


Abstract
Disposition of the lipid-lowering agent ezetimibe (EZ) and its glucuronide (GLUC), which is mainly formed by UDP-glucuronosyltransferase (UGT) 1A1, is influenced by the intestinal efflux transporters P-glycoprotein (P-gp) and multidrug resistance-associated protein (MRP) 2. To evaluate the role of Mrp2 in overall disposition and pharmacodynamic effects of EZ, wild-type and Mrp2-deficient (TR-negative) Lewis.1W rats (eight males each) fed with a cholesterol-enriched diet were orally treated with 5 mg/kg EZ for 14 days. EZ and GLUC in serum, urine, and feces, and cholesterol, campesterol, and sitosterol in serum, were assayed using liquid chromatography (LC)-tandem mass spectrometry and LC-mass spectrometry methods, respectively. Gene expression of Bsep (bile salt exporting pump), multidrug resistance (Mdr) 1a, Mdr1b, Mrp2, Mrp3, Ntcp (sodium taurocholate co-transporting polypeptide), organic anion transporting polypeptides (Oatp) 1, 2, 4, and Ugt1a1 was quantified in several tissues using real-time reverse transcription-polymerase chain reaction. Mrp2 deficiency resulted in lower serum levels and fecal excretion of EZ (1.4 ± 0.4 versus 3.1 ± 1.1 ng/ml; 115 ± 48 versus 361 ± 102 μg/day, both p < 0.01), whereas serum concentrations of GLUC were manyfold increased compared with wild type (196 ± 76 versus 23 ± 25 ng/ml; p < 0.01), associated with elevated renal excretion and decreased intestinal clearance (7.8 ± 3.1 versus 0.4 ± 0.4 μg/day, p < 0.01; 0.3 ± 0.3 versus 15 ± 17 ml/min; p < 0.05). The sterol-lowering effect of EZ was reduced in correlation to EZ serum levels (cholesterol: r = 0.449, p = 0.093; campesterol: r = 0.717, p = 0.003; sitosterol: r = 0.507, p = 0.054), whereas GLUC was inversely correlated (r = -0.743, p = 0.002; r =-0.768, p = 0.001; r =-0.634, p = 0.011). Disposition of EZ may have been additionally influenced by hepatic P-gp, Mrp3, and Ugt1a1, which were expressed significantly higher in Mrp2-deficient rats. Mrp2 deficiency in rats is associated with decreased sterol-lowering effect of ezetimibe, obviously caused by lower intestinal clearance of the glucuronide and decreased enterosystemic and enterohepatic recycling of the parent ezetimibe to the intestinal Niemann-Pick C 1-like 1 sterol-uptake compartment.
The efficacy of drug therapy depends on pharmacokinetic processes that provide pharmacological active concentrations in the vicinity of the respective pharmacological receptor(s) and on expression and function of the effectuation pathways. An example is cholesterol-lowering therapy with ezetimibe that selectively reduces intestinal sterol uptake by inhibition of the recently discovered sterol uptake transporter Niemann-Pick C 1-like 1 protein (NPC1L1) in the brush-border membrane of the jejunum and, to a lower extent, in the duodenum and the ileum (Altmann et al., 2004; Garcia-Calvo et al., 2005). Ezetimibe is widely used in patients with hypercholesterolemia and sitosterolemia. An additional benefit is expected from combinations with low-dose statins (Gagne et al., 2002; Knopp et al., 2003; Salen et al., 2004). After oral administration, ezetimibe is rapidly absorbed from gut lumen and nearly completely conjugated by UDP-glucuronosyltransferases (UGT). It was shown that the parent drug is conjugated to a major phenolic glucuronide and only in traces to a benzylic and ketone glucuronide involving UGT1A1, 1A3, and 2B15, of which UGT1A1 was shown to be the most active isoform and whose activity is highest in jejunal sterol-absorbing enterocytes (Tukey and Strassburg, 2001; Patrick et al., 2002; Shelby et al., 2003; Ghosal et al., 2004). The glucuronide undergoes intestinal and hepatic secretion, thus initiating via colonic cleavage intensive enteral recirculation of ezetimibe back to the side of NPC1L1 action. Therefore, fluctuation of the serum concentration-time curve is observed and the sterol-lowering effect is long-lasting (Ezzet