


Abstract
Human organic cation transporters (hOCTs) are expressed in organs of drug absorption and elimination and play an important role in the uptake and elimination of xenobiotics. The purpose of this study was to evaluate the substrate and inhibitory activity of the H2-receptor antagonists ranitidine and famotidine toward hOCTs and to determine the hOCT isoforms involved in the absorption and elimination of these compounds in humans. Inhibition and substrate specificity of hOCT1, hOCT2, and hOCT3 for ranitidine and famotidine were elucidated in cRNA-injected Xenopus laevis oocytes. Ranitidine and famotidine exhibited similarly potent inhibition of [3H]1-methyl-4-phenyl pyridinium uptake into hOCT1-expressing (IC50 = 33 and 28 μM, respectively) and hOCT2-expressing oocytes (IC50 = 76 and 114 μM, respectively). Famotidine exhibited potent inhibition of hOCT3; in contrast, ranitidine was a moderately weak inhibitor (IC50 = 6.7 and 290 μM, respectively). [3H]Ranitidine uptake was stimulated by hOCT1 (Km = 70 ± 9 μM) and to a much smaller extent by hOCT2. No stimulation of [3H]ranitidine uptake was observed in hOCT3-expressing oocytes. trans-Stimulation and electrophysiology studies suggested that famotidine also is an hOCT1 substrate and exhibits poor or no substrate activity toward hOCT2 and hOCT3. Thus, hOCT1, which is expressed in the intestine and liver, is likely to play a major role in the intestinal absorption and hepatic disposition of ranitidine and famotidine in humans, whereas hOCT2, the major isoform present in the kidney, may play only a minor role in their renal elimination. Famotidine seems to be one of the most potent inhibitors of hOCT3 yet identified.
The H2-receptor antagonists ranitidine and famotidine (Fig. 1) represent a class of weakly basic hydrophilic compounds that exist as partially charged organic cations at physiological pH values. Despite their hydrophilic nature, both ranitidine and famotidine are relatively well absorbed in humans and exhibit oral bioavailability of 40 to 70% (van Hecken et al., 1982; Yeh et al., 1987). They are primarily eliminated unchanged via renal excretion (∼70%) with minor contributions from hepatic metabolism (∼20–30%) and intestinal secretion (Lin, 1991; Gramatte et al., 1994). Biliary excretion of ranitidine and famotidine is insignificant in both rats and humans (Klotz and Walker, 1990; Suttle and Brouwer, 1994). Interestingly, the renal clearance of ranitidine and famotidine greatly exceeds the glomerular filtration rate, suggesting that the kidney actively secretes these compounds into urine (McNeil et al., 1981; Kroemer and Klotz, 1987). Ranitidine also significantly reduces the renal clearance of the organic cations procainamide (Somogyi and Bochner, 1984) and triamterene (Muirhead et al., 1988) in humans, presumably via competition for renal tubular secretion pathways. Such observations clearly suggest the importance of membrane transport processes in the pharmacokinetic behavior of these compounds in humans. However, specific transporters involved in the absorption and elimination of these widely prescribed and extensively used over-the-counter drugs have not been conclusively identified.
Human organic cation transporters (hOCTs) represent a family of recently cloned candidate transporters likely to be involved in the disposition of small hydrophilic organic cations, such as ranitidine and famotidine. hOCT1 (SLC22A1) is predominantly expressed at the sinusoidal membrane of hepatocytes and mediates uptake of organic cations from the blood to the liver (Gorboulev et al., 1997; Zhang et al., 1997). hOCT2 (SLC22A2) is predominantly expressed at the basolateral (BL) membrane of renal proximal tubule cells and mediates the initial step in the renal excretion of organic cations (Motohashi et al., 2002). hOCT3 (SLC22A3) is expressed in a wide range of tissues, although highest expression has been documented in the liver, placenta, kidney, and skeletal muscle (Wu et al., 2000). The subcellular localization of hOCT3 to the apical or BL membrane, however, has not been demonstrated. OCTs mediate substrate transport via a uniport mechanism driven by the inside negative membrane potential, and they also can support electroneutral organic cation exchange via an antiport mechanism (Busch et al., 1996; Zhang et al., 1999). Typical substrates of the human OCT isoforms include small quaternary ammonium organic cations, such as tetraethylammonium (TEA) and 1-methyl-4-phenyl pyridinium (MPP+) as well as endogenous amines such as dopamine, serotonin, and agmatine. A large number of cationic therapeutic agents have also been identified as inhibitors of human OCT family proteins; however, relatively few have been identified as substrates. Thus, the influence of OCT-mediated transport for uptake into human barrier and excretory tissues, such as liver, kidney, and intestine, has not been elucidated for many cationic drugs.

Structures of the H2-receptor antagonists ranitidine and famotidine.
Although the relevance of hOCTs to renal and hepatic disposition has been relatively well established, their potential role in intestinal transport is less well defined. hOCT1 mRNA expression has been reported in human small intestine (Zhang et al., 1999), and mRNA expression of hOCT1, hOCT2, and hOCT3 has been demonstrated in the Caco-2 cell model of intestinal epithelium (Bleasby et al., 2000; Martel et al., 2001; Hayer-Zillgen et al., 2002). The function of these transporters in the intestine or Caco-2 cells, however, is unclear. We have previously observed saturable and inhibitable transport behavior of ranitidine and famotidine across Caco-2 cell monolayers (Lee and Thakker, 1999). Furthermore, the apical uptake of ranitidine in Caco-2 cells is saturable, membrane potential-dependent, and inhibited by a broad range of organic cations, including TEA and MPP+, suggesting that transporters for organic cations (e.g., OCTs) may be involved in the intestinal absorption of ranitidine (Bourdet and Thakker, 2003). Despite functional evidence for involvement of OCTs in their transport across Caco-2 cells, the substrate specificity and isoform selectivity of ranitidine and famotidine for individual hOCTs have not been elucidated.
