


Abstract
Nateglinide (NAT) stimulates insulin secretion from pancreatic β-cells by closing KATP channels. Because KATP channels are widely distributed in cardiovascular (CV) tissues, we assessed the tissue specificity of NAT by examining its effect on KATP channels in enzymatically isolated rat β-cells, rat cardiac myocytes, and smooth muscle cells from porcine coronary artery and rat aorta with the patch-clamp method. The selectivity of known antidiabetic agents glyburide (GLY) and repaglinide (REP) was also studied for comparison. NAT was found to inhibit KATP channels in the cells from porcine coronary artery and rat aorta with IC50s of 2.3 and 0.3 mM, respectively, compared with 7.4 μM in rat β-cells, indicating a respective 311- and 45-fold selectivity (p < .01) for β-cells. With an IC50 of 5.0 nM in β-cells, REP displayed an ∼16-fold (p < .05) selectivity for β-cells over both types of vascular cells. GLY was nonselective between vascular and β-cells. At equipotent concentrations (2× respective IC50s in β-cells), NAT, GLY, and REP all caused 62% reduction of pancreatic KATP current but a respective 39, 55, and 66% inhibition of cardiac KATPcurrent. These data collectively indicate that NAT, when compared with GLY and REP, at concentrations effective in stimulating insulin secretion is least likely to cause detrimental CV effects via blockade of CV KATP channels.
The maintenance of homeostatic blood glucose concentration is an integrated process predominantly regulated by the antihyperglycemic hormone insulin. When blood glucose rises, uptake of glucose into the pancreatic β-cells leads to an elevation in ATP/ADP ratio and closure of KATP channels. The closure of KATP channels and the resultant membrane depolarization lead to the increase in Ca2+influx through voltage-gated Ca2+ channels, which triggers exocytosis and insulin release (Ashcroft et al., 1984; Cook and Hales, 1984). Many agents that are capable of blocking KATP channels in pancreatic β-cells can induce insulin secretion and hence serve as antidiabetic drugs. Representatives of this class of compounds are glyburide (GLY), a sulfonylurea (SU) used for more than 30 years in the treatment of type 2 diabetes; repaglinide (REP), a meglitinide analog introduced recently (Gromada et al., 1995; Malaisse, 1995); and nateglinide (NAT, also known as A-4166), a [scap]d-phenylalanine derivative currently in phase III clinical development. All these agents have been shown to share a common mechanism of action by binding to the SU receptor (SUR) or by displacing labeled SU to inhibit KATP channels in pancreatic β-cells (Rajan et al., 1990; Akiyoshi et al., 1995; Fuhlendorff et al., 1995; Gromada et al., 1995; Fujita et al., 1996). The chemical structures of these compounds are shown in Fig. 1.

Structures of NAT, GLY, and REP.
KATP channels are ubiquitously present in a wide variety of extrapancreatic tissues including cardiac, neuronal, skeletal, and smooth muscle. Native KATP channels appear to be a complex of a regulatory protein containing the SUR and an inwardly rectifying K+ channel (Kir6.2) serving as a pore-forming subunit. Recent cloning of three isoforms of SUR (SUR1, SUR2A, and SUR2B) has led to the understanding of the molecular structure of the KATP channels in β-cells, cardiac cells, and smooth muscle cells, which are a heteromultimeric assembly of SUR1/Kir6.2, SUR2A/Kir6.2, and SUR2B/Kir6.1 (an isomer of Kir6.2), respectively (Yokoshiki et al., 1998). While inhibition of KATP channels in β-cells facilitates insulin secretion, stimulation of cardiovascular (CV) KATP channels mediates vasorelaxation and myocardial protection against ischemia (Cavero et al., 1995; Cleveland et al., 1997a; Hiraoka, 1997). Thus, drugs that cross-react with different members of the KATP channel family to block them could be therapeutic for diabetes but have the potential to cause undesired CV side effects. In fact, numerous studies reported increased risk of CV mortality in patients with type 2 diabetes treated with SU (Pogasta, 1995; Smits and Thien, 1995; Bernauer, 1997;Cleveland et al., 1997b). The mechanism of these actions has been hypothesized as the loss of the cardioprotective mechanism after blockade of KATP channels (University Group Diabetes Program, 1976, 1978; Pogasta, 1995). In contrast, the data from the recent UK Prospective Diabetes Study did not support the suggestion of adverse CV effects from SU (UKPDS, 1998). Because the results on CV actions by SU remain inconclusive, the absence of an obvious pernicious effect of SU on CV outcomes should be reassuring for skeptics (Nathan, 1995, 1998). In this regard, an ideal antidiabetic agent would be one that interacts preferentially with the KATP channels in pancreatic β-cells. In this study, the tissue selectivity of NAT was assessed and compared with those of the two known antidiabetic drugs, GLY and REP, by determining the potencies/efficacies of these three insulinotropic agents in blocking KATP channels in rat pancreatic β-cell as well as in cardiac (rat heart) and vascular [rat aorta (RA) and porcine coronary artery (PCA)] tissues. Our results indicated a favorable in vitro tissue selectivity with NAT compared with GLY and REP.