et al., 2001a; Patrick et al., 2002). Recent data from an in vitro study with transfected MDCKII (Madine Darby canine kidney), LLC (Lewis lung carcinoma) and 2008 (human ovarian carcinoma) cells confirmed that ezetimibe glucuronide is a high-affinity substrate of the multidrug resistance-associated protein (MRP2) 2 and has low affinity to P-glycoprotein (P-gp), whereas ezetimibe interacts moderately with both P-gp and MRP2 (Oswald et al., 2006a). Therefore, we assumed that presystemic elimination of ezetimibe and the glucuronide by intestinal and/or hepatic UGT1A1, P-gp, and MRP2 was the major variable for the bioavailability of ezetimibe. Inhibition of P-gp and MRP2 by gemfibrozil, fenofibrate, and cyclosporine leads consistently to elevation of ezetimibe serum concentrations (Kosoglou et al., 2004; Reyderman et al., 2004; Bergman et al., 2006). On the contrary, up-regulation of intestinal UGT1A1, P-gp, and MRP2 in healthy subjects by the pregnane X receptor (PXR)-ligand rifampicin is associated with markedly decreased serum concentrations and sterol-lowering effect of ezetimibe (Oswald et al., 2006a). However, the rate-limiting elimination pathway in overall disposition and lipid-lowering effect of ezetimibe could not be derived from the results of the so-far published drug interaction studies because P-gp, MRP2, and UGT1A1 are obviously coregulated in humans by the nuclear receptors PXR and constitutive androstane receptor (Tirona and Kim, 2005; Xu et al., 2005).
A suitable approach to differentiate between the role of P-gp, MRP2, and UGT1A1 in disposition and pharmacological effect of ezetimibe is the use of hereditary variant animals such as Mrp2-deficient rats. Such rats do not synthesize Mrp2 because of mutations in the Abcc2 gene, which creates premature termination codons (Paulusma et al., 1996; Keppler and Konig, 1997). However, in studies with Mrp2-deficient rats, complex changes in the expression of other multidrug transporters and drug-metabolizing enzymes must be considered, which may have an additional influence on the disposition and pharmacological effect of ezetimibe (Hirohashi et al., 1998; Ogawa et al., 2000; Kuroda et al., 2004; Newton et al., 2005; Johnson et al., 2006). Overall, we hypothesized that Mrp2 deficiency should result 1) in a substantially reduced intestinal and hepatic secretion of ezetimibe and its glucuronide, and in turn, 2) to a decreased enteral recirculation of the active ezetimibe to the intestinal NPC1L1 receptor compartment and 3) to a reduction of the sterol-lowering effect. The Mrp2-related differences might be 4) augmented by increased organ expression of Mdr1, Mrp3, and Ugt1a1.
Materials and Methods
Chemicals and Reagents. Ezetimibe (Ezetrol) was obtained from MSD, Haar, Germany. Acetonitrile and diethyl ether were purchased from Merck (Darmstadt, Germany), and methyl cellulose, cholesterol, campesterol, and sitosterol were from Sigma-Aldrich (Taufkirchen, Germany). All other reagents were obtained from commercial sources.
Animals. Disposition and sterol-lowering effect of ezetimibe was evaluated in eight male wild-type (236-328 g) and eight male congenic Mrp2-deficient, TR-negative Lew.1W rats (358-390 g), which were purchased from the Department of Pathophysiology of the University of Greifswald. The presence of the premature termination codon in the Mrp2 gene and the absence of Mrp2 were confirmed by genotyping and Western blot analysis, respectively. The animals were housed under standard laboratory conditions in the life island box A 110 (Flufrance, Wissous, France) with mass-air displacement, a temperature of 25°C, and a 12-h light/dark cycle with lights on at 8 AM, with one rat per polycarbonate cage, bedding (ssniff, Soest, Germany), and free access to acidified water and to a sterol-enriched diet containing 16.1% fat, 1% cholesterol, and 20.7% proteins (ssniff). The study was permitted by the Federal Authorities.
