Article Text

Abstract
Objective To evaluate the efficacy and tolerability of add-on treatment with lesogaberan (AZD3355), a novel reflux inhibitor, in patients with persistent gastro-oesophageal reflux disease (GORD) symptoms despite proton pump inhibitor (PPI) therapy.
Methods A double-blind, placebo-controlled, randomised, parallel-group, multicentre phase IIA study was carried out in outpatient clinics. The study group comprised 244 adult patients with persistent GORD symptoms (heartburn and/or regurgitation) of at least mild intensity and for 3 days of 7 days before enrolment, despite ≥6 weeks of continuous PPI therapy. Patients received either lesogaberan (65 mg twice daily) or placebo in addition to PPI therapy for a period of 4 weeks. Symptom intensity, based on the Reflux Disease Questionnaire, was recorded twice daily. Treatment response (defined as at most one 24 h period with heartburn or regurgitation of not more than mild intensity during the last 7 days of treatment). Time to response, proportion of symptom-free days and measures of tolerability were also analysed.
Results A total of 232 (114 lesogaberan- and 118 placebo-treated patients) of the 244 randomised patients were analysed for efficacy. Treatment with lesogaberan, compared with placebo, resulted in a significantly larger proportion of responders to treatment (16% vs 8% of patients; p=0.026) and cumulative proportion of responders over time (log-rank p=0.0195). Lesogaberan was well tolerated: adverse events of mostly mild to moderate intensity were reported in 45% of patients on lesogaberan and in 37% on placebo.
Conclusions Lesogaberan add-on therapy to PPIs significantly improved heartburn and regurgitation symptoms; however, the proportion of responders was small.
Clinical trial number NCT00394472.
- Lesogaberan
- AZD3355
- gastro-oesophageal reflux disease
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
Approximately 20–30% of patients with gastro-oesophageal reflux disease (GORD) continue to experience persistent reflux symptoms despite daily proton pump inhibitor (PPI) therapy.
An unmet medical need for additional treatment options therefore remains for many patients with GORD.
What are the new findings?
The novel reflux inhibitor lesogaberan, administered at a dose of 65 mg twice daily as add-on therapy to a PPI, significantly improved heartburn and regurgitation symptoms, compared with placebo, in patients with persistent GORD symptoms despite daily PPI therapy.
Lesogaberan 65 mg twice daily was well tolerated in this patient population.
How might it impact on clinical practice in the foreseeable future?
Reflux inhibitors may have the potential to address the unmet medical need in patients who experience persistent reflux symptoms despite daily PPI therapy.
Introduction
Gastro-oesophageal reflux disease (GORD) is a common condition that affects 10–20% of adults in the Western world.1 Proton pump inhibitors (PPIs) are the most effective available treatment for this disease.2 However, clinical experience shows that 20–30% of patients with GORD continue to experience persistent reflux symptoms despite daily PPI treatment.3 An unmet medical need therefore remains for many patients with GORD.
Transient lower oesophageal sphincter relaxations (TLOSRs) are a normal physiological response to postprandial gastric distension, and have also been identified as a major contributing mechanism to GORD.4 Reducing the number of TLOSRs by pharmacological agents, referred to as reflux inhibitors, would therefore offer a novel approach to the treatment of GORD. A number of agents, including γ-aminobutyric acid type B (GABAB) receptor agonists and metabotropic glutamate receptor 5 (mGluR5) modulators, hold particular promise in this regard.5 For example, baclofen, a GABAB receptor agonist indicated for the treatment of neurological spasticity, has been shown to reduce the number of TLOSRs6 and reflux episodes6–8 in patients with GORD, including those with a large hiatal hernia.9 This pharmacodynamic profile translates into relief of reflux symptoms in patients with persistent GORD symptoms despite treatment with PPIs.8 However, baclofen is active in the central nervous system (CNS), resulting in CNS side effects such as dizziness, sleepiness and tiredness.7 10 11 Such effects may be difficult to tolerate in patients with GORD, thereby limiting the clinical usefulness of baclofen for this indication.
The reflux inhibitor lesogaberan (AZD3355) is a novel GABAB receptor agonist developed as an add-on treatment to PPIs for symptom relief in patients with GORD who have a partial response to PPI therapy, characterised by persistent GORD symptoms. As this compound predominantly targets peripheral, rather than central, GABAB receptors,12 it is expected to have better CNS tolerability than baclofen. Preclinical evaluations12 and early clinical pharmacodynamic studies13 14 have confirmed its potential utility as a reflux inhibitor.
This proof-of-concept study therefore aimed to assess the efficacy and tolerability of add-on treatment with lesogaberan in patients with GORD with persistent reflux symptoms despite treatment with PPIs.
