Article Text

Abstract
Background: Patients with Crohn’s disease suffer from intestinal bile acid malabsorption. Intestinal bile acid absorption is mediated by the apical sodium dependent bile acid transporter ASBT/IBAT (SLC10A2). In rats, ASBT is induced by glucocorticoids.
Aims: To study whether human ASBT is activated by glucocorticoids and to elucidate the mechanism of regulation.
Patients and methods: ASBT expression in ileal biopsies from patients with Crohn’s disease and from healthy subjects was quantified by western blot. ASBT promoter function was studied in luciferase assays and by electrophoretic mobility shift assay.
Results: In 16 patients with Crohn’s disease, ASBT expression was reduced to 69 (7.5)% compared with healthy controls (mean (SEM); p = 0.01). In 10 healthy male volunteers, ASBT protein expression was increased 1.34 (0.11)-fold (mean (SEM); p<0.05) after 21 days’ intake of budesonide (9 mg/day) whereas expression of the peptide transporter 1 was unaffected. Reporter constructs of the human ASBT promoter were activated 15–20-fold by coexpression of the glucocorticoid receptor (GR) and exposure to the GR ligands dexamethasone or budesonide. Two glucocorticoid response elements in the ASBT promoter, arranged as inverted hexanucleotide repeats (IR3 elements), conferred inducibility by GR and dexamethasone in a heterologous promoter context and were shown to bind GR in mobility shift assays.
Conclusions: Human ASBT is induced by glucocorticoids in vitro and in vivo. Induction of ASBT by glucocorticoids could be beneficial in patients with Crohn’s disease who exhibit reduced ASBT expression. This study identifies ASBT as a novel target of glucocorticoid controlled gene regulation in the human intestine.
- Crohn’s disease
- bile acids
- gene regulation
- intestinal transport
- steroid receptors
- ASBT, apical sodium dependent bile acid transporter
- SLC, solute carrier gene family
- HNF, hepatocyte nuclear factor
- PPAR, peroxisome proliferator activated receptor
- GC, glucocorticoids
- GR, glucocorticoid receptor
- GRE, glucocorticoid response element
- Caco2 cells, colon carcinoma cells
- Huh7 cells, human hepatoma cells
- DMSO, dimethyl sulphoxide
- PEPT1, peptide transporter 1
- UTR, untranslated region
- SXR, steroid/xenobiotic receptor
- PXR, pregnane X receptor
- ER, everted hexanucleotide repeat
- IR, inverted hexanucleotide repeat
- TK, thymidine kinase
- IBD, inflammatory bowel disease
- PCR, polymerase chain reaction
Statistics from Altmetric.com
- ASBT, apical sodium dependent bile acid transporter
- SLC, solute carrier gene family
- HNF, hepatocyte nuclear factor
- PPAR, peroxisome proliferator activated receptor
- GC, glucocorticoids
- GR, glucocorticoid receptor
- GRE, glucocorticoid response element
- Caco2 cells, colon carcinoma cells
- Huh7 cells, human hepatoma cells
- DMSO, dimethyl sulphoxide
- PEPT1, peptide transporter 1
- UTR, untranslated region
- SXR, steroid/xenobiotic receptor
- PXR, pregnane X receptor
- ER, everted hexanucleotide repeat
- IR, inverted hexanucleotide repeat
- TK, thymidine kinase
- IBD, inflammatory bowel disease
- PCR, polymerase chain reaction
Intestinal absorption of bile acids is a specific function of the ileum that is mediated primarily by the apical sodium dependent bile acid transporter ASBT (SLC10A2). Human ASBT is a 348 amino acid protein that transports conjugated and unconjugated bile acids with high efficiency and is abundantly expressed in the ileum.1 Mutations in the ASBT gene have been described in primary bile acid malabsorption,2 an intestinal disorder that presents with congenital chologenic diarrhoea.3 A more common clinical entity associated with intestinal bile acid malabsorption is inflammatory bowel disease (IBD), particularly Crohn’s disease.4–8 Intestinal bile acid absorption is a major determinant of the amount of bile acids that return to the liver and consequently of the bile acid pool size.9 Faecal bile acid loss can be partly compensated for by de novo hepatic synthesis; however, patients with Crohn’s disease have increased bilirubin secretion rates into bile and an increased risk of pigment gall stone formation, presumably due to increased colonic bile salt levels, which solubilise unconjugated bilirubin, prevent calcium complexing, and promote its absorption and enterohepatic cycling.10 ASBT has emerged as a pharmaceutical target for the treatment of lipid disorders but also for drug targeting strategies based on the coupling of drugs to bile acids in order to enhance intestinal absorption and hepatic uptake.11,12
Ileal bile acid transport is a highly regulated process at the level of gene transcription. The rat ASBT gene promoter contains an AP-1 element that binds and is activated by c-Jun.13 The human ASBT gene binds and is transactivated by the hepatocyte nuclear factor HNF1α and by the peroxisome proliferator activated receptor PPARα.14,15 In rats, a major inducing effect on ASBT function is exerted by glucocorticoids (GC). Intraperitoneal injection of corticosterone over three days increases ASBT protein expression eightfold.16 Subcutaneous injection of dexamethasone (0.1 μg/g body weight) over four days increases ASBT mRNA 20-fold.17 Treatment with either corticosterone or methylprednisolone significantly stimulates Na+ dependent taurocholate transport in ileal brush border membrane vesicles.16,18 Conversely, adrenalectomy performed in adult rats reduces active ileal taurocholate transport without changing the Km, suggesting a decrease in ASBT expression.19 The pharmacological effect of GC on ileal bile acid transport is of major interest in view of the central role of GC for the treatment of Crohn’s disease. GC can exert their biological activity through the glucocorticoid receptor (GR), a nuclear steroid hormone receptor with a high level of expression in the ileum and colon.20 GR are present in an inactivated form in the cytoplasm as part of a chaperone multiprotein system.21 After interaction with GC, the ligand associated GR complex travels as a homodimer to the nucleus to activate transcription through interaction with a short palindromic DNA consensus sequence called glucocorticoid response element (GRE).21 After contact with DNA, GR can recruit an array of cofactors that influence the assembly of the pre-initiation complex of transcription and the basal transcription machinery.