Experimental Protocol. The study was performed in eight wild-type and eight Mrp2-deficient rats. After adaptation for 14 days, approximately 0.5 ml of blood was obtained by puncture of the retrobulbar venous plexus in ether narcosis. After that, the animals were orally treated with 5 mg/kg ezetimibe for 14 days. The drug was suspended in 0.5% methyl cellulose suspension and administered orally via a gavage (administration volume, 5 ml/kg). To measure intestinal and renal excretion of ezetimibe and its glucuronide at steady state, feces were collected between the 9th and 14th treatment days and urine on the 14th treatment day and stored at least at -80°C until analysis. In the morning of the 15th study day (24 h after last drug administration), animals were anesthetized with diethyl ether for blood sampling from the retrobulbar plexus (approximately 0.5 ml) and for sacrifice by cervical dislocation and dissection. Aliquots of liver, kidney, duodenum, jejunum, ileum, and colon were immediately frozen with liquid nitrogen to measure mRNA expression. The further storage of the samples until analysis was at least at -80°C.
Quantification of Gene Expression. Approximately 20 to 30 mg of the respective frozen tissue were homogenized (Mikro-Dismembrator S; B. Braun, Melsungen, Germany) in the presence of a guanidine isothiocyanate containing buffer, and total mRNA was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany) and assayed for content and purity using the spectrometry (Biophotometer; Eppendorf, Hamburg, Germany). Reverse transcription of 200 ng of mRNA was performed with random hexamers and TaqMan Reverse Transcription Reagents (Applied Biosystems, Foster City, CA). Abcb1a mRNA and Abcc2 mRNA expression were quantified by real-time reverse transcriptase-polymerase chain reaction according to the TaqMan technology using the ABI Prism 7700 cycler and the TaqMan Universal PCR MasterMix (Applied Biosystems). For Abcb1a, 5′-CCCATGGCCGGAACAGT was used as forward primer, 5′-ATGGGCTCCTGGGACACA-3′ as reverse primer, and 5′-FAMTGGCTCCGCGCCCACCTGT TAMRA-3′ as probe; and for Abcc2, 5′-TGTGGGCTTTGTTCTGTCCA-3′ as forward primer, 5′-CAGCCACAATGTTGGTCTCG-3′ as reverse primer, and 5′-FAM-CTCAATATCACACAAACCCTGAACTGGCTGT TAMRA-3′ as probe (Molbiol, Berlin, Germany). To quantify mRNA expression of Bsep, Mdr1b, Mrp3, Ntcp, Oatp1, Oatp2, Oatp4, and Ugt1a1, TaqMan Gene Expression Assays were used according to the recommendations of the manufacturer (Applied Biosystems). The reference gene for all quantifications was 18S rRNA by use of the ΔΔCT (cycles of threshold) method (Livak and Schmittgen, 2001).
Western Blot Analysis. For protein preparation, liver tissue (20 g) of wild-type and TR-negative rats was homogenized by 20 strokes (1000 rpm) using the Potter S homogenizer (B. Braun). Thereafter, the homogenate was centrifuged at 100,000g for 30 min, and the resulting membrane pellets were resuspended in Tris/HCl buffer (5 mM, pH 7.4). Protein concentration of these crude membrane fractions was analyzed by the bicinchoninic acid method.
For Western blot analysis, 50 μg of each sample were loaded onto a 7.5% sodium dodecylsulfate-polyacrylamide gel after incubation in Laemmli buffer at 95°C for 10 min. After electrophoretic separation for 2 h at 180 V, the immunoblotting on a nitrocellulose membrane (Schleicher and Schüll, Dassel, Germany) was performed using a tank blotting system (BioRad, Hercules, CA). After a protein control staining with Ponceau S, unspecific binding sites were blocked overnight at 4°C using a blocking solution containing 5% milk powder and 5% fetal calf serum in TBST (Tris-buffered saline containing 0.05% Tween 20). For primary antibody incubation, membranes were washed with TBST and incubated with the respective antibody diluted in TBST containing 5% bovine serum albumin. The detection of Mrp2 was performed using the EAG15 anti-Mrp2 antibody (dilution, 1:5000; kindly provided by Dr. D. Keppler, Deutsches Krebsforschungszentrum Heidelberg, Germany); for P-gp, with the C219 monoclonal mouse antibody (dilution, 1:500; Alexis, Grünberg, Germany); for Mrp3, with FDS anti-Mrp3 rabbit antiserum (dilution, 1:1000; kindly provided by Dr. D. Keppler) (Konig et al., 1999); and for UGT1a, a cross-reactive human UGT1A family antibody from BD Gentest (dilution, 1:500; Heidelberg, Germany) was used as recently described (Johnson et al., 2006). The secondary horseradish peroxidase-conjugated goat anti-rabbit and anti-mouse antibodies (BioRad) were used at a 1:2000 dilution. Finally, the immobilized antibodies were stained using an enhanced chemiluminescence system (Amersham Biosciences, Freiburg, Germany) and exposed to X-ray films. For quantification, the blots were scanned, and the respective banddensity was calculated using the SigmaGel software (Jandel Scientific, San Rafael, CA).