The primary objective of this study was to evaluate the substrate and inhibitory activity of ranitidine and famotidine toward the human organic cation transporters hOCT1, hOCT2, and hOCT3. Thus, we examined the ability of ranitidine and famotidine to serve as both inhibitors and substrates of each OCT isoform expressed in Xenopus laevis oocytes. Ranitidine and famotidine exhibited similar inhibitory potencies toward hOCT1 and hOCT2; however, each displayed striking differences in their potency toward hOCT3. The H2-receptor antagonists were substrates of hOCT1, but they exhibited little or no substrate activity toward hOCT2 and hOCT3. This report highlights the differential handling of hydrophilic organic cations by human organic cation transporter isoforms and provides a mechanistic basis for potential drug interactions involving ranitidine and famotidine in their absorption, hepatic uptake, and renal excretion in humans.
Materials and Methods
Materials. [3H]MPP+ (85 Ci/mmol) was obtained from American Radiolabeled Chemicals (St. Louis, MO). [3H]Ranitidine (7 Ci/mmol) was obtained as a gift from GlaxoSmithKline (Research Triangle Park, NC) and originally custom-synthesized by GE Healthcare (Little Chalfont, Buckinghamshire, UK). [3H]Ranitidine was purified immediately before experiments by reverse-phase-high-performance liquid chromatography, and radiochemical purity exceeded 95% as verified by radio-high-performance liquid chromatography detection (Flow Scintillation Analyzer 500TR Series; PerkinElmer Life and Analytical Sciences, Boston, MA). TEA, quinidine, and metformin were obtained from Sigma-Aldrich (St. Louis, MO). Famotidine was purchased from ICN Biomedicals Inc. (Aurora, OH). Unlabeled ranitidine was purchased from Sigma/RBI (Natick, MA). hOCT1 cDNA was provided by Dr. Kathleen Giacomini (University of California, San Francisco, CA) cloned in the pEXO vector. hOCT2 cDNA was provided by Dr. John Pritchard (National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC) cloned in the pcDNA3.1/V5-His-TOPO vector (Invitrogen, Carlsbad, CA). hOCT3 cDNA was provided by Dr. Vadivel Ganapathy (Medical College of Georgia, Augusta, GA) cloned in the pSPORT1 vector (Invitrogen).
cRNA Synthesis. Capped hOCT1, hOCT2, and hOCT3 cRNA was synthesized after linearization of the pEXO (hOCT1), pcDNA3.1/V5-His-TOPO (hOCT2), and pSPORT1 (hOCT3) plasmids downstream of the cDNA inserts with BamH1 (hOCT1 and hOCT3) and Pme1 (hOCT2), respectively. Transcription was carried out with T7 RNA Polymerase using mMESSAGE mMACHINE in vitro transcription kit (Ambion, Austin, TX). RNA yield and concentration were determined by UV absorbance at 260 nm on a Beckman DU 640 UV spectrophotometer (Beckman Coulter, Fullerton, CA).
Oocyte Isolation and Injection. Adult female X. laevis were cooled in an ice bath, anesthetized by immersion in tricaine methanesulfonate, pithed, and decapitated. All animal procedures were conducted according to protocols approved by the National Institute of Environmental Health Sciences Animal Care and Use Committee. Mature stage V and VI oocytes were isolated and defolliculated by treatment with collagenase A (Roche Diagnostics, Indianapolis, IN). Oocytes were maintained at 18°C in oocyte Ringer's (OR-2) buffer (82.5 mM NaCl, 2.5 mM KCl, 1 mM Na2HPO4, 3 mM NaOH, 1 mM CaCl2, 1 mM MgCl2, 1 mM pyruvic acid, and 5 mM HEPES, pH 7.6) supplemented with 0.05 mg/ml gentamicin sulfate, 1.5 mM sodium pyruvate, and 5% heat-inactivated horse serum. Oocytes were injected with 20 ng of hOCT1 or hOCT2 cRNA and 50 ng of hOCT3 cRNA 1 or 2 days following isolation. Oocytes injected with water (16.1 nl) served as controls.
Oocyte Uptake Studies. Uptake of 1 μM [3H]MPP+ (1 or 2 μCi/ml) or [3H]ranitidine (0.5 μCi/ml) was evaluated at various time points in OR-2 buffer at room temperature 3 days after injection of hOCT cRNA. Inhibition or ranitidine uptake studies included the indicated concentrations of unlabeled inhibitors or cold substrate. Uptake was arrested by aspiration of the uptake solution followed by washing of the oocytes three times with 2 ml of ice-cold OR-2 buffer. Individual oocytes were dissolved in 300 μl of 1 N NaOH and neutralized with 300 μl of 1 N HCl, and the radioactivity associated with each oocyte was determined by liquid scintillation counting using a Packard 1600TR liquid scintillation counter (PerkinElmer Life and Analytical Sciences).
trans-Stimulation Studies. Oocytes were preloaded for 2 h with 1 μM [3H]MPP+ (1 μCi/ml). After loading, the uptake solution was aspirated, and oocytes were washed three times with 2 ml of ice-cold OR-2 buffer. The OR-2 wash solution was replaced with OR-2 buffer or a high K+ OR-2 buffer (82.5 mM KCl, 2.5 mM NaCl, 1 mM Na2HPO4, 3 mM NaOH, 1 mM CaCl2, 1 mM MgCl2, 1 mM pyruvic acid, and 5 mM HEPES, pH 7.6) containing ranitidine (1 mM), famotidine (1 mM), or TEA (1 mM), and preloaded [3H]MPP+ was allowed to efflux at room temperature for 1 h. Radioactivity in the efflux buffer and individual oocytes was determined by liquid scintillation counting using a Packard 1600TR liquid scintillation counter. Individual oocytes were dissolved in 300 μl of 1 N NaOH and subsequently neutralized with 300 μl of 1 N HCl before liquid scintillation counting. The percentage of [3H]MPP+ effluxed from the oocytes was calculated from the amount in the efflux medium and remaining intracellular [3H]MPP+.