Although GC have been shown to induce ASBT function and expression in rat models, it is unknown whether human ASBT is regulated by GC. To address this question, we first measured ASBT expression in patients with Crohn’s disease and investigated whether the GR ligand budesonide, that is commonly used for the treatment of active Crohn’s disease, affects ASBT expression in human subjects. We then studied the effect of GC on the human ASBT gene promoter and searched for potential GREs. Our results identified GR as a novel regulator of the ASBT gene and defined an important pharmacodynamic effect of GC on the major human intestinal bile acid uptake system.
MATERIALS AND METHODS
Western blot analysis
Total protein was extracted from ileal biopsies obtained from patients with Crohn’s disease or healthy volunteers. Chemiluminescent western blot analysis (Perkin Elmer, Boston, Massachusetts, USA) was performed with ASBT antibody, raised as a GST fusion protein against the C terminal 40 amino acids (kindly provided by Dr PA Dawson, Department of Internal Medicine, Wake Forest University School of Medicine, Winston-Salem, North Carolina, USA), in a 1:5000 dilution (8 mg/ml). Antibody against the peptide transporter PEPT1 (kindly provided by Dr W Sadée, Department of Biopharmaceutical Sciences, University of California, San Francisco, USA) was affinity purified (AminoLink Plus Immobilization Kit; Pierce, Perbio Science, Lausanne, Switzerland) and used in a 1:25 dilution (0.1 mg/ml). For normalisation of expression, blots were stripped (62.5 mM Tris/HCl, pH 6.8, 0.2% sodium dodecyl sulphate, 110 mM mercaptoethanol) and reprobed with a 1:1000 dilution of villin antibody (Chemicon, Juey Supply Gmbtt, Lucerne, Switzerland). For quantification, blots were scanned with a TLC Scanner II using the CatS3 software (Camag, Muttenz, Switzerland).
Human study
The study protocol was approved by the local ethics committee and the Federal Office of Public Health. Ten healthy male volunteers completed the trial after giving written informed consent. Mean age was 25 years (range 23–29) and mean body mass 75 kg (range 57–92). Medical history, physical examination, and standard clinical chemistry and haematology parameters were normal. No medications were allowed for at least five days before the study and throughout the study period. Subjects underwent ileocolonoscopy without requiring sedation on study days 1 and 22 following peroral colonic lavage with macrogol solution (Fordtran; Streuli Pharma, Uznach, Switzerland). Eight ileal biopsies were immediately frozen in liquid nitrogen and stored at −70°C, and another four biopsies were taken for histopathological evaluation. Beginning on study day 2, subjects took budesonide (Entocort CIR; Astra Zeneca, Zug, Switzerland) at a dose of 3×3 mg/day for 21 days. The study period ended with the second ileocolonoscopy on study day 22. Ileal biopsies were analysed by western blot for expression of ASBT, PEPT1, and villin.
Plasmid construction
The ASBT reporter constructs −1688/UTR-Luc, −1492/UTR-Luc, and UTR-Luc have been reported previously.15 Two additional deletional constructs of the 5′ region of the human ASBT gene were amplified by polymerase chain reaction (PCR) using the plasmid −1688/UTR-Luc as template, upstream primers ASBT/−830for or ASBT/−492for, downstream primer ASBT/+526rev (table 1), and PfuTurbo DNA polymerase (Gibco Invitrogen, Basel, Switzerland). The resulting PCR products were ligated into the luciferase reporter gene vector pGL3-Basic (Promega Catalys AG, Wallisellen, Switzerland), yielding promoter constructs −830/UTR-Luc and −492/UTR-Luc. The −735/UTR-Luc construct was generated by excising an AflII/SacI fragment from −1688/UTR-Luc. Sequence identity of the constructs with the ASBT gene was verified by sequence analysis.
Oligonucleotides used for chimeric plasmid construction and mobility shift assays
Cell culture and reporter assays
Colon carcinoma (Caco2) and human hepatoma (Huh7) cell lines were cultured as described previously.15 Caco2 cells were transfected with Lipofectamin 2000 reagent (Gibco Invitrogen). Plasmid DNA for reporter assays comprised 400 ng luciferase promoter construct, 50 ng pSV-β-galactosidase plasmid, and either 50 ng pCMX-GR expression plasmid (kindly provided by Dr K Yamamoto, Department of Cellular and Molecular Pharmacology, University of California, San Francisco, USA) or 50 ng pBluescript vector (pB-SKII; Clontech, Basel, Switzerland) as carrier DNA to ensure that the total DNA amount transfected remained constant. Eighteen hours after transfection, cells were treated with either dexamethasone or budesonide (Sigma, St Louis, Missouri, USA) dissolved in water or dimethyl sulphoxide (DMSO), or with water or DMSO alone, as indicated, for 24 hours. Luciferase and β-galactosidase reporter assays were performed as described previously.15
Electrophoretic mobility shift assays
Gel mobility shift assays were performed as described previously using nuclear extracts from Caco2 and Huh7 cells and the oligonucleotide probes shown in table 1.15,22 For competition assays, 500 fmol unlabelled dimerised oligonucleotide was added (table 1). For supershift experiments, 2 μl of antibody against GR (E-20, sc1003×; Santa Cruz Biotechnology Inc., Heidelberg, Germany) were employed.