Materials and Methods
Enzymatic Isolation of Rat Pancreatic β-Cells.
Male Sprague-Dawley rats weighing 250 to 275 g were anesthetized with Na pentobarbital i.p. at 60 mg/kg before the operative procedure. Islets of Langerhans were isolated from pancreas by librase digestion (0.5 mg/ml; Boehringer Mannheim, Mannheim, Germany) followed by a Ficoll gradient centrifugation. The islets were then dissociated into single cells by treatment with protease (0.5 mg/ml, type IX; Sigma, St. Louis, MO). The buffer used in the entire isolation procedure consisted of 5 mM NaCl, 140 mM KCl, 2 mM MgCl2, 10 mM HEPES, 2 mM CaCl2, and 5 mM glucose (pH 7.4). The individual cells were seeded in Connaught Medical Research Lab medium (Life Technologies, Gaithersburg, MD) supplemented with 1% fetal calf serum, 1% antibiotic-antimycotic, and 10 mM glucose and incubated at 37°C in an atmosphere of 95% air/5% CO2 for 2 to 5 days before the electrophysiological recording. Given that the islet cell culture contained both α- and β-cells, only cells with a diameter greater than 10 μm and well-preserved granulation were used for the current recording, because β-cells were reported to have a volume usually 2- to 3-fold larger than that of α-cells (Pipeleers et al., 1985).
Enzymatic Isolation of Smooth Muscle Cells from PCA and RA.
Left circumflex and left anterior descending PCAs were dissected from porcine hearts that were obtained from a local slaughterhouse. The arteries were isolated, cleaned of fat, and cut open longitudinally. The endothelium was removed by gentle abrasion of the lumen of the tissue with a cotton swab. The media intima was peeled from the vessels and placed into preoxygenated Ca2+-free Tyrode's solution of the following composition: 140 mM NaCl, 5 mM KCl, 10 mM HEPES, 10 mM glucose, 10 mM Na pyruvate, and 2 mM EGTA.
Sprague-Dawley rats were sacrificed by induction of hypercapnia with dry ice, and the descending thoracic aortas were carefully excised. The aortas were cleaned of adhering connective tissue and cut open along the lumen. After removal of the endothelium, the media intima was dissected out and placed in the saline described above.
Single PCA and RA smooth muscle cells were prepared by digesting the muscle preparations with collagenase (1.6 mg/ml), papain (1.4 mg/ml), and dl-dithiothreitol (0.4 mg/ml) for 55 min at 36°C. Individual cells were released into the Ca2+-containing (2-mM) Tyrode's solution by gentle agitation of the digested tissue through a blunt-tipped glass pipette.
Isolation of Single Rat Cardiac Myocytes.
Sprague-Dawley rats (250–275 g) were sacrificed by i.p. injection of an overdose of Na pentobarbital (120 mg/kg). The chest was opened, and the heart was quickly excised and immersed into preoxygenated and prewarmed Ca2+- and Mg2+-free Tyrode's solution. The left ventricle of the heart was dissected and cut into small strips. The muscle strips were digested in a Ca2+- and Mg2+-free Tyrode's solution containing collagenase (2 mg/ml) and BSA (4 mg/ml) for 50 min. Single rat cardiac myocytes were released by gentle shaking in Ca2+- and Mg2+-free Tyrode's solution.
