
The mean concentration of morphine in perfusate vs. time profiles for the three groups of single-dose IPK studies are presented semilogarithmically in figure 1. Profiles were biphasic in nature with an initial rapid distribution phase, lasting for approximately 5 min, followed by a slower elimination phase. The terminal elimination phases for the three treatment groups were observed to be parallel. Data on the renal excretory clearance of morphine in each urine collection interval from the single-dose experiments are shown in figure 2, and pharmacokinetic parameters for these kidneys are presented in table2. CLR M was 0.8 to 2 ml/min (fig. 2A) and was greater than fu · GFR during each time interval (fig. 2B). There were no significant differences among the three groups (P > .05) in the CLR M/fu · GFR ratio and CLR M estimates for each time period and in CLR(0–90) M. However, there was a significant decrease with time in CLR M and the CLR M/fu · GFR ratio for the low-dose and medium-dose groups. For each of the three groups, there was no statistically significant difference between the two clearance estimates, CLR(0–90) M and CLM (P > .05); however, upon combining the data for the three groups we detected a small but significant difference (P = .02) between these two clearance estimates. The mean VK M values ranged between 25 and 31 ml/g kidney weight, and there were no significant differences among the three groups (P > .05) for VK M and T1/2.

Concentration of morphine in perfusate vs.time profile for single-dose studies; low (○; n = 4), medium (□; n = 4) and high (▵; n = 4). Data are presented as mean and S.D.

CLR M (panel A) and index of renal tubular transport, CLR M/fu · GFR (panel B) for the three groups of the single-dose studies; low (□;n = 4), medium (▪; n = 4) and high (░; n = 4). Data are presented as mean and S.D.
Pharmacokinetic parameters for morphine disposition observed in single-dose IPK studies, for the low, medium and high morphine concentrations