Statistical analysis
ASBT expression in 16 patients with Crohn’s disease was compared with 10 normal volunteers using the Mann-Whitney U test. Intraindividual changes in ASBT versus PEPT1 expression before and after budesonide intake were compared using the paired Wilcoxon signed rank test. Reporter gene activities are expressed as the mean (SD) of at least three individual transfection experiments. All data were reproduced at least once using two different preparations of plasmid DNA.
RESULTS
ASBT expression is reduced in Crohn’s disease
To investigate whether the known association of Crohn’s disease with intestinal bile acid malabsorption is attributable to decreased expression of ASBT,4–8 ASBT protein levels were quantified in ileal biopsies from 16 patients with Crohn’s disease. Patient biopsies were taken from the Zurich IBD tissue bank and selected on the basis of histologically intact ileal mucosa with no inflammatory infiltrate. As shown in fig 1, ASBT expression was reduced in the majority of Crohn patients compared with controls (ratio ASBT/villin 1.27 (0.06) in normal volunteers v 0.88 (0.10) in Crohn patients (mean (SEM)); p = 0.01). Only patient No 5 had mild inflammation histologically, indicating that decreased ASBT expression in Crohn patients was not correlated with local inflammation. These data support the hypothesis that bile acid malabsorption in Crohn’s disease is caused by decreased expression of ASBT.

Analysis of apical sodium dependent bile acid transporter (ASBT) expression in patients with Crohn’s disease. Ileal biopsies from 16 patients with Crohn’s disease were selected from the Zurich inflammatory bowel disease tissue bank on the basis of histologically documented normal mucosa with no or only mild (patient No 5) inflammation. Homogenates (20 μg) of ileal biopsy specimens were analysed by western blotting. ASBT expression was normalised to expression of villin and was found to be decreased to 69 (7.5)% (p = 0.01) compared with ASBT expression in controls (normal volunteers in fig 2, before budesonide treatment).
The GR ligand budesonide induces ileal ASBT expression in human subjects
In rats, GC have been shown to upregulate taurocholate transport in the ileum through induction of ASBT expression.16 We therefore investigated whether ASBT expression in the human ileum is induced in response to the GR ligand budesonide. We chose budesonide for three reasons: (i) budesonide is a potent glucocorticoid that is well absorbed in the intestine but undergoes extensive hepatic first pass metabolism, thereby eliminating systemic side effects in human subjects23,24; (ii) budesonide has a >9-fold binding affinity to GR compared with dexamethasone25; and (iii) budesonide is commonly used in the treatment of Crohn’s disease and characterisation of its effects on ileal ASBT expression is therefore of pharmacological interest.26 Ten healthy male volunteers underwent ileal biopsy before and after 21 days of treatment with oral budesonide (Entocort CIR 9 mg/day). Ileal disease was excluded by endoscopic and routine histological evaluation in all cases. Total RNA and protein were isolated from two pooled biopsies from each volunteer before and after treatment. ASBT protein expression was quantified by western blot analysis and was normalised for expression of villin, a 95 kDa protein required for the cytoskeletal and membrane organisation of the brush border epithelium.27 Compared with ASBT expression before budesonide treatment, ASBT protein levels were induced 1.25–2.13-fold in six of 10 volunteers (fig 2). To test for the specificity of ASBT induction by budesonide, expression of PEPT1 was analysed. PEPT1 has the same intestinal distribution as ASBT and has been shown to be unaffected by three days of treatment with dexamethasone in rats.28 Whereas PEPT1 protein levels after budesonide treatment were 1.024 (0.04) in relation to pretreatment values (mean (SEM), calculated as the ratio PEPT1/villin), ASBT expression increased to 1.34 (0.11) (mean (SEM); p<0.05 versus the change in PEPT1 expression), indicating that the budesonide effect was specific for ASBT (fig 2). Moreover, reporter constructs of the human PEPT1 gene promoter were not affected by coexpression of GR and incubation with dexamethasone in transfected Caco2 cells (D Jung and GA Kullak-Ublick, unpublished data). These data indicate that human ASBT is induced in vivo by budesonide treatment.

Induction of ileal apical sodium dependent bile acid transporter (ASBT) expression by budesonide in human subjects. Ileal biopsies were taken from 10 male volunteers (P1–P10) before (b) and after (a) treatment with budesonide (9 mg/day) for 21 days. Homogenates (20 μg) of ileal biopsy specimens were analysed by western blotting. Molecular size markers are shown on the left. ASBT expression was normalised for expression of villin and was found to be increased 1.25–2.13-fold in six of 10 subjects. For control purposes, expression of the peptide transporter PEPT1 was analysed on a separate membrane and was unchanged by budesonide treatment. The factor of ASBT induction in all 10 subjects was 1.34 (0.11) (mean (SEM); p<0.05 versus the change in PEPT1 expression).