Methods
Patients and study design
This was a double-blind, randomised, placebo-controlled, parallel-group, proof-of-concept study (http://ClinicalTrials.gov/ identifier: NCT00394472; study code: D9120C00011) conducted between November 2006 and June 2007 at 45 centres in eight countries (Australia, Belgium, France, Germany, Hungary, The Netherlands, Norway and Romania).
Adult patients (aged 18–70 years; upper age limit selected to limit the number of concomitant diseases that could confound the safety evaluation of lesogaberan) with a body weight of 60–100 kg (limits selected to minimise the variation in lesogaberan exposure) whose GORD symptoms persisted despite ≥6 weeks of continuous PPI therapy at approved doses (based on country-specific labels for any GORD indication) were screened for study inclusion. Patient eligibility was based on the following criteria assessed by symptom recall over the last 7 days: at least 3 days with ‘a burning feeling behind the breastbone’ (heartburn) and/or ‘unpleasant movement of material upwards from the stomach’ (regurgitation) of at least mild intensity, as determined using the Reflux Disease Questionnaire (RDQ).15 Patients with the dominant symptom of ‘pain in the centre of the upper stomach’ (intensity equal to or greater than a burning feeling behind the breastbone), without fulfilling the regurgitation inclusion criteria, were excluded from participation. Other exclusion criteria included: a history of clinically significant cardiovascular, respiratory, renal, hepatic, neurological, mental or gastrointestinal (GI) disease other than GORD (patients with uncomplicated and well-controlled diabetes mellitus or hypertension could be included); serum creatinine levels >1.2 times greater than the upper limit of normal; history of clinically significant orthostatic reactions; systolic blood pressure <110 mm Hg; prior surgery to the GI tract; history of severe allergy or hypersensitivity; or history of drug addiction or alcohol abuse. Patients who required concomitant treatment with the following medications were also excluded: drugs that may interfere with the pharmacodynamic effect of lesogaberan (eg, baclofen), drugs that may influence GI symptoms; drugs with a narrow therapeutic index (eg, warfarin, digoxin, phenytoin or carbamazepine); or drugs that may prolong the QT interval. Women were only included if postmenopausal (at least 12 months without a menstrual period and >50 years of age), or if they had been surgically sterilised (ie, bilateral oophorectomy or hysterectomy).
This study was approved by an independent Institutional Review Board/Research Ethics Committee within each country, and was performed in accordance with the ethical principles outlined in the Declaration of Helsinki. All patients provided written, informed consent prior to initiation of the study.
Treatments
After screening, eligible patients entered an 8–12 day run-in period with symptom and rescue medication recording, during which they continued their existing PPI treatment (figure 1). Thereafter, patients were randomised to 4 weeks of oral treatment with placebo or lesogaberan 65 mg twice daily (taken 30 min before breakfast and dinner) in addition to existing PPI treatment. Patients were randomised (1:1 ratio) to treatment groups sequentially based on a treatment allocation list generated by the study sponsor using a validated computer program. Treatment allocation was blinded for the patients, for study site personnel and for AstraZeneca personnel involved in the conduct of the study. Lesogaberan and placebo were packaged in identical capsules and were labelled identically to maintain blinding. Rescue antacid medication was provided at the enrolment visit and throughout the study.

Study design. GERD, gastro-oesophageal reflux disease; PPI, proton pump inhibitor.
Assessments
During the run-in period patients reported the intensity of symptoms, as well as use of rescue medication, twice daily using an electronic diary (eDiary), based on the RDQ, in the morning (for the preceding night) and at bedtime (for the day) over a period of at least 7 days. Symptom intensity was rated on a 6-point Likert scale (ranging from ‘did not have’ to ‘severe’), to provide an assessment of baseline symptom load.
Patients continued to evaluate symptoms and use of rescue medication on a twice-daily basis, using the eDiary, throughout the 4 week treatment period. The primary variable was treatment response, defined as at most one 24 h period with ‘burning feeling behind the breastbone’ (heartburn) or ‘unpleasant movement of material upwards from the stomach’ (regurgitation) of not more than mild intensity during the last 7 days of treatment. The responder status was set to missing if intensity was missing for more than two of the seven 24 h periods. There was an Ethics Committee (Germany)-requested reduction in sample size at an early stage of the study (the initial sample size of 400 patients was considered to be too large for a phase IIA study) and, as a result, the primary outcome variable was specified to account for the smaller sample size at that early stage without knowledge of efficacy data.
Secondary variables assessed for heartburn and/or regurgitation symptoms (as defined above) included: time to treatment response (defined as time from the day of the first dose to the first of seven consecutive days meeting the responder definition); the more stringent criterion of time to sustained absence of symptoms (defined as time from the day of the first dose to the first of seven consecutive symptom-free days); and the proportion of symptom-free days. The change from baseline in Gastrointestinal Symptom Rating Scale scores16 was explored and patients' consumption of rescue antacid medication over the 4 week treatment period was also investigated.