Quantitative Assay for Ezetimibe. Ezetimibe and its glucuronide in serum, urine, and feces were determined using liquid chromatography-tandem mass spectrometry as recently described (Oswald et al., 2006b). Thereby, ezetimibe glucuronide was quantified indirectly after hydrolysis using β-glucuronidase. The validation ranges of ezetimibe were 0.5 to 500 ng/ml for serum, 5 to 1000 ng/ml for urine, and 0.1 to 15 μg/ml for feces. Within-day and between-day accuracy and precision of calibration values and quality control for serum were lower than 10.7% of the nominal values and means, respectively, and lower than 15% for urine and feces.
Quantitative Assay for Cholesterol, Campesterol, and Sitosterol. Serum concentrations of total cholesterol and the plant sterols campesterol and sitosterol were quantified by means of a validated LC-MS method with 4-hydroxychalcone as internal standard. In brief, 0.1 ml of serum was diluted with 0.9 ml of water. Of this, 0.2 ml was mixed with 25 μl of internal standard solution (50 μg/ml) and incubated with 2 ml of ethanol (96%, v/v) and 0.4 ml of potassium hydroxide (10 M) for 3 h at room temperature (22-25°C) to hydrolyze any sterol esters. After neutralization with formic acid, the samples were extracted with chloroform that was evaporated to dryness under a gentle nitrogen stream at 60°C. The residue was dissolved in acetonitrile/0.1% formic acid (90:10, v/v) for quantification using a liquid chromatography-mass spectrometry system consisting of the pump series 1100 (Hewlett Packard, Waldbronn, Germany), the autosampler series 200 (Perkin Elmer,Überlingen, Germany), the column thermostat L-5025 (Merck-Hitachi, Darmstadt, Germany), and the PE Sciex API 2000 mass spectrometer equipped with the Analyst 1.2 software (Applied Biosystems, Darmstadt, Germany). The chromatography was performed isocratically using acetonitrile/0.1% formic acid (90:10; v/v) as mobile phase (flow rate 200 μl/min) and the column XTerra MS (C8, 2.1 × 50 mm, particle size, 3.5 μm; Waters, Milford, MA). The mass spectrometer was used with the Heated Nebulizer (atmospheric pressure chemical ionization) interface in the positive ion mode. The following m/z values were monitored for quantification: 396.3 for cholesterol, 384.3 for campesterol, 397.3 for sitosterol, and 225.1 for 4-hydroxychalcone. The method was validated for 10 to 500 μg/ml cholesterol and for 0.1 to 7.5 μg/ml campesterol and sitosterol. Within-day and between-day accuracy and precision for all sterols were within -7.8 and 10% of the nominal values and were 14.3% lower than the mean values, respectively.
Pharmacokinetic and Statistical Evaluation. Trough serum concentrations of ezetimibe and its glucuronide at steady state (Css) were taken from the study data. Renal clearance (CLR), metabolic clearance (CLM), and fecal clearance (CLF) were derived from the amounts (Ae) excreted into the urine and feces (average values from 9th to 14th study days) over the respective product of Css × 24 h [represents approximately the area under the plasma concentration time curve from time 0 to 24 h (AUC0-24h)] of ezetimibe and the glucuronide, respectively. Samples are presented as arithmetic means ± S.D. or medians and 75% percentiles. Mann-Whitney, Wilcoxon, Jonckheere-Terpstra, and Spearman's rank test were used for statistical analysis as appropriate.