Human ASBT (SLC10A2) gene promoter is activated by the glucocorticoid receptor
Because the biological activities of GC are mediated by GR, we studied whether the ASBT gene promoter contains GREs that confer induction by GC. A luciferase promoter construct of the human ASBT gene containing 1688 nucleotides of the 5′ flanking sequence and a major part of the 5′ untranslated region (UTR) conferred significant baseline luciferase activity in transfected Caco2 cells compared with the promoterless control vector pGL3-Basic (fig 3). Cotransfection of a GR expression plasmid induced ASBT promoter activity ∼30-fold in the presence of the GR ligand dexamethasone (100 nmol/l) whereas coexpression of GR in the absence of dexamethasone, or incubation with dexamethasone in the absence of coexpressed GR, had no effect (fig 3). These data indicate that the human ASBT promoter is activated by GR.
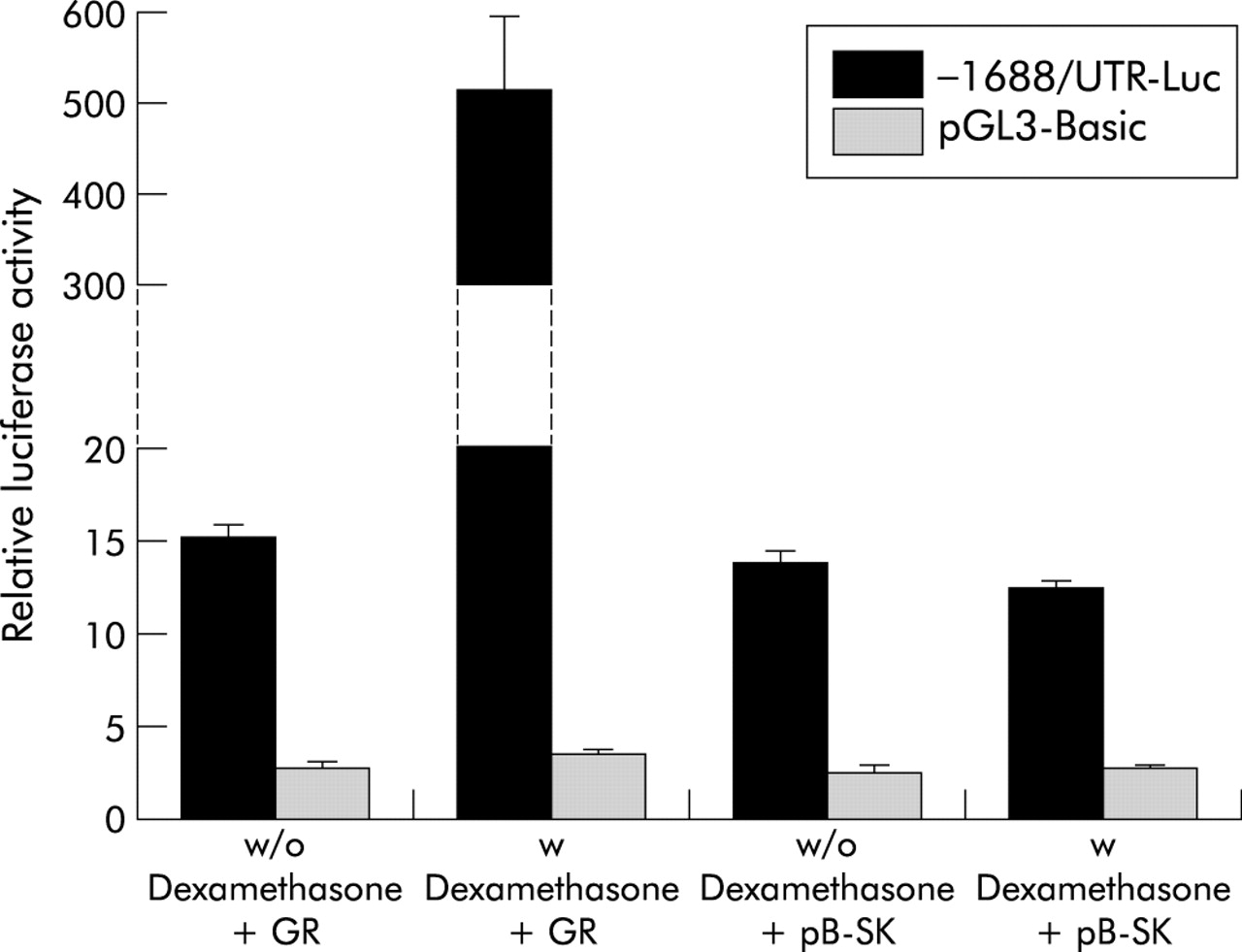
Activation of the human apical sodium dependent bile acid transporter (ASBT) promoter by the glucocorticoid receptor (GR) in colon carcinoma (Caco2) cells. Caco2 cells were transfected with the −1688/UTR-Luc construct or the promoterless control vector pGL3-Basic, in combination with either GR expression plasmid (+GR) or carrier DNA (+pB-SK). Eighteen hours after transfection, cells were treated with 100 nmol/l dexamethasone (w Dexamethasone) or water (w/o Dexamethasone). Coexpression of GR in the presence of the ligand dexamethasone led to a ∼30-fold induction in ASBT promoter activity. Transfection efficiency was normalised by cotransfection of pSV-β-galactosidase and promoter activity is expressed as relative light units of luciferase per unit of β-galactosidase. Data are expressed as the mean (SD) of 6–8 transfections.