The mean concentration of morphine in perfusate vs. time profiles for the multiple-dose IPK studies are shown in figure3. As expected, a 10-fold increase in perfusate concentration was observed after each dose of morphine. CLR M was 1 to 2 ml/min (fig.4A) and was greater than fu · GFR during each time interval (fig. 4B). Both CLR M (P = .003) and the CLR M/fu · GFR ratio (P = .010) were lower at a concentration of 0.2 μM (5–15 min) compared with the medium (20–30 min), high (35–45 min) and extra-high (50–60 min) morphine concentrations.

Concentration of morphine in perfusate vs.time profile for multiple-dose studies (n = 6), 0.2 to 200 μM). Data are presented as mean and S.D.
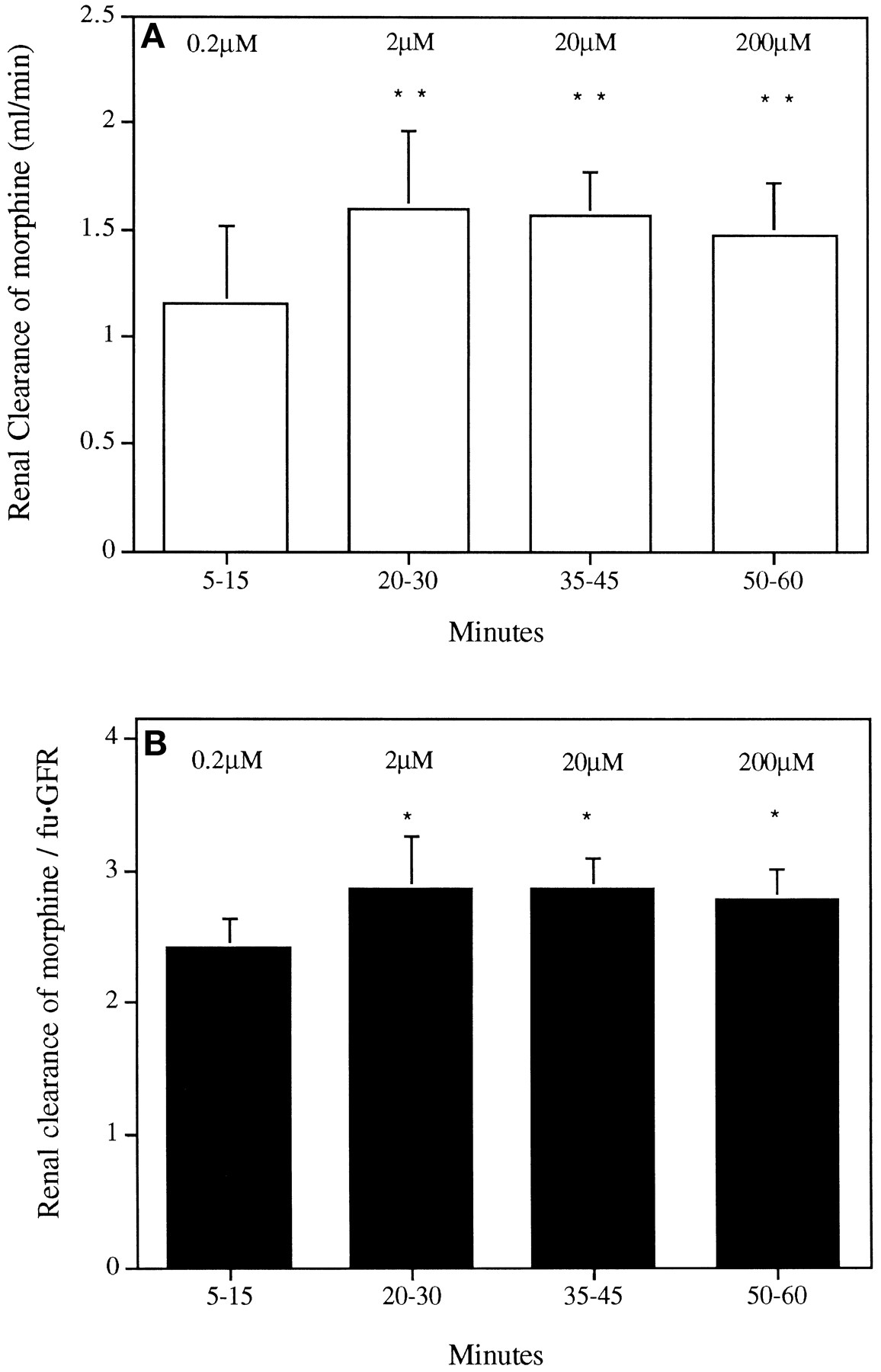
CLR M (panel A) and index of renal tubular transport, CLR M/fu · GFR (panel B) for the multiple-dose studies (n = 6) in the presence of sequentially increasing morphine concentration (0.2–200 μM). Data are presented as mean and S.D. Statistically significant difference observed between the low concentration (5–15 min) and each of the other three concentrations of morphine, *P < .05 **P < .01.
A highly significant positive relationship was observed between CLR M and GFR and between CLR M − fu · GFR and GFR for both the single-dose and the multiple-dose IPK studies (table 3). Significant positive relationships were also observed between CLR Mand UFR or %TR of water and between CLR M − fu · GFR and UFR or %TR of water for the single-dose IPK studies.
Bivariate regression analyses for the combined low, medium and high groups of the single-dose IPK studies and multiple-dose IPK studies
Discussion
Previous studies in the rat IPK have shown that morphine undergoes renal tubular secretion (Ratcliffe et al., 1985; van Crugtenet al., 1991), and it has been postulated that morphine also undergoes carrier-mediated reabsorption (van Crugten et al., 1991). Until now, however, there has been no information about the effect of increasing concentration on the renal excretory clearance of morphine and the relative contribution of the various renal clearance mechanisms. Moreover, there is controversy in the literature regarding the possible role of the rat kidney in the metabolism of morphine. Hence, the aims of the present studies in the IPK were to investigate the effect of alterations in morphine concentration on the renal disposition of the drug and to resolve the uncertainty about the contribution of the rat kidney to the metabolism of morphine.