Results
Gene and Protein Expression in Mrp2-Deficient Rats.Mrp2 mRNA as well as protein was not expressed in organs of Mrp2-deficient rats (Figs. 1 and 2). In wild-type animals, Mrp2 mRNA levels were highest in the liver. Compared with liver, intestinal expression was markedly lower with a minimum in the colon. Regional differences in intestinal mRNA expression were also observed for Mdr1a and Ugt1a1 in wild type as well as TR-negative animals. Mdr1a mRNA was expressed with significantly increasing levels along the small intestine. Maximal levels were found in the ileum. In contrast, Ugt1a1 mRNA content was highest in duodenum and decreased significantly toward distal parts of the gut. In the colon, Mdr1a and Ugt1a1 were expressed in significantly lower extent. Mrp3 mRNA was well expressed in all parts of the intestine with highest levels in the colon but failed to reach level of significance caused by the huge standard deviations. Wild-type and Mrp2-deficient rats were not different in intestinal mRNA content of Mdr1a, Mrp3, and Ugt1a1 with exception of Mdr1a in the ileum. In the liver of Mrp2-deficient rats, however, Mdr1a, Mdr1b, Mrp3, and Ugt1a1 expression was significantly higher that in wild-type animals (Fig. 1). Expression of the hepatic uptake transporters Oatp1, Oatp2, Oatp4, Ntcp, and the major bile salt elimination pump Bsep was not changed in the Mrp2-deficient rats (Table 1).
Hepatic mRNA expression of Oatp1, Oatp2, Oatp4, and of Bsep and Ntcp relative to 18S rRNA of eight wild-type and eight Mrp2-deficient TR-negative Lew.1W rats
Arithmetic means ± S.D. are given.
Western blot analysis revealed significantly higher hepatic protein expression of Mrp3 (5.8-fold, p = 0.02) and Ugt1a (4.2-fold, p = 0.02) in Mrp2-deficient animals, whereas P-gp content was only slightly increased (1.3-fold, p = 0.24) (Fig. 2).
Disposition of Ezetimibe.Mrp2 deficiency resulted in an approximately 8.5-fold increase of the glucuronide serum concentrations but in markedly lower levels of unchanged ezetimibe (Table 2). This was associated with a manyfold higher renal but lower fecal excretion of the glucuronide and a significantly elevated metabolic clearance of ezetimibe into the urine. The amount of glucuronide in feces was significantly lower in Mrp2-deficient rats, whereas the amount excreted into urine was manyfold higher. Renal clearance and urinary excretion of ezetimibe were also markedly increased in TR- animals (Table 2).
Pharmacokinetic characteristics of ezetimibe in wild-type and Mrp2-deficient TR-negative Lew.1W rats (each n = 8) after oral administration of 5 mg/kg ezetimibe for 14 days
Arithmetic means ± S.D. are given.
Sterol-Lowering Effect. Mrp2 deficiency was associated with significantly lower baseline serum concentrations of cholesterol (119 versus 177 mg/dl, p < 0.05) and the plant sterols campesterol and sitosterol (39.8 versus 51.2 mg/dl and 10.4 versus 12.7 mg/dl, both p < 0.05). Treatment of wild-type rats with ezetimibe resulted in significant lowering of the mean cholesterol and plant sterol serum concentrations by approximately 40 to 50%. In Mrp2-deficient rats, the sterol-lowering effect of ezetimibe was markedly reduced and even failed to reach the level of statistical significance referring to cholesterol (Fig. 3). Furthermore, the effect of ezetimibe on sterol absorption was correlated to serum concentrations of ezetimibe (significant only according to campesterol) but significantly inversely correlated to the serum concentration of the glucuronide (Fig. 4).

Expression of Mdr1a, Mdr1b, Mrp2, Mrp3, and Ugt1a1 mRNA relative to 18S rRNA in liver and intestine of eight wild-type (white columns) and eight Mrp2-deficient TR-negative Lew.1W rats (gray columns). Columns and bars indicate medians ± 75% quartile (*, p < 0.05; **, p < 0.01 relative to wild-type, Mann-Whitney test; †, p < 0.05 Jonckheere-Terpstra trend test for wild type and ‡, p < 0.05 for Mrp2-deficient animals).