To determine the dexamethasone concentration range at which activation occurs, the effect of 1 pmol/l to 1 μmol/l dexamethasone on ASBT promoter activity was assayed in Caco2 cells cotransfected with the ASBT reporter construct and GR expression plasmid. Maximal induction (∼20-fold) by dexamethasone was observed at 0.1–1 μmol/l (fig 4A). At 1 nmol/l, the factor of induction was still 10-fold. The GR ligand budesonide also induced ASBT promoter activity, with maximal induction (∼15-fold) occurring at 0.1–1 μmol/l (fig 4B). In contrast with dexamethasone, significant activation (∼7-fold) was still seen at 0.1 nmol/l, suggesting a higher affinity of GR for budesonide than for dexamethasone. Induction of the human ASBT promoter occurs at concentrations of ligand that typically activate GR but are too low to activate the steroid or pregnane X receptor (SXR/PXR).29–31

Concentration dependence of apical sodium dependent bile acid transporter (ASBT) promoter induction by glucocorticoids. Colon carcinoma (Caco2) cells were cotransfected with glucocorticoid receptor expression plasmid and either the −1688/UTR-Luc construct or pGL3-Basic control vector and treated with dexamethasone or budesonide at the indicated concentrations. (A) The −1688/UTR-Luc construct was activated by dexamethasone at concentrations ranging from 10−9 mol/l (1 nmol/l) to 10−6 mol/l (1 μmol/l) whereas pGL3-Basic showed no induction. (B) Activation by budesonide occurred at concentrations as low as 10−10 mol/l (0.1 nmol/l). Promoter activity is shown as the ratio luciferase/β-galactosidase and is expressed in relation to values measured in water treated cells (0). Data represent the mean (SD) of 6–8 transfections.
Identification of glucocorticoid response elements in the ASBT promoter
To identify potential GREs in the ASBT promoter, the sequence from nt −1688 to +598 relative to the transcription initiation site was analysed using a weighted matrix based computational approach.32 GREs are short palindromic DNA consensus sequences (TGTTCT as half site) with which ligand associated GR complexes interact.21 A functional GRE consists of two hexamer half sites, each half site recognised by a single subunit of the GR homodimer.33 Next to the consensus GRE originally identified in the mouse mammary tumour virus promoter,34 GR has been shown to bind to and promote transactivation from specific variants of the GRE,33,35 including direct (DR), everted (ER), and inverted (IR) repeats of TGTTCT with different spacings between half sites.35,36 Palindromic GREs arranged as IR3 elements are established GR binding motifs.37 Analysis of the human ASBT promoter identified two highly conserved IR3 motifs, termed IR3-A and IR3-B (fig 5A). IR3-A (AAAACAcgtTGTTCT) is located at nt −781 to −795 and IR3-B (AGGACAatcTCTTCT) at nt −549 to −563. In addition, a cluster containing overlapping ER7, IR2, and IR5 motifs was identified at nt −1062 to −1091 (fig 5A).