The rat IPK is a suitable model for studying the renal handling of drugs, because tubular transport systems, in particular those involved in secretion, remain viable while making possible the study of renal disposition in the absence of confounding factors such as extrarenal metabolism (Bekersky, 1983). Reabsorption of water, which occurs along the entire tubule, is decreased in the IPK, resulting in a diminished driving force for passive reabsorption of organic solutes, whereas carrier-mediated reabsorption, which is confined largely to the proximal tubule, is well preserved (Maack, 1980, 1986; Besseghir and Roch-Ramel, 1987).
The kidney is known to be involved in the metabolism of xenobiotics (Jones et al., 1979; Anders, 1980). Glucuronidative metabolism of 1-naphthol (Redegeld et al., 1988) and oxidative metabolism of meperidine (Acara et al., 1981) have been observed previously in the rat IPK. The lack of normorphine, M3G and M6G in perfusate and urine samples collected in the present series of IPK studies indicates that the rat kidney is not capable of metabolizing morphine to these metabolites. Indeed, judging by the limit of quantification of the respective assays, the degree of conversion of morphine to normorphine, M3G or M6G must have been less than 0.4%. The apparent absence of glucuronidation of morphine in the IPK confirms the findings of Ratcliffe et al. (1985) and van Crugten et al. (1991) in IPKs from unspecified and male Hooded-Wistar rats, respectively, and is consistent with studies performed with microsomes prepared from the kidney of Fischer 344 rats (Rush et al., 1983; Rush and Hook, 1984).
Horton and Pollack (1991), using female Sprague-Dawley rats, hypothesized a role for the kidney in the metabolism of morphine after performing dispositional studies in intact rats and rats in which the bile duct was cannulated and renal pedicles were ligated. These investigators estimated that renal metabolic clearance accounted for 28.5% of the total systemic clearance of morphine. Their conclusion concerning renal metabolism of morphine was based on a number of assumptions, perhaps the most important being that the hepatic clearance of morphine remained constant upon renal ligation. Violation of this or any other of their assumptions may explain their unsubstantiated finding on the role of the kidney in morphine metabolism.
The study by Horton and Pollack (1991) involved the use of female Sprague-Dawley rats, whereas the current studies were performed using male Sprague-Dawley rats. Hence gender differences may explain the contrasting findings for the renal metabolism of morphine. Interestingly, Chhabra and Fouts (1974) observed a 2-fold greater rate of glucuronidation of p-nitrophenol in female rat renal microsomes of the CD strain than in male rat renal microsomes. This observation was supported by Rush et al. (1983) for the glucuronidation of p-nitrophenol by renal microsomes from female and male Fischer 344 rats. However, Rush et al.(1983) did not observe glucuronidation of morphine in renal microsomes from either female or male Fischer 344 rats.
Using the isolated perfused liver preparation, we have previously shown that normorphine is formed in significant quantities in the male rat, but not the female rat (Evans and Shanahan, 1995b). However, oxidative metabolism of morphine in the kidney has not been assessed previously. In the current study, there was no evidence for the metabolism of morphine to normorphine in the male rat IPK.
Further evidence for the lack of a significant role for the rat kidney in morphine metabolism comes from the finding, in each of the three groups of the single-dose studies, that CLR(0–90) M was similar in magnitude to CLM (P > .05). It is interesting to note, however, that when the data from all three groups were combined, CLM was significantly greater than CLR(0–90) M (P < .05), and this may suggest that a metabolic route other than those assessed in the present study plays a role in morphine metabolism. An alternative explanation, however, may be that extensive tissue uptake of morphine, with subsequent slow release into perfusate, led to an overestimate of the total clearance value calculated as dose divided by AUC0 ∞. Previous studies in the chicken kidney (May et al., 1967; Watrouset al., 1970) have shown that morphine undergoes renal metabolism to morphine ethereal sulfate; however, this pathway for metabolism of morphine is minimal in humans (Yeh et al., 1977) and rats (Smith et al., 1973).