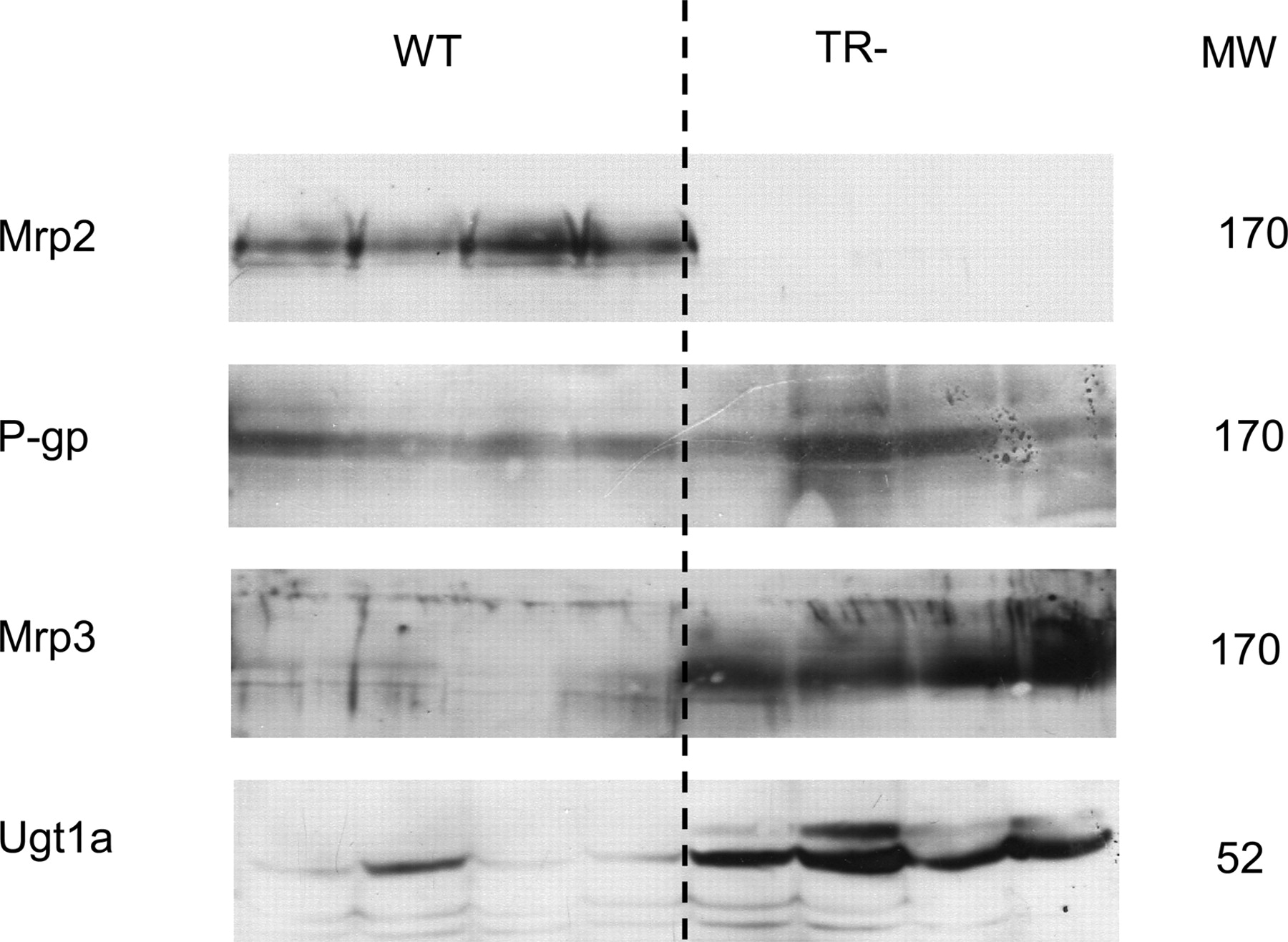
Western blots of Mrp2, Mrp3, P-gp, and Ugt1a of liver homogenates from wild-type (WT) and TR- animals. Blots were prepared as described under Materials and Methods in each case from four separate animals. Approximate molecular weights (MW) are shown.