Electrophysiology Studies. Electrophysiology studies were carried out at room temperature 5 to 7 days postinjection as described previously (Aslamkhan et al., 2003). In brief, water-, hOCT1-, hOCT2-, or hOCT3-injected oocytes were voltage-clamped at –60 mV and superfused with OR-2 buffer or OR-2 buffer containing the test compounds. Compound applications were followed by washout with OR-2 buffer before application of the next compound. The inward electrical currents induced by application of potential substrates to the oocytes was measured and recorded. Electrophysiology recordings were acquired and analyzed using the pClamp 9.0 software acquisition package (Molecular Devices, Sunnyvale, CA).
Data Analysis. Values are expressed as mean ± S.E. Statistical significance was evaluated using unpaired t tests with p < 0.05 considered statistically significant. Kinetic constants (Jmax and Km) for transporter-mediated [3H]ranitidine uptake were obtained by fitting the Michaelis-Menten model to the ranitidine uptake data. Inhibition of transporter-mediated uptake was determined by subtraction of the uptake observed in water-injected oocytes from that observed in transporter-injected oocytes. IC50 values were determined by fitting a sigmoidal inhibition model to the inhibition data using the following equation: V = Vo/[1 + (I/IC50)n], where V is the uptake rate of [3H]MPP+ in the presence of inhibitor, Vo is the uptake rate of [3H]MPP+ in the absence of inhibitor, I is the concentration of inhibitor, and n is the Hill coefficient. WinNonlin (Pharsight, Mountain View, CA) was used for estimation of kinetic constants and IC50 values. Uptake clearance (CLup) of [3H]ranitidine and [3H]MPP+ was determined from the rate of uptake into the oocytes divided by the initial extracellular concentration. Rate of uptake for [3H]ranitidine and [3H]MPP+ was linear over a 30-min time course. Normalization of [3H]ranitidine CLup to [3H]MPP+ CLup was accomplished by dividing the [3H]ranitidine CLup by the [3H]MPP+ CLup.
Results
Functional Expression of hOCT1, hOCT2, and hOCT3 inX. laevisOocytes. Uptake of [3H]MPP+ (1 μM), a model substrate for hOCT1, hOCT2, and hOCT3, was examined in oocytes injected with water or with hOCT1, hOCT2, or hOCT3 cRNA as a function of time. Uptake of [3H]MPP+ in oocytes injected with hOCT1, hOCT2, or hOCT3 cRNA was significantly greater at each time point than in oocytes injected with water (Fig. 2). Uptake of [3H]MPP+ was linear over the time course investigated (0–60 min) for each transporter. The significantly increased uptake of the OCT substrate [3H]MPP+ in transporter cRNA-injected oocytes demonstrates functional expression of the transport proteins on the oocyte cell membrane.
Inhibition of hOCT1, hOCT2, and hOCT3 by Ranitidine and Famotidine inX. laevisOocytes. Interaction of ranitidine and famotidine with human organic cation transporters was probed by evaluating their ability to inhibit uptake of the model OCT substrate MPP+. Additional organic cations were examined based on their expected inhibitory potency toward human organic cation transporters [quinidine (strong), TEA (moderate), and metformin (weak)] (Zhang et al., 1998; Dresser et al., 2002) to provide comparison with ranitidine and famotidine. Uptake of [3H]MPP+ into hOCT1-, hOCT2-, and hOCT3-expressing oocytes was inhibited in a concentration-dependent manner by each of the investigated organic cations (Fig. 3). The potency of inhibition, as measured by IC50 values, is displayed in Table 1. As expected, quinidine was a relatively potent inhibitor of all three hOCT isoforms (IC50 < 20 μM), whereas TEA and metformin were decidedly less potent. TEA exhibited significantly weaker affinity for hOCT3 than for hOCT1 and hOCT2, as has been observed previously (Wu et al., 2000). The H2-receptor antagonists ranitidine and famotidine inhibited hOCT1 and hOCT2 with potency that was ∼7-fold (hOCT1) and 2- to 3-fold (hOCT2) greater than that of TEA and 5- to 15-fold less than that of quinidine. Inhibition potency of ranitidine and famotidine for hOCT1 was similar (IC50 of ∼30 μM). Inhibition potency of ranitidine and famotidine for hOCT2 also was similar (IC50 of ∼75–100 μM). Ranitidine and famotidine thus displayed approximately 2- to 3-fold greater inhibition potency for hOCT1 than for hOCT2. Interestingly, ranitidine was a significantly less potent inhibitor of hOCT3 than of hOCT1 and hOCT2, whereas famotidine was a significantly more potent inhibitor of hOCT3 than the other two hOCTs (Table 1). In fact, famotidine was almost a 3-fold more potent inhibitor of hOCT3 than quinidine (Table 1) (famotidine IC50 = 6.7 ± 2.0 μM) and may be one of the most potent hOCT3 inhibitors identified to date.
IC50 values for inhibition of hOCT1-, hOCT2-, or hOCT3-mediated [3H]MPP+ uptake in X. laevis oocytes
IC50 values were determined from three separate experiments, and they represent mean ± S.E.

Functional expression of hOCT1, hOCT2, and hOCT3 in X. laevis oocytes. Water-injected or hOCT1-, hOCT2-, and hOCT3-expressing oocytes were incubated with [3H]MPP+ (1 μM) for the indicated time. Data represent mean ± S.E. from a representative experiment. Experiments were repeated in oocytes from multiple frogs. Six to eight oocytes were used per time point.