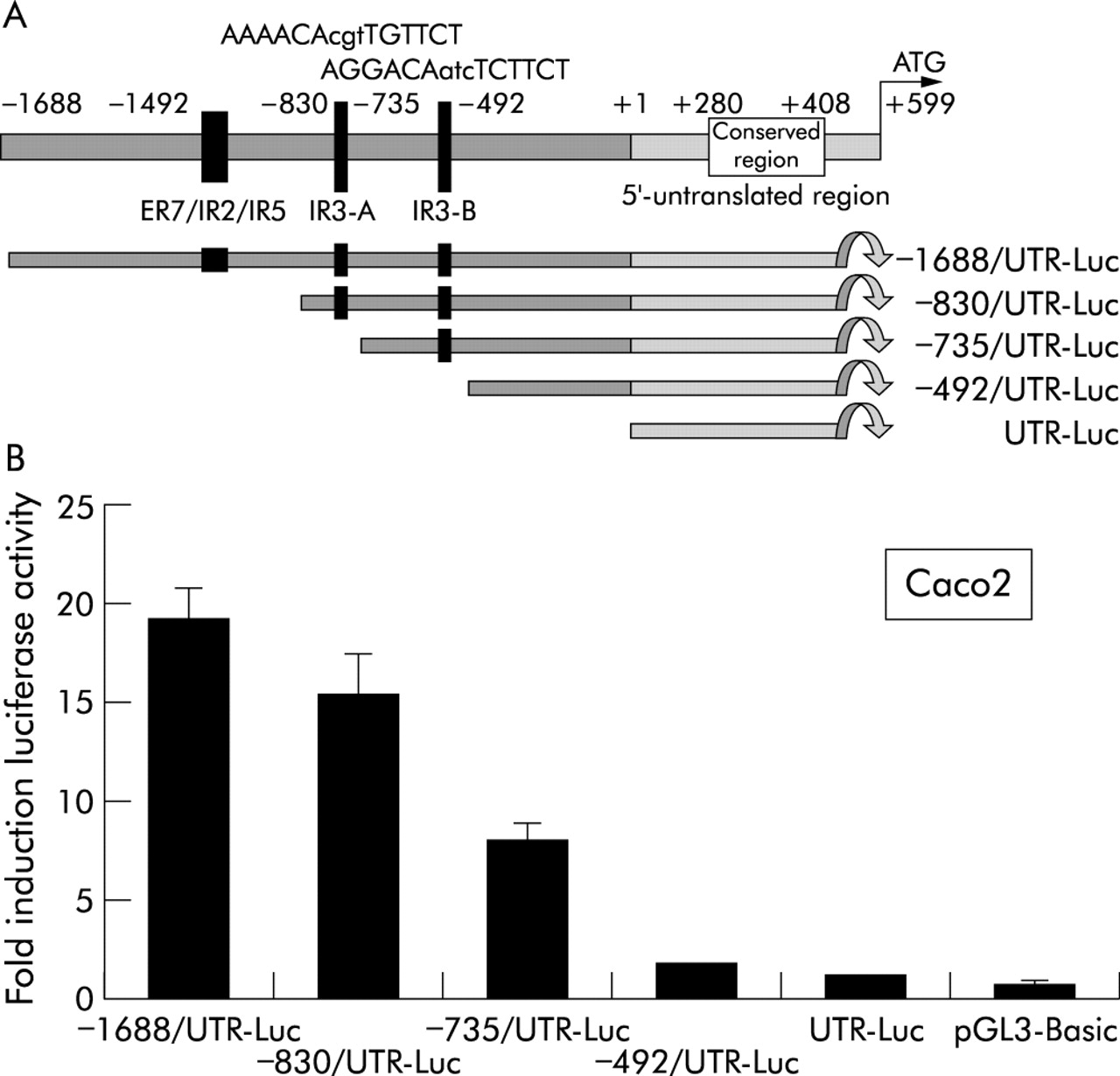
Functional analysis of consensus glucocorticoid response elements in the human apical sodium dependent bile acid transporter (ASBT) promoter. (A) Sequence analysis of the ASBT promoter identified two inverted hexanucleotide repeat IR3 elements termed IR3-A and IR3-B and a more distal cluster of overlapping consensus motifs termed ER7/IR2/IR5. Deletional constructs were designed to study the functional role of each of these elements. (B) Colon carcinoma (Caco2) cells were cotransfected with glucocorticoid receptor expression plasmid and deletional ASBT promoter constructs. UTR-Luc contained only the untranslated region of the ASBT gene without the 5′ flanking sequence. Eighteen hours after transfection, cells were incubated with 100 nmol/l dexamethasone. Promoter activity was induced in all constructs containing at least 735 nt of the 5′ flanking sequence. Progressive induction was observed in constructs containing the IR3-B and IR3-A motifs (constructs −735/UTR-Luc and −830/UTR-Luc, respectively), whereas the ER7/IR2/IR5 motif conferred no additional inducing effect. Promoter activity is shown as the ratio luciferase/β-galactosidase and is expressed in relation to promoter activity of each construct measured in the absence of dexamethasone. Data represent the mean (SD) of 6–8 transfections.
To determine which of these elements could represent a functional GRE, Caco2 cells were cotransfected with deletional ASBT reporter constructs and GR expression plasmid. ASBT promoter activity was assayed in the presence of 100 nmol/l of the GR ligand dexamethasone. Construct −735/UTR-Luc, that contained the IR3-B motif, was activated eightfold in relation to promoter activity measured in the absence of dexamethasone (fig 5B). Inclusion of the IR3-A motif in construct −830/UTR-Luc led to a 15.5-fold induction. The −1688/UTR-Luc construct that contained the ER7/IR2/IR5 cluster was not induced further compared with the −830/UTR-Luc construct, suggesting that the IR3-A and IR3-B motifs are functional response elements for the GR.
The IR3-A and IR3-B motifs confer activation by glucocorticoids
To further confirm the role of the IR3-A and IR3-B motifs in activation by the GR, both IR3 and the ER7/IR5/IR2 elements were cloned in front of the thymidine kinase (TK) promoter of the luciferase reporter gene vector TK-Luc. The resulting reporter plasmids contained single copies of the respective elements as present in the sequence of the human ASBT gene. In GR cotransfected Caco2 cells, all TK plasmids had similar basal luciferase activities attributable to the TK promoter (fig 6). Treatment with dexamethasone (100 nmol/l) resulted in a 2.9-fold induction in IR3-B-TK-Luc and a fourfold induction in IR3-A-TK-Luc, whereas native TK-Luc and ER7/IR5/IR2-TK-Luc showed unchanged activity in the presence of the GR ligand. These data verified the functional role of both IR3 motifs as GR response elements.