Electrophysiological Recording of KATP Currents.
The KATP currents were recorded at 22°C with the whole-cell configuration of the patch-clamp technique (Hamill et al., 1981) in the primary culture of rat β-cells, PCA and RA smooth muscle cells, and rat cardiac myocytes.
The KATP currents in β-cells and vascular smooth muscle cells were elicited by a voltage ramp ranging from −120 to +40 mV over a 1.5-s period from a holding potential of −60 mV. At very negative voltages, where the voltage-dependent K+ channels and Ca2+-activated K+ channels were all inactivated, the only remaining current component was the voltage-independent KATP current. In addition, the inward KATP current was further magnified by using high K+ (140 mM) symmetrical bath and pipette solutions to shift the K+ reversal potential from the conventional −80 to 0 mV. Thus, the KATP currents at negative potentials could be measured in the absence of interference of any other ion current components. This protocol was given to the cells under investigation every 30 s repeatedly, and the current amplitude was measured every time until maximal drug effects were achieved. To determine the concentration response, drugs were applied to the cells at concentrations in an ascending order. At each concentration, 10 to 20 min were allowed for a full development of inhibitory effect. The bath solution was composed of 5 mM NaCl, 140 mM KCl, 2 mM MgCl2, 10 mM HEPES, 2 mM CaCl2, and 5 mM glucose, pH 7.4; the pipette solution had 5 mM NaCl, 140 mM KCl, 2 mM MgCl2, 10 mM HEPES, 0.1 mM CaCl2, 0.6 mM EGTA, 2 mM Na2UDP, 2 mM (for β-cells) or 0.5 mM (for vascular cells) K2ATP, pH 7.4.
In the case of cardiac KATP currents, a positive voltage ramp to +60 mV from a holding potential of −40 mV was applied to myocytes to inhibit calcium currents. Thereafter, a negative voltage ramp from +60 to −100 mV was applied, and the resulting currents were recorded. The total duration of voltage pulses was 9 s. This protocol was given to the cells under investigation every 30 s repeatedly, and the current amplitude was measured every time until maximal drug effects were achieved.
Because the basal KATP currents were rather small in all types of cells, manipulations were adopted to induce the currents, such as intracellular dialysis of low ATP (0.5 mM) or extracellular application of the channel opener cromakalim (30 μM) in CV cells or diazoxide (30 μM) in β-cells, so that the degree of blockade of the current by the antidiabetic agents could be measured. The reasons for using different channel openers were that the cardiac and vascular KATP channels are known to be sensitive to cromakalim but to a much lesser extent to diazoxide, and the opposite was true with the pancreatic KATPchannels (Yokoshiki et al., 1998). The glucose concentration in all experiments was maintained at a physiological level of 5 mM (90 mg/dl) to mimic a normoglycemic condition.
The currents recorded were amplified by a List EPC-7 amplifier (Adams & List Assoc., Darmstadt, Germany), digitized at 4 kHz with a TL-1–125 DMA interface (Axon Instruments, Foster City, CA), and stored on a Compaq Microcomputer for later analysis with software pClamp version 6.03 (Axon Instruments). The junction potential between the electrodes and the bath solution was compensated by the d.c. offset on the amplifier. No leak subtraction was applied. Patch-clamp electrodes were pulled from Kimax-51 capillary tubes. The resistance of electrodes after fire polishing was between 3 and 5 MΩ.
Data Analysis.
The current amplitude at −90 mV (approximately 300 ms from the beginning of the voltage-ramp pulse) in β-cells and vascular smooth muscle cells and the current at +56 mV during the descending portion of the ramp pulse (approximately 3.2 s from the beginning of the voltage-ramp pulse) in cardiac myocytes were measured as indices of KATP for all quantitative analysis. Baseline currents present at the beginning of the experiments were subtracted to separate KATP currents from background currents. The residual currents after blockade by drugs at various concentrations as fractions of the maximal level induced by channel openers were used to construct sigmoidal concentration-response curves, which were fit to the logistic equation Y = 1/[1 + (X/a)b] with a general nonlinear, least-squares analysis. In the equation,X and Y were, respectively, the drug concentration and the residual current in the presence of drugs as fraction of the control. a was the IC50(defined as the concentration for a half-maximal blockade) andb was the slope coefficient. Statistical significance of the data at a given concentration was determined with ttest (single tailed). All data are expressed as means ± S.E.