In the present study, the binding of morphine to albumin in perfusate was low (table 1) and comparable to that observed in human plasma (Olsen, 1974; Milne et al., 1992) and IPK perfusate (van Crugten et al., 1991). The fu of morphine was independent of morphine concentration over a 1000-fold range, a result comparable to findings in human plasma (Olsen, 1974). It is not surprising that the fu of morphine was concentration-independent, because the concentration of BSA (≈950 μM) utilized in the IPK perfusate was nearly 5-fold greater than that of the highest morphine concentration studied (200 μM). Hence the concentration of the binding sites exceeded that of the ligand, and under these circumstances the unbound fraction would be expected to be independent of ligand concentration.
The disposition of morphine in the IPK system was characterized by accumulation in the kidney and by a renal clearance that involves net tubular secretion. Morphine was shown to distribute rapidly and extensively into kidney tissue with a resultant VK M of approximately 30 ml/g kidney tissue, which was independent of perfusate morphine concentration over the 100-fold range used in the single-dose studies (table 2). A large volume of distribution of morphine was previously observed in rats (11–27 l/kg) (Iwamoto and Klaassen, 1977;Bhargava et al., 1991), which supports the large VK M observed in the present study. Extensive uptake and distribution within the kidney were also observed in a study by Mulliset al. (1979) in which radiolabeled morphine was administered to rats; uptake of morphine by the kidney was substantially greater than that by other organs, including the liver. In addition, extensive uptake of morphine by kidney slices from the dog (Hug, 1967) and mouse (Teller et al., 1976) has been observed, and the findings are consistent with the involvement of carrier-mediated mechanisms of renal uptake.
The CLR M values (figs. 2A; 4A) observed in the present study are similar to values reported from previous studies in the rat IPK (van Crugten et al., 1991) and the intact rat (Horton and Pollack, 1991). For all time intervals in each kidney in the single- and multiple-dose studies, the CLR M/fu · GFR ratio was greater than unity, with no ratio less than 1.64, a result that indicates net renal tubular secretion of morphine. This finding is consistent with the renal handling index observed for morphine in vivo in humans (Milne et al., 1992;Somogyi et al., 1993) and sheep (Milne et al., 1995) and in previous studies using the rat IPK (Ratcliffe et al., 1985; van Crugten et al., 1991).
In the kidney, separate transport systems exist for the secretion of organic anions (Møller and Sheikh, 1983) and cations (Rennick, 1981) across the contraluminal and luminal membranes of the proximal tubule cells, and each transport system has a broad and overlapping substrate specificity. Contraluminal organic cation transport is driven by an electrical potential difference, whereas luminal organic cation transport occurs via electroneutral H+/organic cation exchange (Holohan and Ross, 1980; Somogyi, 1987; Ullrich, 1994). P-glycoprotein exists in proximal cells (Dutt et al., 1994), and this transporter has been shown to be involved in the transport of morphine across the luminal membrane of renal proximal tubule cells of mice and humans (Schinkel et al., 1995).
Tubular secretion, like any other carrier-mediated transport event, has a limited capacity, so saturation of secretion may occur. Cimetidine, which like morphine also exists as an organic cation at physiological pH, undergoes active tubular secretion that is saturated with increasing concentration of cimetidine over the range of 8 to 793 μM in rats in vivo (Weiner and Roth, 1981) and 10 to 40 μM in the rat IPK model (Boom et al., 1994). Saturation of cimetidine tubular transport is possible because cimetidine has a relatively high affinity for secretory transport in the rat kidney (Boom et al., 1994). In the present study, the CLR M/fu · GFR ratio for morphine remained constant over the concentration range 0.2 to 20 μM used in the single-dose experiments. In the multiple-dose study, the CLR M/fu · GFR ratio was marginally but significantly higher at concentrations of 2, 20 and 200 μM compared with that at 0.2 μM. The small change in the ratio cannot be explained by saturation of renal tubular secretion, because this would have resulted in a decrease in renal clearance. Hence the results of the present studies indicate that the renal secretion of morphine was not saturated over the 1000-fold range of concentrations investigated.