Discussion
This study in Mrp2-deficient rats clearly demonstrates that the activity of the multidrug transporter Mrp2 is a major determinant for the disposition and sterol-lowering effect of ezetimibe. Mrp2 is colocalized with P-gp to the luminal membrane of the villous enterocytes, the canalicular membrane of hepatocytes, the proximal tubular cells in the kidney, and the luminal surface of blood capillaries (Chan et al., 2004; Takano et al., 2006). Mrp2 serves as an export pump for a wide range of conjugated and unconjugated anionic compounds and shares some degree of overlapping substrate specificity with P-gp (Keppler and Konig, 1997, 2000). Ezetimibe has low affinity to MRP2, whereas the glucuronide is a high-affinity substrate (Oswald et al., 2006a). After oral administration, ezetimibe is intensively conjugated by intestinal and/or hepatic UGTs, resulting in a major phenolic glucuronide that undergoes intestinal and/or hepatic secretion followed by bacterial hydrolysis in the colon and reuptake of the parent ezetimibe into systemic blood (van Heek et al., 2000; Patrick et al., 2002; Ghosal et al., 2004). The mechanism behind intestinal secretion of the glucuronide seems to be MRP2-mediated efflux, as up-regulation of intestinal MRP2 by chronic treatment with the PXR-ligand rifampicin leads to a markedly increased intestinal clearance and a reduction of glucuronide serum concentrations for approximately 70% (Oswald et al., 2006a). In this study, the absence of intestinal and hepatic Mrp2 in our TR-negative rats resulted in manyfold increased serum glucuronide concentrations, a significantly increase of urinary excretion at unchanged renal clearance, and a nearly abolished intestinal clearance. From this pattern of pharmacokinetic changes, we conclude that Mrp2 is the major pathway for intestinal and hepatic secretion of the glucuronide. Furthermore, it seems very likely that that the glucuronide is the source for the long-lasting circulation of ezetimibe via hepatic and intestinal secretion of the glucuronide, bacterial hydrolysis in the colon, and reabsorption of the parent drug into the systemic circulation. This recycling scheme yields in humans characteristic multiple serum concentration peaks that are, to our interpretation, amplified by a gastro-ileo-cecal reflex that results in bolus propulsion of chyme from the terminal ileum to the hydrolytic environment of the colon after meals (Schiller et al., 2005). Furthermore, food-triggered emptying of the gall bladder most likely enforces this effect by delivering glucuronized drug to the upper intestine.

Sterol-lowering effects of ezetimibe after oral administration of 5 mg/kg for 14 days in percentage of the pretreatment (baseline) serum concentrations of wild-type (white columns) and Mrp2-deficient TR-negative Lew.1W rats (gray columns). Columns and bars indicate arithmetic means ± S.D. (*, p < 0.05 compared with baseline, Wilcoxon test; ††, p < 0.01 compared with wild type, Mann-Whitney test).
With respect to P-gp and Mrp2, the physiological preconditions for colonic absorption of the parent drug ezetimibe are well comparable for wild-type and Mrp2-deficients rats. Ezetimibe is a substrate of P-gp and less of Mrp2, but P-gp expression is low in the colon of wild-type and TR- rats; Mrp2 expression is low in wild-type rats and missing in TR- rats. We assume that the ezetimibe permeability across the mucosa of the colon is much better than in distal parts of the small intestine in which P-gp and Mrp2 are expressed highest. Furthermore, presystemic glucuronidation in the colon is low because Ugt1a1, for which ezetimibe is a high-affinity substrate, is lowest expressed in colonic enterocytes (Tukey and Strassburg, 2001; Ghosal et al., 2004). Therefore, the reduced serum concentrations of ezetimibe assessed in Mrp2-deficient animals may be the consequence of diminished glucuronide secretion in the intestine and increased biliary ezetimibe secretion by P-gp, which was found to be slightly up-regulated in our congenic rats. Interestingly, the renal clearance of parent ezetimibe was markedly increased in TR-negative rats. This cannot be addressed to glucuronide cleavage during the 24-h urine collection period, because corresponding stability tests revealed that only a negligible amount (0.9%) of the glucuronide was degraded to free ezetimibe. Due to this, the Mrp2-deficient rats seem to exhibit an enhancement of other renal elimination pathways, because Mdr1 was not found to be induced (data not shown) in accordance to findings of Johnson et al. Alternatively, differences in expression of other renal drug-transporting systems like organic anion transporters, organic cation transporters, or Mrp3 and Mrp4, which were shown to be markedly higher expressed in the kidney of Mrp2-deficient rats, may be responsible to this phenomenon (Chen et al., 2005; Johnson et al., 2006).