Results
Inhibition of KATP Currents in β-Cells.
The effects of the antidiabetic agents NAT, GLY, and REP on KATP channel activity were examined in rat pancreatic β-cells at a physiological glucose level of 5 mM. Because the basal level of KATP currents in β-cells is usually rather low, diazoxide (100 μM), a known effective opener of the KATPchannels in insulin-secreting cells, was applied to enhance KATP currents before the addition of the channel blockers. Figure 2 illustrates typical recordings showing that NAT (0.3–300 μM), GLY (3–300 nM), and REP (1–300 nM), respectively, produced an inhibitory effect on KATPcurrents in a concentration-dependent manner. Five to seven such experiments for each drug were pooled and averaged to form concentration-response curves.

Typical records of KATP currents demonstrating a concentration-dependent inhibition by NAT (A), GLY (B), and REP (C) in rat pancreatic β-cells. The experiments were performed in 5 mM glucose. KATP currents elicited by a voltage ramp (shown at the top of each figure) in control, in 100 μM diazoxide (DIA), and in DIA with NAT, GLY, or REP at various concentrations as indicated in the figures. The currents at positive voltage are ignored, whereas those at negative voltage (indicated by an arrow) are composed of KATP currents. Dotted lines indicate the zero-current level. The amplitude of currents at −90 mV was measured as an index to perform the quantitative analysis.
The kinetics of the effects on KATP channels by these agents differed considerably. The effect by NAT had a rapid onset and was largely or completely reversed shortly after the drug was removed. In contrast, the duration of KATPchannel-blocking action by GLY and REP was long lasting. This was especially true with REP, whose action often outlasted the duration of drug presence by severalfold. In most cases, a complete recovery from REP effect on its removal was not seen within 3 h, a result in agreement with a previous report (Gromada et al., 1995). A summary ofT1/2 on (the time required to reach a half-maximal inhibition) and T1/2 off (the time required to obtain a 50% recovery from the maximal inhibition after withdrawal of drugs) for all three drugs at equipotent concentrations (2 × respective IC50s) is shown in Table 1. TheT1/2 off values of GLY and REP, being two and five times that of NAT, suggest that most KATP channels remained at a closed state long after the drugs were removed.
Time course of KATP channel-blocking effect
Inhibition of KATP Currents in Vascular Smooth Muscle Cells.
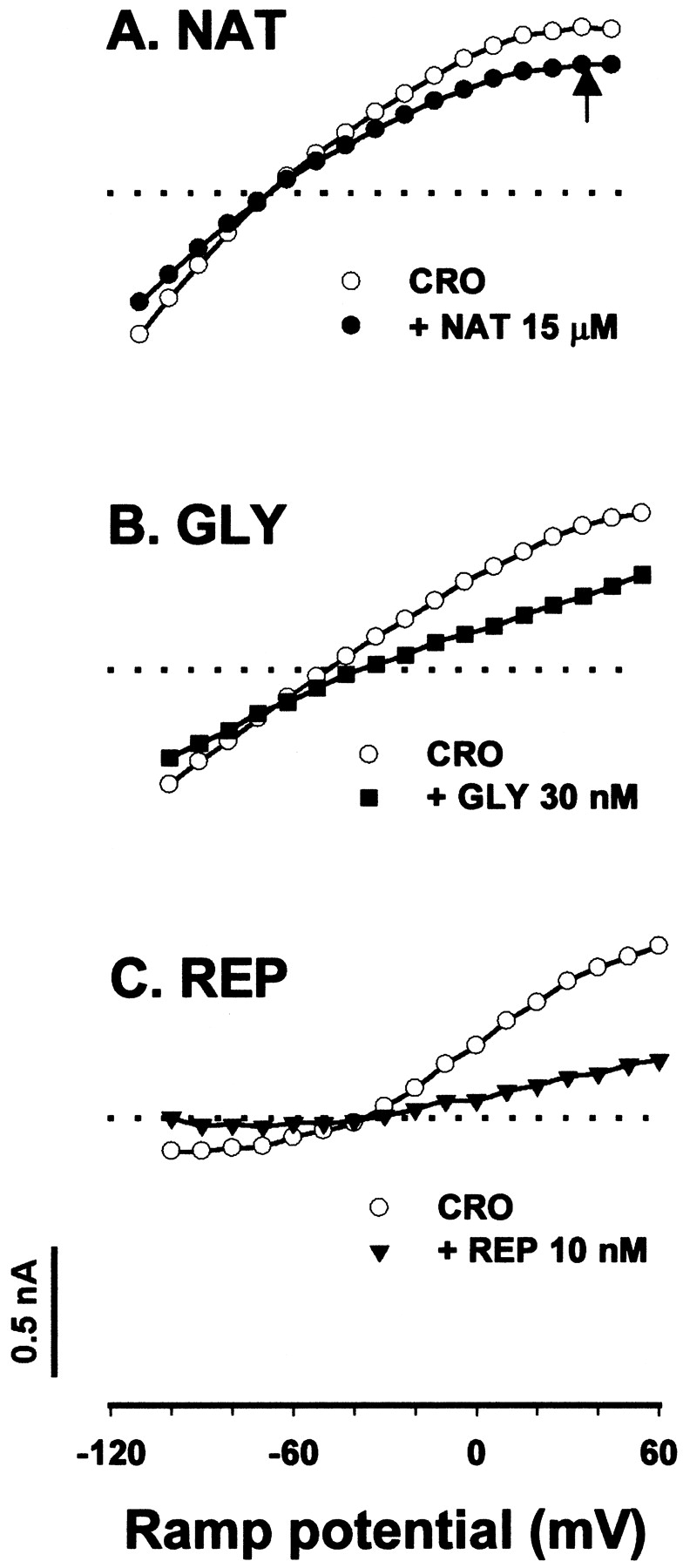