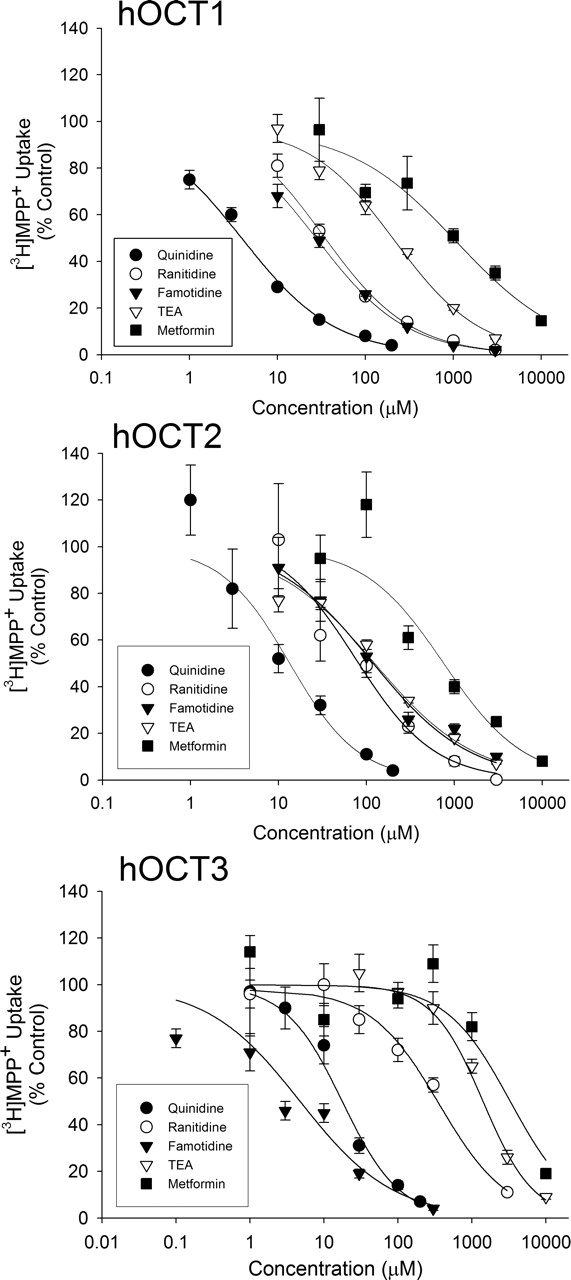
Concentration-dependent inhibition of hOCT1, hOCT2, and hOCT3 in X. laevis oocytes by organic cations. Uptake of [3H]MPP+ in water-injected or hOCT1-, hOCT2-, and hOCT3-expressing oocytes was determined in the absence or presence of increasing concentrations of quinidine (•), ranitidine (○), famotidine (▾), TEA (▿), and metformin (▪) for 45 min. Data represent inhibition of the transporter-mediated portion of [3H]MPP+ uptake. Data are expressed as mean ± S.E. of experiments containing six to eight oocytes per data point.
Uptake of [3H]Ranitidine by hOCT1-, hOCT2-, and hOCT3-ExpressingX. laevisOocytes. Numerous organic cations exhibit inhibition of organic cation transporters, yet they are not transported substrates. To determine whether ranitidine is a substrate of human organic cation transporters, uptake of [3H]ranitidine (10 μM) was evaluated in water-injected and hOCT1-, hOCT2-, and hOCT3-expressing oocytes. Uptake of [3H]ranitidine was ∼50-fold greater in hOCT1-expressing oocytes compared with water-injected controls (Fig. 4A). In contrast, uptake of [3H]ranitidine was only ∼2-fold greater in hOCT2-expressing oocytes compared with water-injected controls (Fig. 4A). Uptake of [3H]ranitidine was similar in hOCT3-expressing oocytes and water-injected controls, suggesting that ranitidine is not a substrate of hOCT3 (Fig. 4A). Because variability in transporter expression renders a direct comparison between hOCT1- and hOCT2-mediated [3H]ranitidine transport difficult, [3H]ranitidine uptake was normalized relative to the uptake of [3H]MPP+ in the same batch of oocytes. Efficiency of uptake was expressed as CLup, as described under “Data Analysis”. Reported Km values of MPP+ for hOCT1 and hOCT2 are 15 and 19 μM, respectively (Gorboulev et al., 1997; Zhang et al., 1997). Thus, uptake of 1 μM [3H]MPP+ ([S] ≪ Km) should reflect the intrinsic efficiency (Vmax/Km) of the uptake process for both hOCT1 and hOCT2 and account for differences in transporter expression between hOCT1- and hOCT2-expressing oocytes. The CLup of [3H]ranitidine in hOCT1-expressing oocytes was ∼9-fold greater in hOCT1-expressing oocytes than in hOCT2-expressing oocytes when normalized to the CLup of [3H]MPP+ (Fig. 4B). The results indicate that hOCT1-mediated [3H]ranitidine uptake is ∼10% as efficient as [3H]MPP+ uptake. hOCT2-mediated [3H]ranitidine uptake, however, is only approximately 1% as efficient as hOCT2-mediated [3H]MPP+ uptake (Fig. 4B). The results clearly demonstrate that ranitidine is a substrate of hOCT1 and hOCT2, although the efficiency of translocation by hOCT2 is poor in comparison with hOCT1.

Uptake of [3H]ranitidine by hOCT1-, hOCT2-, and hOCT3-expressing X. laevis oocytes. A, uptake of [3H]ranitidine (10 μM) was determined in water-injected or hOCT1-, hOCT2-, and hOCT3-expressing oocytes. B, comparison of [3H]ranitidine uptake efficiency by hOCT1 and hOCT2. CLup of [3H]ranitidine (10 μM) was determined in hOCT1- and hOCT2-expressing oocytes and normalized to the CLup determined for [3H]MPP+ (1 μM) in the same batch of oocytes. CLup for [3H]ranitidine in hOCT1- and hOCT2-expressing oocytes was 1.34 ± 0.07 and 0.06 ± 0.01 μl/h/oocyte, respectively. CLup for [3H]MPP+ in hOCT1- and hOCT2-expressing oocytes was 10.86 ± 0.33 and 4.11 ± 0.35 μl/h/oocyte, respectively. Data represent mean ± S.E. of a representative experiment containing six to eight oocytes per condition. *, p < 0.05 compared with water-injected uptake.