The regioselective expression of Mdr1, Mrp2, Mrp3, and Ugt1a1 along the rat intestine observed in our study is in well accordance with published data (Tukey and Strassburg, 2001; Brady et al., 2002; Rost et al., 2002; Dietrich et al., 2003; Ho et al., 2003). One might speculate that a high colonic expression of the basolateral efflux transporter Mrp3 may contribute to the reabsorption of ezetimibe because it shares a considerable overlap in substrate specificity with P-gp and Mrp2 (Chan et al., 2004). However, the systemic concentration of ezetimibe in blood significantly predicts the extent of the sterol-lowering effect, which results from inhibition of NPC1L1 in the small intestinal epithelium (Ezzet et al., 2001b). Thereby, glucuronide exposure is inversely correlated to the pharmacological effect. That means, according to our hypothesis, that the decrease or the absence of intestinal (and hepatic) “first-pass” secretion of the glucuronide caused by Mrp2 deficiency reduces or even interrupts recycling of the active compound ezetimibe to the NPC1L1 receptor compartment. In the Mrp2-defcient rats, the nonsecreted portion of the glucuronide is now available in the systemic circulation, yielding very high serum concentrations that cause a significantly increased amount to be excreted into urine by renal filtration. Renal clearance is consequently not influenced as shown by our data.

Correlations between trough serum concentrations of ezetimibe (left) and ezetimibe glucuronide (right) and sterol-lowering effects after oral administration of 5 mg/kg ezetimibe for 14 days in percentage of the pretreatment (baseline) levels. Open circles indicate data from wild type, and solid circles from Mrp2-deficient TR-negative Lew.1W rats. Spearman rank correlations (r) and significance levels (p) are given.
The magnitude of changes in ezetimibe disposition caused by the absence of Mrp2 may have been augmented by particularities in the expression of hepatic drug transport and metabolism in Mrp2-deficient rats. In our TR-negative rats, the expression of hepatic mRNA of Mdr1a, Mdr1b, Mrp3, and Ugt1a1 as well as protein content of Mrp3 and Ugt1a, was significantly increased, probably by adaptation of liver cells to deficient Mrp2-mediated canalicular export. Similar upregulation of Mrp3 and Ugt1a was recently shown in GY/TR- Wistar rats (Johnson et al., 2006). A compensatory up-regulation of Mrp3 was also observed in patients with the Dubin-Johnson syndrome (Konig et al., 1999). Mrp3 serves in hepatocytes as a basolateral efflux transporter whose activity is enhanced during pathophysiological situations of hepatic elimination, such as cholestasis leading to a boosted urinary detoxification pathway (Keppler and Konig, 2000; Chan et al., 2004). Increased activity of hepatic Ugt1a1 may have additionally lowered ezetimibe availability in blood but may have increased exposure with the glucuronide synergistically to the effects of Mrp2 deficiency and Mrp3 overexpression. Because there was no specific Ugt1a1 rat antibody available, we used for our Western blot analysis a cross-reactive human UGT1A family antibody and could show a markedly higher expression of the according isoenzyme family.
The molecular mechanism behind P-gp, Mrp3, and Ugt1a1 up-regulation in Mrp2-deficient animals may result from induction of the nuclear farnesoid X receptor, PXR, and constitutive androstane receptor by bile acids that are elevated in Mrp2-deficient animals and known inducers of the mentioned nuclear receptors (Takikawa et al., 1991; Xie et al., 2001; Guo et al., 2003).
In conclusion, Mrp2 deficiency in rats is associated with a decreased sterol-lowering effect of ezetimibe that is most likely caused by a reduced intestinal clearance of the glucuronide and a decreased enterosystemic recycling of the parent drug ezetimibe to the intestinal NPC1L1 sterol-uptake transporter compartment.
Footnotes
-
The work was supported by the German Federal Ministry for Education and Research Grant 01ZZ0403 and an institutional research grant of MSD Sharp and Dohme.
-
doi:10.1124/jpet.106.104018.
-
ABBREVIATIONS: NPC1L1, Niemann-Pick C 1-like 1; UGT, UDP-glucuronosyltransferase; MRP/Mrp, multidrug resistance-associated protein; P-gp, P-glycoprotein; PXR, pregnane X receptor; CAR, constitutive androstane receptor; Mdr, multidrug resistance; TR-, Mrp2-deficient; Bsep, bile salt exporting pump; Ntcp, sodium taurocholate transporting polypeptide; OATP/Oatp, organic anion transporting polypeptide; TBST, Tris-buffered saline containing 0.05% Tween 20; LC-MS, liquid chromatography-mass spectrometry; CL, clearance; Lew.1w, Lewis.1w.
- Received March 3, 2006.
- Accepted June 8, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}