The instantaneous current-voltage relationship of the KATP current in vascular smooth muscle cells was measured with a ramp voltage protocol ascending from −120 to +40 mV. The basal KATP current was increased by intracellular dialysis of a pipette solution containing 0.5 mM ATP supplemented with an extracellular application of 30 μM cromakalim. Figure3 shows, respectively, representative recordings of the KATP currents from single RA smooth muscle cells in the presence of NAT (3μM to 1 mM), GLY (1 nM to 1 μM), and REP (1 nM to 1 μM). All these compounds were able to inhibit the KATP current in a concentration-dependent manner. Similarly, all three antidiabetic agents inhibited KATP currents in PCA smooth muscle cells (data not shown). Unlike in β-cells, the actions of all drugs tested in vascular tissues had a rapid onset, which was comparable with that with NAT in β-cells, and were readily reversible on withdrawal of drugs.

Typical records of KATP currents demonstrating a concentration-dependent inhibition by NAT (A), GLY (B), and REP (C) in RA smooth muscle cells. The experiments were performed in 5 mM glucose. Basal KATP currents were negligibly small and are therefore not displayed. Traces are the KATPcurrents elicited by a voltage ramp (shown at the top of each figure) in 30 μM cromakalim (CRO) and in cromakalim with NAT, GLY, or REP at various concentrations as indicated in the figures. The currents at positive voltage are ignored, whereas those at negative voltage (indicated by an arrow) are composed of KATP currents. Dotted lines indicate the zero-current level. The amplitude of currents at −90 mV was measured as an index to perform the quantitative analysis.
Concentration Response of KATP Channel-Blocking Actions.
Concentration-response curves of KATPchannel-blocking action were constructed by sigmoidal fitting with a least-squares analysis of the data obtained from β-cells and vascular smooth muscle cells (Fig. 4). The IC50s of all drugs in β-cells and vascular cells are summarized in Tables 2 and3, in which a comparison of the drug potencies in PCA cells versus β-cells or RA cells versus β-cells was made and indexed by the ratios of the IC50s. The data in the tables show: 1) NAT was, in general, a less potent KATP channel blocker than GLY and REP in β-cells and vascular smooth muscle cells; 2) GLY inhibited KATPchannels in all three tissues with similar potencies (16.6, 46.8, and 38.8 nM, respectively, in β-cell, PCA, and RA cells); 3) the potency of NAT in PCA and RA cells was reduced by 311- and 45-fold, respectively, from that in β-cells; and 4) the potency of REP in PCA and RA cells was reduced by 17- and 16-fold, respectively, from that in β-cells. Thus, GLY was a nonselective KATP channel blocker, whereas REP and NAT, to a greater extent, preferentially inhibited the channels in β-cells.