Kinetics of hOCT1-Mediated Uptake of [3H]Ranitidine inX. laevisOocytes. The time course of [3H]ranitidine uptake in hOCT1-expressing X. laevis oocytes was investigated over 45 min. Uptake of [3H]ranitidine was greater in hOCT1-expressing oocytes compared with water-injected controls at each time point investigated (Fig. 5A). Uptake of [3H]ranitidine also was linear over the entire 45-min time course, and hence, the 30-min time point was chosen for further kinetic characterization (Fig. 5A). To determine the kinetics of hOCT1-mediated ranitidine transport, [3H]ranitidine uptake was determined in water-injected and hOCT1-expressing oocytes over a range of [3H]ranitidine concentrations (1–500 μM) for 30 min ([3H]ranitidine uptake was linear with respect to time over 30 min for each tested concentration). The hOCT1-mediated uptake of [3H]ranitidine was saturable as a function of concentration with an estimated Km of 70 ± 9 μM. (Fig. 5B).

Uptake of [3H]ranitidine as a function of time (A) and concentration (B) in hOCT1-expressing X. laevis oocytes. Uptake of [3H]ranitidine (10 μM or indicated concentration) was determined in water-injected (•) or hOCT1-expressing (○) oocytes for the indicated time points (A) or 30 min (B). Data represent mean ± S.E. from six to eight oocytes per time point or concentration.
trans-Stimulation andtrans-Inhibition of MPP+Efflux from hOCT1-, hOCT2-, and hOCT3-ExpressingX. laevisOocytes.trans-Stimulation studies have been previously used to investigate whether known inhibitors of human organic cation transporters also are substrates (Zhang et al., 1998, 1999). The potential for unlabeled famotidine to serve as an OCT substrate was evaluated by preloading hOCT1-, hOCT2-, or hOCT3-expressing oocytes with [3H]MPP+ (1 μM) for 2 h and by evaluating the amount effluxed in the absence and presence of famotidine (1 mM). As a positive control, similar experiments were performed with TEA (1 mM), a known hOCT substrate, and ranitidine (1 mM), which has been evaluated as a substrate for hOCTs by direct radiolabeled uptake (Fig. 5). TEA stimulated efflux of [3H]MPP+ in hOCT1-(∼2-fold) and hOCT2-expressing oocytes (∼2-fold), consistent with its role as an hOCT1 and hOCT2 substrate (Fig. 6, A and B) (Gorboulev et al., 1997); however, TEA did not stimulate efflux of [3H]MPP+ in hOCT3-expressing oocytes (Fig. 6C). Similar to TEA, both famotidine and ranitidine trans-stimulated efflux of [3H]MPP+ from hOCT1-expressing oocytes, suggesting that famotidine also is a substrate of hOCT1 (Fig. 6A). Interestingly, famotidine and ranitidine exhibited trans-inhibition of [3H]MPP+ efflux from hOCT2-expressing oocytes, suggesting that these compounds are not appreciably transported by hOCT2 (Fig. 6B). Famotidine also trans-inhibited efflux of [3H]MPP+ from hOCT3-expressing oocytes; however, ranitidine did not significantly affect the [3H]MPP+ efflux (Fig. 6C). Similar patterns of trans-stimulation and trans-inhibition were obtained for efflux of [3H]MPP+ into a depolarizing buffer (i.e., high K+ concentration). However, efflux of [3H]MPP+ was higher under depolarizing conditions, perhaps due to the reduction in the inside negative membrane potential that provides a more favorable electrochemical environment for organic cation efflux (see Fig. 6 legend). These results confirm that the observed trans-stimulation and trans-inhibition results for hOCT1 and hOCT2 do not result from nonspecific effects of the applied compounds on the membrane potential.
Induction of Electrical Currents in Voltage-ClampedX. laevisOocytes. Electrophysiology studies were undertaken to confirm the results from the trans-stimulation studies. Significant inward currents were induced in voltage-clamped hOCT1-expressing oocytes when superfused with ranitidine (50 μM), famotidine (50 μM), and the positive control TEA (200 μM) (Fig. 7A). None of the ligands induced significant currents in water-injected oocytes, suggesting that the currents induced in hOCT1-injected oocytes result from hOCT1-mediated uptake of the organic cations (Fig. 7A).
Superfusion of voltage-clamped hOCT2-expressing oocytes with high concentrations of ranitidine (1 mM) and famotidine (1 mM) resulted in minimal currents (∼5 nA) that were similar to those observed in water-injected oocytes (Fig. 7B). TEA (1 mM), however, did induce significant currents in hOCT2-expressing oocytes, as expected (Fig. 7B). The small currents observed in water-injected oocytes for ranitidine, famotidine, and TEA may result from endogenous uptake and/or a nonspecific effect of high cation concentrations (1 mM) on the membrane potential. High concentrations were used in an attempt to maximize the signal from inefficiently transported substrates. Additional studies using lower concentrations of ranitidine and famotidine (200 μM) exhibited no significant inward currents in water-injected or hOCT2-injected oocytes (data not shown). These results suggest that ranitidine and famotidine are not appreciably transported by hOCT2 and are consistent with the results of the trans-stimulation studies.
Superfusion of voltage-clamped hOCT3-expressing oocytes with ranitidine (1 mM) and famotidine (1 mM) resulted in no significant inward currents. Similar results were obtained in oocytes injected with water, suggesting that none of the tested compounds was significantly transported by hOCT3. Unfortunately, TEA, a substrate for hOCT3, also did not produce inward currents, thus failing to provide a positive control for the electrophysiology results obtained with hOCT3-expressing oocytes. However, uptake of [3H]MPP+ into the hOCT3-expressing oocytes was significantly greater (2–3-fold) than the observed uptake into water-injected oocytes, indicating that the hOCT3 transport protein was functional during the course of these studies (Fig. 2). On weight of evidence, it is clear that hOCT3 is not capable of transporting ranitidine and famotidine. Clearly, hOCT3 did not support uptake of labeled ranitidine (Fig. 4). Thus, the inability of ranitidine to elicit a current is to be expected. Furthermore, ranitidine and famotidine each inhibited both uptake (Fig. 4) and efflux (Fig. 6) of MPP+ in hOCT3-expressing oocytes. Thus, the evidence indicates that both drugs may bind to— and inhibit— hOCT3 but that neither drug is itself transported by it.