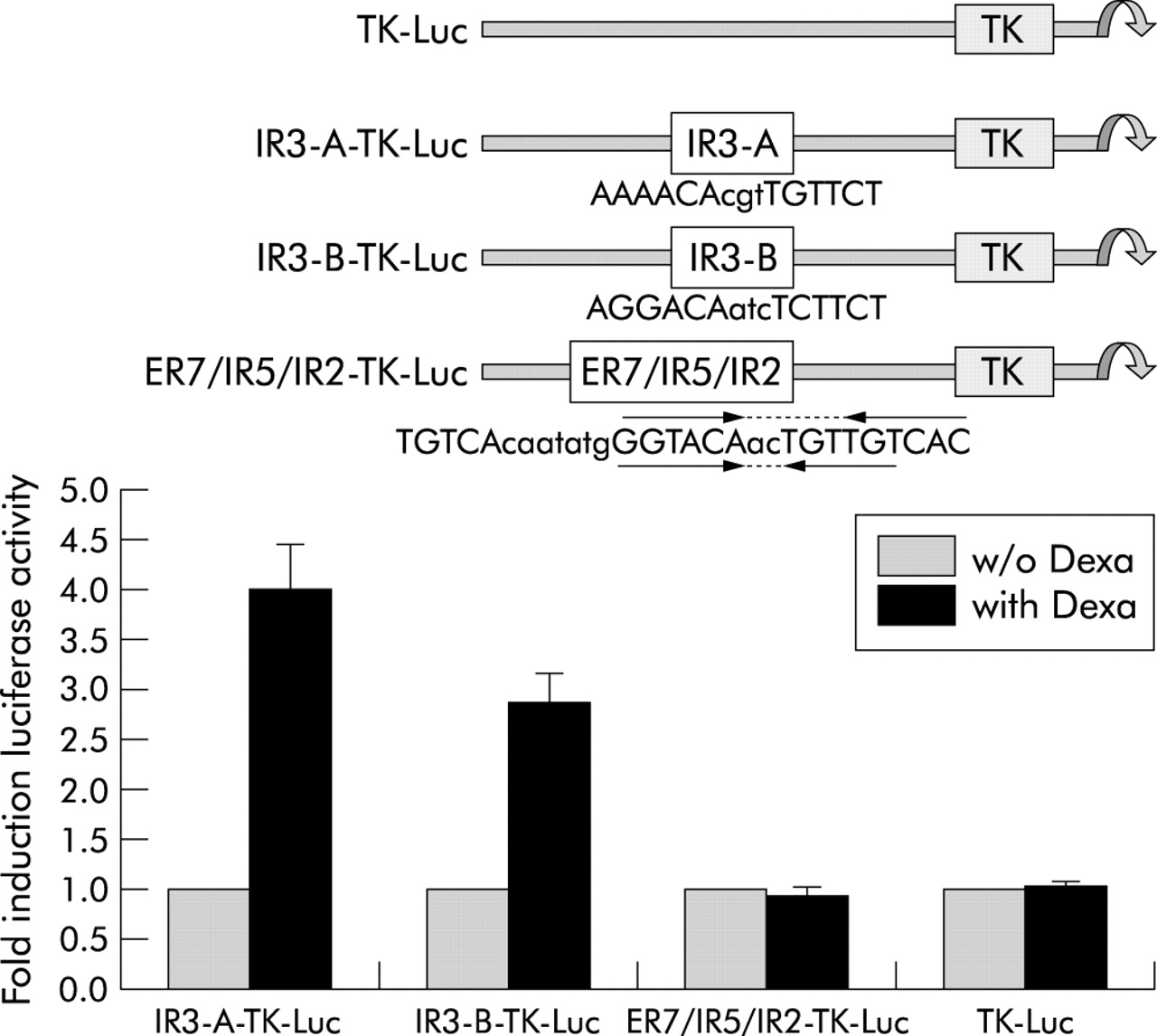
Role of the inverted hexanucleotide repeat IR3 elements in glucocorticoid receptor (GR) activation of the apical sodium dependent bile acid transporter (ASBT) promoter in a heterologous promoter context. Colon carcinoma (Caco2) cells were transfected with GR expression plasmid and either the TK-Luc, IR3-A-TK-Luc, IR3-B-TK-Luc, or ER7/IR5/IR2 plasmid. Treatment with 100 nmol/l dexamethasone resulted in a fourfold induction in IR3-A-TK-Luc and a 2.9-fold induction in IR3-B-TK-Luc, whereas ER7/IR5/IR2-TK-Luc and TK-Luc associated luciferase activity was unchanged. Promoter activity of each construct is expressed in relation to values obtained in the absence of dexamethasone (w/o Dexa).
The IR3 elements in the ASBT gene bind the GR
To determine whether the GR homodimer indeed binds to the predicted GREs in the ASBT gene promoter, electrophoretic mobility shift assays were performed. 32P labelled dimerised oligonucleotides corresponding to the IR3-A, IR3-B, and ER7/IR5/IR2 elements of the ASBT gene (table 1) were incubated with nuclear extracts prepared from Caco2 cells. Both the IR3-A and IR3-B sequences formed a visible DNA-protein complex whereas no protein binding was observed using the ER7/IR5/IR2 oligonucleotide (fig 7A). To confirm that the protein bound to the IR3 motifs was the GR, the IR3-A and IR3-B oligonucleotides were incubated with nuclear extracts from Huh7 and Caco2 cells in the absence or presence of antibody against GR. The GR antibody competed off binding to both the IR3-A and IR3-B elements, confirming that the bound protein was GR (fig 7B). To further test the specificity of binding to the IR3 sequences, an excess of unlabelled oligonucleotide was added to the reaction mix. Protein binding to the IR3-A element was inhibited by addition of excess unlabelled IR3-A and to a lesser extent IR3-B oligonucleotide (fig 7C). Binding to the IR3-B element was inhibited to a similar degree by both IR3-A and IR3-B competitor oligonucleotides (fig 7C), suggesting that the IR3-A sequence binds nuclear protein with higher affinity than IR3-B. Taken together, the data show that both IR3 motifs bind GR, indicating that the transactivating effect of GR on the human ASBT gene is attributable to these two GREs.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Binding of the glucocorticoid receptor (GR) to the inverted hexanucleotide repeat IR3 elements in the apical sodium dependent bile acid transporter (ASBT) promoter. (A) Electrophoretic mobility shift assays were performed using nuclear extracts from colon carcinoma (Caco2) cells and 32P labelled IR3-A, IR3-B, or ER7/IR5/IR2 oligonucleotides that corresponded to the sequence of the wild-type binding sites in the ASBT gene (table 1). Both the IR3-A and IR3-B oligonucleotides formed a visible DNA-protein complex whereas no detectable complex was formed using the ER7/IR5/IR2 oligonucleotide. (B) Addition of antibody against GR competed off binding of nuclear extracts from human hepatoma (Huh7) and Caco2 cells to the IR3-A sequence (top panel), implicating GR as the major factor that binds to this region. In the case of IR3-B, competition by GR antibody was detectable but less pronounced (lower panel). (C) Binding of nuclear proteins from Caco2 cells to the IR3-A sequence was competed off by an excess of unlabelled IR3-A and to a lesser extent IR3-B oligonucleotide (left panel). Binding to the IR3-B sequence (right panel) was competed off to a similar degree by both the IR3-A and IR3-B oligonucleotides, indicating that the IR3-A sequence binds GR with higher affinity.