Concentration-response curves for the inhibition of KATP currents in rat β-cells (●) and in smooth muscle cells from RA (▴) and PCA (▪) by NAT (top), GLY (middle), and REP (bottom). The points are mean amplitude of KATP currents at −90 mV from five to seven experiments. The ordinates are the relative KATP currents as fraction of the maximal values activated by diazoxide or cromakalim, and the abscissas indicate the concentration of the drugs (M) in a logarithmic scale. The slope coefficients of the curves are 0.7, 0.6, and 0.4, respectively, for β-cell, RA, and PCA with NAT (top); 0.9, 0.4, and 0.5 with GLY (middle); and 0.7, 0.7, and 0.5 with REP (bottom).
IC50s of inhibition of KATP in porcine artery cells versus β-cells
IC50s of inhibition of KATP in aorta cells versus β-cells
Inhibition of KATP Current in Rat Cardiac Myocytes.
Because of the instability and rapid rundown of the KATP current in rat cardiac myocytes, we have not had much success in experimenting a full range of concentration response in an individual cell. As an alternative, the efficacy of these antidiabetic compounds at an equipotent concentration (approximately 2-fold of respective IC50s in β-cells) on cardiac myocytes was determined. Figure 5 shows typical recordings of the inhibitory effect of NAT (15 μM), GLY (30 nM), and REP (10 nM) on cromakalim-induced KATP currents in cardiac myocytes. At these equipotent concentrations for β-cells, NAT, GLY, and REP, all inducing 62% reduction of pancreatic KATPcurrents, caused 39 ± 2, 55 ± 4, and 66 ± 10% inhibition of the cardiac KATP current (n = 4–6), respectively. REP appeared to be the most effective blocker of the cardiac KATP currents. The onset and washout of the effect by all three drugs in cardiac myocytes were relatively fast and comparable with those of NAT in pancreatic β-cells but considerably more rapid than those of GLY and REP in β-cells.

Inhibition of KATP currents in rat cardiac myocytes by NAT (15 μM; A), GLY (30 nM; B), and REP (10 nM; C) at equipotent concentrations in β-cells. The experiments were performed in 5 mM glucose. Typical records of cromakalim-induced KATP currents in response of a voltage ramp, whose scale is shown at the bottom of the figure. The currents at +56 mV (indicated by the arrow) were used as an index to calculate percent inhibition. Dotted lines indicate the zero-current level. Currents in each panel are recorded from a same cell.
To depict the differential ability of NAT, GLY, and REP to block the KATP currents in β-cells and all CV tissues studied, the percentage inhibition of the currents by drugs at concentrations 2-fold of the respective IC50s in β-cells was collectively illustrated in Fig.6. The data of β-cells and vascular cells at this particular concentration were extrapolated from the concentration-response curves (Fig. 4), because, in the experiments determining the IC50s in these cell types, the drug concentrations were chosen evenly over a range in an ascending order but did not necessarily include the concentration of 2× IC50s. At this fixed concentration, NAT, GLY, and REP, being equally effective with a 62% blockade of the β-cell KATP channels, caused a respective 15, 44, and 25% reduction in the KATP currents in PCA cells, a 13, 47, and 20% inhibition in RA cells, and 39, 55, and 66% inhibition in rat cardiac cells.

Summary of percent inhibition of KATPchannels by NAT (15 μM), GLY (30 nM), and REP (10 nM) in rat β-cells (empty column), smooth muscle cells from RA (grid columns), and PCA (filled columns), and rat cardiac myocytes (hatched columns). The chosen concentrations of the drugs were approximately twice their respective IC50s in β-cells. Ordinate denotes percent inhibition at the fixed concentration for each drug. The data of β-cells and vascular cells at this fixed concentration were extrapolated from the concentration-response curves in Fig. 4, whereas the data of cardiac myocytes were measured directly.