Discussion
In the present study, the interaction of the H2-receptor antagonists ranitidine and famotidine with the human organic cation transporters hOCT1, hOCT2, and hOCT3 has been evaluated. Consistent with the polyspecific nature of this family of transporters, ranitidine and famotidine displayed significant inhibition of each of the investigated transporters. However, their inhibitory potency for the three hOCTs differed markedly. For example, ranitidine and famotidine displayed moderately potent inhibition against hOCT1 with IC50 values of approximately 30 μM (Table 1). Their inhibition of hOCT2, however, was somewhat less potent (IC50 values of ∼75–100 μM). Interestingly, famotidine was an excellent inhibitor of hOCT3 (IC50 = 6.7 μM) and represents one of the most potent inhibitors of hOCT3 among currently marketed therapeutic agents. Ranitidine, however, displayed relatively poor affinity for this transporter (IC50 = 290 μM), thus suggesting that famotidine contains structural features that significantly increase its affinity for hOCT3 compared with ranitidine. One possible explanation is that hOCT3 binds with higher affinity to the guanidinium cationic moiety of famotidine as opposed to the N-dimethyl cationic group of ranitidine (see Fig. 1 for structures). However, the relatively poor affinity of guanidine itself for hOCT3 (Ki = 6200 μM) (Wu et al., 2000) as well as the relatively weak inhibition by guanidinium-containing metformin (IC50 = 2332 μM) (Table 1) would suggest that the presence of the guanidinium group itself does not confer high affinity to hOCT3 and that structural differences residing outside of the cationic moiety of ranitidine and famotidine most likely dictate the high affinity of famotidine toward hOCT3. Such interpretation is consistent with reports of increased affinity of n-tetraalkylammonium compounds for hOCT1 with increasing alkyl chain length, suggesting that the affinity of n-tetraalkylammonium to hOCT1 is defined by structural motifs other than the charged center of these compounds (Zhang et al., 1999). Identification of the specific structural motifs that confer such strong affinity of famotidine to hOCT3, however, is beyond the scope of this study.
The observed inhibition of hOCT1, hOCT2, and hOCT3 by ranitidine and famotidine raises the possibility of drug-drug-interactions involving these agents and coadministered organic cations. Ranitidine is actively secreted by the kidney and has been reported to reduce the renal clearance of the actively secreted organic cations procainamide and triamterene in humans (Somogyi and Bochner, 1984; Muirhead et al., 1988). It is plausible that inhibition of highly expressed renal hOCT2 by ranitidine may explain the observed decrease in renal clearance for procainamide and triamterene in humans. Typically observed Cmax plasma concentrations of ranitidine after a 150-mg oral dose are relatively low (∼1–2 μM) (van Hecken et al., 1982). The IC50 determined for inhibition of hOCT2 by ranitidine (76 μM) (Table 1) in vitro was approximately 35-fold higher than its maximal plasma concentrations, however, suggesting that inhibition of hOCT2 may not be the mechanism behind the reduced renal clearance of procainamide and triamterene in vivo. However, higher local ranitidine concentrations at the hOCT2 substrate binding site or differences in inhibition potency toward triamterene may complicate such in vitro-in vivo predictions. Unlike ranitidine, famotidine exhibits virtually no known drug interactions and does not reduce the renal clearance of procainamide in humans (Klotz et al., 1985). This is most likely a function of its 8-fold greater H2-receptor potency over ranitidine, which results in lower effective doses and subsequently lower plasma concentrations that do not interfere with renal tubular secretion.

trans-Stimulation and trans-inhibition of [3H]MPP+ efflux from hOCT1-(A), hOCT2-(B), and hOCT3-expressing (C) X. laevis oocytes. Oocytes were preloaded with [3H]MPP+ (1 μM) for 2 h and washed three times, and intracellular [3H]MPP+ was allowed to efflux into OR-2 buffer in the absence (control) or presence of the indicated compounds (1 mM) for 1 h. Efflux of [3H]MPP+ into a high K+ OR-2 buffer (depolarizing conditions) also was examined, and it displayed a similar pattern. Percentage of [3H]MPP+ effluxed under depolarizing conditions for hOCT1 was 7.1 ± 0.04 (control), 25.4 ± 6.1 (ranitidine), 26.2 ± 5.1 (famotidine), and 19.3 ± 4.4 (TEA). For hOCT2, percentage of [3H]MPP+ effluxed under depolarizing conditions was 11.1 ± 2.2 (control), 5.3 ± 1.8 (ranitidine), 7.2 ± 2.8 (famotidine), and 15.6 ± 2.4 (TEA). For hOCT3, percentage of [3H]MPP+ effluxed under depolarizing conditions was 7.9 ± 2.6 (control), 8.3 ± 2.1 (ranitidine), 4.5 ± 0.7 (famotidine), and 8.6 ± 1.5 (TEA). Data are presented as the amount of [3H]MPP+ found in the efflux buffer expressed as a percentage of the total amount loaded. Data represent mean ± S.E. from at least three separate experiments; six to eight oocytes were used per condition per experiment. *, p < 0.05 compared with control.