DISCUSSION
Our study has demonstrated induction of the human ileal bile salt transporter gene ASBT (SLC10A2) by glucocorticoids. ASBT protein levels were reduced in ileal biopsies from patients with Crohn’s disease (fig 1). In healthy volunteers, ASBT expression was induced by three weeks of treatment with the GR ligand budesonide, an effect that could not be shown for the peptide transporter PEPT1 (fig 2). The promoter of the ASBT gene was induced in vitro by GR and GC (figs 3, 4) and contained two glucocorticoid response elements arranged as inverted hexanucleotide repeats (IR3 elements) (fig 5). Both elements were shown to bind the GR and conferred inducibility by dexamethasone in transfected Caco2 cells (figs 6, 7). Our data showed conclusively that the ASBT gene binds and is transactivated by GR, thereby adding an important target gene to the spectrum of GC regulated genes in the human intestine.
GR is the third transcription factor next to HNF1α and PPARα shown to transactivate the human ASBT gene.15 The clinical implications of ASBT activation by GC should be considered in view of the central role of corticosteroids for the treatment of inflammatory bowel diseases. In Crohn’s disease, controlled release formulations of budesonide are an established treatment modality for ileal and right colonic disease.23,24 Bile salt malabsorption in Crohn’s disease is a well characterised clinical entity.4,5,7,8 Patients with IBD have increased faecal excretion of bile salts and decreased absorption of the orally ingested radiolabelled bile acid 75Se-homotaurocholic acid.6 In a rabbit model, intestinal inflammation decreased ileal ASBT mRNA levels and experimental ileitis in hamsters decreased active bile acid uptake by 84%.38,39 The rat ASBT promoter is suppressed by cytokines through binding of a c-fos/c-jun heterodimer to an AP-1 element.40 The results of this study showed that patients with Crohn’s disease have reduced ASBT protein levels in ileal biopsies (fig 1). The beneficial effect of GC in Crohn’s disease could be related not only to suppression of inflammation and mucosal injury but also to increased bile acid absorption in the ileum through induction of ASBT expression. Reduced spillover of bile acids into the colon could improve the chologenic component of diarrhoea in these patients and might even reduce the risk of colon cancer imposed by increased exposure of the colonic mucosa to bile acids.41
GR belongs to the group of classic nuclear steroid hormone receptors that bind to DNA as homodimers following activation by steroid hormones. A second nuclear receptor that is activated by corticosteroids is the human steroid/xenobiotic receptor SXR and its rodent orthologue PXR.29–31 SXR/PXR belong to the orphan nuclear receptors that function as heterodimers with the retinoid X receptor RXR.42 They induce expression of intestinal multidrug resistance gene product 1 and cytochrome P450 3A4, which are important in the metabolism and elimination of xenobiotics and steroids.29,30 However, the affinity of corticosterone for SXR/PXR is in the range 10–30 μmol/l, compared with 30 nmol/l for GR.20 In this study, GR induction of the ASBT promoter occurred at a dexamethasone concentration of 1 nmol/l and at a budesonide concentration of 0.1 nmol/l (fig 4A, 4B), which is below the threshold for PXR activation. Moreover, no conserved binding motifs for PXR could be identified in the ASBT promoter sequence, suggesting that GR activation of the ASBT gene fully accounts for the inducing effect of GC.
In summary, in this study we have shown that the human ASBT gene is activated by the GR ligands dexamethasone and budesonide through direct binding of ligand activated GR to two IR3 motifs in the promoter region. Activation of the major human intestinal bile acid uptake system by glucocorticoids has major implications in view of the central role of GC for the treatment of inflammatory bowel disorders. The beneficial effect of GC could be related in part to increased ASBT expression and consequently reduced spillover of bile acids into the colon. GR mediated regulation of the ASBT gene provides an important new link between the pathways that govern bile acid homeostasis, lipid metabolism, and inflammation control.
Acknowledgments
This work was supported by grants 32-59155.99 and 632-062773 from the Swiss National Science Foundation, Bern, Switzerland (to GK), grant 2001-99 from the Roche Research Foundation (to GK), a research grant from the University of Zurich (to DJ) and Astra Zeneca AG, Zug, Switzerland (AF). The authors thank L Mazzucchelli (Department of Pathology, Inselspital Bern, Switzerland) for histological evaluation of ileal biopsies, M Kaelin (Enzym-Labor Dr Weber AG, St. Gallen, Switzerland) for routine laboratory testing in human subjects, M Wissmeyer (Institute of Nuclear Medicine, Inselspital Bern, Switzerland) and Streuli Pharma (Uznach, Switzerland) for providing macrogol colonic lavage solution (Fordtran Streuli). The expert technical assistance of Claudia Seitz is gratefully acknowledged.
REFERENCES
Footnotes
-
↵* D Jung and A C Fantin contributed equally to this work.