Discussion
We investigated, at the molecular level, the tissue selectivity of the antidiabetic drugs NAT, GLY, and REP to assess the potential for the occurrence of adverse CV activities. KATPchannels are not only present in β-cells of endocrine pancreas but also with high density in extrapancreatic cell types such as the smooth muscle cells of vascular systems and the cardiac muscle cells. Whereas the principal role of KATP channels in β-cells is to link blood glucose and insulin secretion (Ashcroft and Rorsman, 1989), the roles in extrapancreatic tissues are less well characterized. It is likely that the channels in the heart open in response to metabolic stress, as occurs during cardiac ischemia (Nichols and Lederer, 1991). KATP channels are also important in the control of vascular tone and therefore of blood pressure (Quayle et al., 1997). Given that all three drugs tested share a common mechanism of action—blockade of KATP channels—their potencies/efficacies on the channels in β-cells and CV cells have been determined and used as indices to evaluate the tissue selectivity.
Our concern for the physiological relevance of the study was reflected in the design of the experiments. The studies were directed on KATP current from freshly isolated and fully metabolically viable cells instead of their cloned counterparts SUR1/Kir6.2 and SUR2A/Kir6.2 or SUR2B/Kir6.1. To preserve cell integrity and intracellular nucleotide, whole-cell KATP current of intact β-cells and CV cells were investigated instead of single KATP channel activity from a detached membrane patch, which is seemingly less physiological.
Our data regarding the KATP channels in native β-cells and CV cells in response to the known antidiabetic drug GLY are generally consistent with those in the literature. The IC50 of 16.6 nM in adult rat β-cells from our study was comparable with the reported 5 nM in the same cell type (Gillis et al., 1989), 27 nM in CRI-G1 insulinoma cells (Sturgess et al., 1988), and 0.4 to 0.6 nM in mouse β-cells (Zunkler et al., 1988; Panten et al., 1989). However, a considerably lower value of 47 pM has been reported in newborn rat β-cells (Gromada et al., 1995). The IC50s of 38.8 and 46.8 nM in aorta and coronary artery cells, respectively, were well within the range of 20 to 100 nM reported for arterial cells (Xu and Lee, 1994; Nelson and Quayle, 1995; Quayle et al., 1995). Based on the observed 54.6 ± 4.1% inhibition of the KATP channels in rat cardiac cells by 30 nM GLY, the IC50 should be lower than 30 nM, which was reasonably close to the reported 6 to 9 nM (Findlay, 1992; Krause et al., 1995).
Our results showed that the concentration-response curve of GLY from pancreatic β-cells largely overlapped those from arterial vascular cells (Fig. 4), confirming a poor tissue selectivity of the drug. In contrast, the concentration-response curves for both NAT and REP effects on vascular smooth muscle cells had a significant rightward shift from their respective ones with the β-cells. The greater magnitude of shift with NAT than with REP in PCA (311- versus 17-fold) and RA (45- versus 16-fold) clearly suggested a more preferred in vitro tissue specificity with NAT as an antidiabetic drug. In cardiac myocytes, NAT, GLY, and REP at equally effective concentrations (2 × IC50s) in β-cells blocked cardiac KATP channels with an efficacy in the order of REP 66% > GLY 55% > NAT 39%. Thus, NAT at therapeutic concentrations produced an overall weaker inhibitory effect on KATP channels in CV tissues than GLY and REP did. Although in vitro results at cellular or molecular levels are indicative of tissue selectivity of drugs, cautions are necessary to directly extrapolate these results for in vivo selectivity, because multiple signaling pathways coexist and react to the drugs in the whole body.