Because inhibition of transporter activity does not necessarily translate to substrate activity, a multiexperimental approach was used to evaluate the substrate specificity of ranitidine and famotidine for hOCT1, hOCT2, and hOCT3. Direct radiolabeled uptake represents the preferred method for evaluation of substrate specificity, and [3H]ranitidine uptake was stimulated by hOCT1- and hOCT2-expressing oocytes, indicating that it is a substrate of these transporters (Fig. 4). Uptake of ranitidine by hOCT2, however, was relatively weak, especially compared with that observed for MPP+. trans-Stimulation studies suggested that famotidine also was translocated by hOCT1; however, a trans-inhibition effect was observed for both ranitidine and famotidine in inhibiting MPP+ efflux from hOCT2-expressing oocytes (Fig. 6). This apparent trans-inhibition is consistent with the poor substrate activity of ranitidine for hOCT2 and suggests that famotidine also is not well translocated by hOCT2. The electrophysiology studies provide further evidence supporting this conclusion because even high concentrations (1 mM) of ranitidine and famotidine failed to induce significantly greater currents in hOCT2-expressing oocytes than those observed in water-injected controls (Fig. 7). No evidence was obtained that ranitidine or famotidine is transported by hOCT3, suggesting this transporter does not play a role in the disposition of these agents in humans.
The finding that ranitidine and famotidine are poor hOCT2 substrates is surprising, because both ranitidine and famotidine exhibit active renal secretion that may be presumed to occur via highly expressed renal hOCT2. However, the observed poor substrate activity of ranitidine and famotidine toward hOCT2 is also consistent with a recent report, suggesting that famotidine is not appreciably translocated by hOCT2 (Motohashi et al., 2004). Interestingly, cimetidine, another H2-receptor antagonist, seems to exhibit the reverse selectivity because it is a relatively good substrate of hOCT2 but reportedly exhibits weak substrate activity toward hOCT1 (Zhang et al., 1998; Urakami et al., 2002). Although hOCT2 likely contributes to the renal excretion of ranitidine and famotidine to some extent in humans, it may not be the dominant transport mechanism. Recent investigations suggest that cimetidine and famotidine are substrates of the highly expressed renal organic anion transporter hOAT3 (SLC22A8) (Cha et al., 2001; Motohashi et al., 2004), whereas ranitidine has been identified as a substrate of the rat isoform rOAT3 (Nagata et al., 2004). Although it is not known whether ranitidine is also a substrate of hOAT3, probenecid, an organic anion transporter inhibitor, reduces the renal clearance of ranitidine in the dog (Boom et al., 1998) as well as that of famotidine in humans (Inotsume et al., 1990). Therefore, multiple transporters, including hOCT2 and hOAT3, are likely involved in the renal excretion of ranitidine and famotidine in humans.
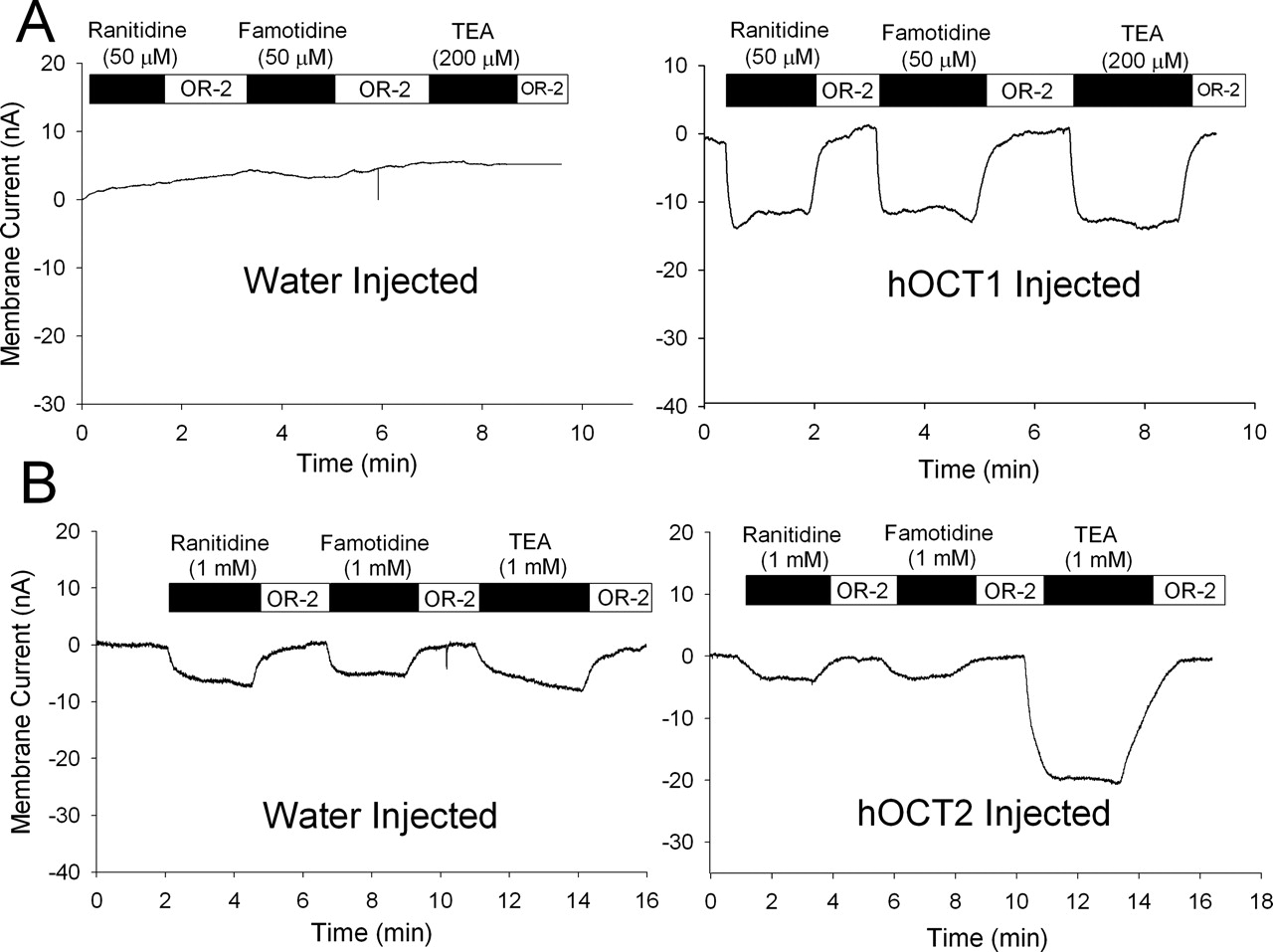
Electrical recordings of induced currents in water-injected and hOCT1-(A) and hOCT2-expressing (B) X. laevis oocytes. Oocytes were voltage-clamped at –60 mV 5 to 7 days postinjection and perfused with OR-2 buffer. Voltage-clamped oocytes were exposed to ranitidine, famotidine, or TEA at the indicated concentrations. Compounds were removed in between applications by washing with OR-2 buffer. Recordings are representative of results observed in at least three oocytes isolated from three different animals.