The lack of saturation suggests that morphine, like some other organic cations, has a low affinity for the transport system(s) involved in renal secretion. Transport of the organic cation N1-methylnicotinamide across the contraluminal renal tubular membrane occurs with a Km of 540 μM, whereas movement of N-methylphenylpyridinium from the lumen into the cell occurs with a Km of 200 μM in the rat proximal tubule (Ullrich, 1994). Ullrich and Rumrich (1995), using the stop-flow microperfusion method in the rat proximal tubule, determined a somewhat lower affinity for morphine transport, with Ki values of 780 μM and 1150 μM for the inhibition of the transport of N1-methylnicotinamide and N-methylphenylpyridinium across the contraluminal and luminal membranes, respectively.
van Crugten et al. (1991) suggested that morphine undergoes carrier-mediated tubular reabsorption in the rat kidney. The basis of this hypothesis was an increase in the CLR M/fu · GFR ratio for morphine after the co-administration of M3G and M6G to an IPK perfusion system. In the present study it was not possible to quantitate the degree of net tubular reabsorption, but reabsorption may have contributed to the decrease observed in the CLR M/fu · GFR ratio for the low and medium groups of the single-dose studies as well as to the slightly lower CLR M/fu · GFR ratio observed for the 0.2 μM morphine concentration in the multiple-dose studies.
A highly significant positive relationship was observed between the renal clearance of morphine and GFR (table3), which is not unexpected given that filtration is an important component of renal clearance. Upon removal of the filtration component (i.e., subtraction of fu · GFR from CLR M), a significant positive relationship with GFR remained, which suggests that the net renal tubular transport of morphine and GFR are related. Possible reasons for this relationship are, first, that tubular function and net secretory transport are related to GFR and, second, that an increase in GFR produces an increase in UFR that indirectly results in a decrease in the fractional reabsorption of morphine.
The CLR M/fu · GFR ratio observed in the present study was approximately half the corresponding ratio observed by van Crugten et al. (1991), which indicates that in the present study, either secretion was lower or reabsorption was more extensive. Comparison of the %TR for water for the two studies supports the latter of these two hypotheses. Thus, the tubular reabsorption of water observed by van Crugten et al. (1991) was approximately 50% less than that observed in the current study. Hence the passive reabsorption of morphine, which is driven by the concentration gradient created by the reabsorption of water, may have been higher in the current studies. Morphine is a relatively lipophilic molecule with an octanol to pH 7.4 phosphate buffer partition coefficient for the unchanged species of approximately 6 (Milne et al., 1996); hence it may be expected to be passively reabsorbed from urine into the peritubular capillary.
In conclusion, the rat IPK did not mediate the glucuronidation or oxidative metabolism of morphine. The disposition of morphine in the kidney involved tubular secretion and intracellular accumulation, both of which were unaffected by changes in morphine concentration.
Footnotes
-
Send reprint requests to: Dr. Allan M. Evans, Centre for Pharmaceutical Research, School of Pharmacy and Medical Sciences, University of South Australia, GPO Box 2471 Adelaide, South Australia, 5001.
-
↵1 This study was supported by the National Health and Medical Research Council of Australia, grant number 940330.
- Abbreviations:
- Ae090
- total amount of morphine excreted unchanged in urine between 0 and 90 min
- AUC090
- area under the perfusate morphine concentrationvs. time curve from 0 to 90 min
- AUC0∞
- area under the perfusate morphine concentration vs. time curve from time zero to infinity
- BSA
- bovine serum albumin
- CLM
- total organ clearance of morphine
- CLRM
- renal excretory clearance of morphine in a urine collection interval
- CLR(0–90)M
- renal excretory clearance of morphine from 0 to 90 min
- fu
- fraction of morphine unbound in perfusate
- IPK
- isolated perfused kidney
- M3G
- morphine-3-glucuronide
- M6G
- morphine-6-glucuronide
- %TR
- percent tubular reabsorption
- T1/2
- half-life
- UFR
- urine flow rate
- VKM
- volume of distribution of morphine within the kidney
- Vp
- perfusate volume
- Received November 18, 1996.
- Accepted May 6, 1997.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}