KATP channels are formed of a pore-forming subunit, Kir6.2, and a SUR, which coassemble in a 4:4 stoichiometry. Among the two genes encoding SUR (SUR1 and SUR2), SUR1 serves as the regulatory subunit of β-cell KATP channels (Aguilar-Bryan et al., 1995), and the splice variants of SUR2, SUR2A and SUR2B, act, respectively, as the cardiac and vascular SUR subunits (Chutkow et al., 1996; Inagaki et al., 1996). Native KATP channels in various tissues exhibit different sensitivities to SU and KATP channel openers (Inagaki et al., 1996). These differences in pharmacological profile are believed to be conferred by the distinct intrinsic properties of different isoforms/subunits of SUR (Gribble et al., 1998;Yokoshiki et al., 1998). While the molecular mechanism(s) underlying the differential activities of these antidiabetic agents in different tissue types remain to be elucidated, the distinct molecular structure of the KATP channel protein (especially SUR subunit) present in each preparation and their differential interaction with each drug are likely to underlie the varying tissue selectivity of these drugs.
The time course of in vivo hypoglycemic effect of an antidiabetic agent may largely depend on the intrinsic characteristics of the mechanism of action as well as its pharmacodynamic profile. The observed mechanism-based rapid reversibility of the effect of NAT and the slower recovery of the action of GLY and REP in rat pancreatic β-cells may be important factors that contribute to the differential duration of in vivo hypoglycemic action of these compounds. Although GLY and REP are capable of blocking both β-cell and CV KATP channels, the mechanism of this blockade appears to be different, because the inhibition was hard to reverse in β-cells in this and other studies (Gromada et al., 1995) but was readily reversible in the CV preparations. Similar observations with GLY have been reported between their cloned counterparts SUR1/Kir6.2 (pancreatic type) and SUR2A/Kir6.2 (cardiac type; Gribble et al., 1998). These observations might be accounted for by the speculation that GLY and REP bind to SUR1 at two sites (i.e., a tolbutamine and benzamino site) but only a single site (benzamino) on SUR2A/SUR2B (Gribble et al., 1998). If this were the case, then both halves of the drug molecule would need to dissociate from SUR1 simultaneously to reverse their effect, which is likely to occur with a low probability or long time lag. On the other hand, unbinding from SUR2A/SUR2B would take place more readily, because the drugs need to dissociate only from a single site. In the case with NAT, the washout times in both β-cell and CV KATP channels were comparable and shorter than with GLY and REP in β-cells, which may suggest a single binding site for NAT in all these tissues. Future studies are required to validate these hypotheses.
The clinical relevance of this study is obvious, because millions of type 2 diabetic patients who are treated with these drugs are in the age group where CV risks are extremely high. Given that KATP channels constitute an important cardioprotective mechanism against ischemic damage (Cavero et al., 1995; Cleveland et al., 1997b; Hiraoka, 1997), closure of CV KATP channels by antidiabetic drugs could reduce coronary blood flow and/or exacerbate ischemia-induced myocardial damage either by direct action or through diminished cardioprotective preconditioning. The lack of selectivity with GLY between CV and pancreatic β-cells observed in this study seems to be in line with the reported increased incidence of CV diseases in type 2 diabetic patients treated with SU drugs (Pogasta, 1995; Smits and Thien, 1995;Leibowitz and Cerasi, 1996; Bernauer 1997; Cleveland et al., 1997b). Recent data from the UK Prospective Diabetes Study (1998), however, did not support the suggestion of adverse CV effect from SU. Because these data disputed most of the earlier studies, a consensus over the CV action by SU is yet to be reached (Nathan 1995, 1998). Whereas the data on clinical evaluation of NAT and REP with respect to their CV actions are currently unavailable, our data showing an overall higher in vitro selectivity for endocrine pancreas over CV preparations with NAT than with GLY and REP may warrant the application of NAT as an antidiabetic agent with a relatively lower likelihood of adverse CV activities.
Footnotes
-
Send reprint requests to: Shiling Hu, Metabolic and Cardiovascular Diseases, Novartis Institute for Biomedical Research, 556 Morris Av., Summit, NJ 07901-1027. E-mail:hiling.hu{at}pharma.novartis.com
- Abbreviations:
- GLY
- glyburide
- NAT
- nateglinide
- REP
- repaglinide
- CV
- cardiovascular
- SU
- sulfonylurea
- SUR
- sulfonylurea receptor
- RA
- rat aorta
- PCA
- porcine coronary artery
- Received June 16, 1999.
- Accepted August 23, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}