Although hepatic metabolism does not play a major role in the overall clearance of ranitidine and famotidine in humans (Lin, 1991), the mechanism of hepatic uptake is likely mediated via hOCT1. The Km value determined for ranitidine (70 μM; Fig. 5B) would dictate that the hOCT1-mediated hepatic uptake would not be saturated at typical therapeutic doses. Because biliary excretion of ranitidine and famotidine is reported as low in humans (<2.6% of dose) (Klotz and Walker, 1990), unmetabolized ranitidine and famotidine may accumulate in the liver before subsequent efflux back into blood followed by their eventual renal excretion. Studies using rat-isolated perfused liver systems seem to support the uptake and accumulation of H2-receptor antagonists by the liver as perfusate concentrations significantly decline over time, indicating efficient liver extraction (Mihaly et al., 1982; Hughes et al., 1995). Ranitidine and famotidine also may compete for hepatic uptake with other organic cations, potentially leading to drug-drug interactions at the level of hepatic uptake. Since hOCT1 controls access to drug-metabolizing enzymes for its substrates, such interactions may affect the hepatic elimination of certain drugs. Interestingly, a reduction in the hydroxylation clearance of the organic cation triamterene has been reported when coadministered with ranitidine, despite the noted lack of effect of ranitidine upon cytochrome P450-mediated metabolism at therapeutic doses (Muirhead et al., 1988; Smith and Kendall, 1988). Such events may potentially be explained by reduced hepatic uptake of organic cations by hOCT1 in the presence of ranitidine and highlight the potential interplay between drug transporters and drug-metabolizing enzymes.
The role of hOCTs in the intestinal handling of organic cations is not clear. Although the rat isoforms Oct1 and Oct3 clearly demonstrate intestinal expression (Grundemann et al., 1994; Kekuda et al., 1998), little information is available regarding their human expression. Zhang et al. (1999) reported intestinal expression of hOCT1 as determined by reverse-transcriptase-polymerase chain reaction, suggesting that this isoform is present in the human intestine. A number of studies have also reported expression of hOCT1, hOCT2, and hOCT3 in the Caco-2 cell model of intestinal epithelium (Zhang et al., 1999; Bleasby et al., 2000; Martel et al., 2001; Hayer-Zillgen et al., 2002). Functional studies suggest a lack of saturable OCT-mediated uptake of TEA along the basolateral membrane of Caco-2 cells (Lee et al., 2002). However, uptake of TEA and MPP+ across the apical membrane of Caco-2 cells is saturable and consistent with OCT-mediated processes (Ng et al., 2002). Interestingly, ranitidine and famotidine also display saturable transport and uptake behavior in the apical-to-BL direction across Caco-2 cells, suggesting that carrier-mediated processes may be involved in their absorption (Lee and Thakker, 1999). The finding that ranitidine and famotidine serve as hOCT1 substrates (Figs. 4, 6, and 7) thus identifies at least one isoform of the OCT family as a candidate transporter to mediate saturable uptake of these compounds into Caco-2 cells. Additional organic cation transporters, such as hOCTN1, hOCTN2, or other unidentified transporters, may also potentially be involved in the uptake of these compounds in Caco-2 cells.
In conclusion, ranitidine and famotidine have been identified as excellent substrates of hOCT1. hOCT1 likely mediates intestinal transport and hepatic uptake of ranitidine and famotidine in humans and presents a potential site for drug-drug interactions among hOCT1 substrates. Ranitidine and famotidine are poor substrates of hOCT2, and neither compound serves as a substrate of hOCT3, suggesting that other transporters may be involved in their renal secretion. Both ranitidine and famotidine display inhibition of all three hOCT isoforms, and famotidine has been identified as one of the most potent marketed therapeutic agents that inhibits hOCT3. Therefore, only hOCT1 likely plays a role in the disposition and elimination of ranitidine and famotidine in humans and may represent a potential site of drug-drug interaction.
Acknowledgments
We gratefully acknowledge the excellent technical assistance of Laura Hall (National Institute of Environmental Health Sciences) with oocyte isolation and microinjection and Dr. Amy Aslamkhan (National Institute of Environmental Health Sciences) for assistance with the electrophysiology studies. The assistance of Xin Ming (University of North Carolina at Chapel Hill) in obtaining selected ranitidine uptake results is gratefully acknowledged. cDNA for hOCT1 and hOCT3 was kindly provided by Dr. Kathleen Giacomini (University of California, San Francisco) and Dr. Vadivel Ganapathy (Medical College of Georgia), respectively.
Footnotes
-
D.L.B. was supported by a Pharmaceutical Research and Manufacturers of America Foundation predoctoral fellowship in pharmaceutics.
-
doi:10.1124/jpet.105.091223.
-
ABBREVIATIONS: hOCT, human organic cation transporter; OCT, organic cation transporter; BL, basolateral; TEA, tetraethylammonium; MPP+, 1-methyl-4-phenyl pyridinium; OR-2, oocyte ringer's 2; CLup, uptake clearance.
- Received June 22, 2005.
- Accepted August 15, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}