Patient compliance with treatment was determined by counts of returned unused capsules at each visit during the period of randomised treatment.
The safety of lesogaberan was evaluated by assessment of adverse events (AEs), vital signs, electrocardiography and laboratory tests. The intensity of AEs was categorised as being mild, moderate or severe by the investigators.
Statistical analysis
A one-sided χ2 test was used to compare the primary variable (treatment response) between lesogaberan- and placebo-treated patients. The difference between the treatment groups was calculated with a two-sided 95% CI, based on standard normal approximation. Survival curves with Kaplan–Meier estimates and a log-rank test were used to investigate the time (in days) to response and the time to sustained absence of symptoms. In addition, the mean maximum symptom intensity (heartburn and/or regurgitation) was calculated at baseline and at the last week of treatment, and treatments were compared at the last week of treatment using a Wilcoxon rank sum test.
As an exploratory analysis, the primary variable was analysed using logistic regression with a number of covariates thought to be potentially important for treatment response. These covariates included: treatment; maximum symptom intensity of heartburn and regurgitation at screening; heartburn or regurgitation being the symptom with highest intensity at screening (heartburn/regurgitation/the same intensity); sex; age; body mass index; history of reflux (erosive) oesophagitis (yes/no); history of hiatal hernia (yes/no); and duration of reflux disease (years). Due to the explorative nature of this study, no corrections for multiplicity were used.
Determination of sample size
Assuming a response rate to placebo of 20% based on the outcome in previous GORD studies, a sample size of 115 randomised patients per treatment group would allow for 90% statistical power to detect an increase in response of 15 percentage points difference (ie, a lesogaberan treatment response of 35%) in the primary variable between the treatment groups at the 10% significance level using a one-sided χ2 test. This significance level was considered sufficient for a phase IIA study with the primary objective to estimate the efficacy of lesogaberan 65 mg twice daily as add-on treatment to a PPI.
Analysis sets
All randomised patients who had received at least one dose of study medication were included in the safety analysis set. The efficacy analysis set consisted of patients who had received at least one dose of study medication and did not violate any major inclusion/exclusion criteria relevant to the evaluation of the efficacy variables such as not receiving correct study medication, not fulfilling symptom frequency and intensity requirements at inclusion, having undergone previous upper GI tract surgery, not taking protocol-prescribed PPI treatment or taking concomitant medication that could interfere with the pharmacodynamic effect of lesogaberan or influence GI symptoms. The evaluability of the data was determined and documented before any statistical analyses were performed.
Results
Patients
A total of 306 patients were enrolled and, of these, 62 patients were withdrawn prior to randomisation for reasons of incorrect enrolment or failure to meet inclusion criteria. Two hundred and forty-four patients (66% men; mean age 49.9 years) were randomised and received at least one dose of study medication (lesogaberan n=122; placebo n=122); premature discontinuations occurred in 9 and 11 of the patients in the lesogaberan and the placebo treatment group, respectively. The flow of patients through this study is summarised in figure 2.

Patient flow through the study.
Eight patients in the lesogaberan group were excluded from the efficacy analysis set due to the following reasons: significant GI disease other than GORD (n=1); intake of disallowed concomitant medication (n=4); PPI treatment stopped or changed prior to or during the study (n=2); and disallowed concomitant medication and change in PPI treatment (n=1). Four patients in the placebo group were excluded from the efficacy analysis set due to: intake of disallowed concomitant medication (n=2) and PPI treatment stopped or changed prior to or during the study (n=2). Thus, the number of patients in the efficacy analysis set was 232 (lesogaberan n=114; placebo n=118). Due to the handling of missing values in the calculation of the primary outcome variable (the responder status was set to missing if >2 out of the 7 days were missing) the number of patients contributing to the primary analysis was 209 (lesogaberan, n=104; placebo, n=105).
All randomised patients were Caucasian, and ∼28% were infected with Helicobacter pylori. More than half of the randomised patients reported a history of GORD of ≥5 years (58%) and/or previous reflux oesophagitis (57%). The most common PPIs used at baseline were esomeprazole 20 mg (23% of patients), omeprazole 20 mg (17%) and pantoprazole 40 mg (15%) once daily (table 1). The study population was notable for a high symptom load despite daily PPI therapy for at least 6 weeks. Seventy-six per cent of the randomised patients had daily heartburn and/or regurgitation, and 87% had moderate to severe symptoms (maximum intensity) during the run-in period (table 1). The treatment groups were well matched in terms of baseline characteristics, with the exception that fewer male patients were randomised to treatment with lesogaberan (58%) than placebo (75%) and the proportion of patients with a maximum of moderate to severe symptom intensity based on the primary RDQ items (heartburn and regurgitation) was lower in the lesogaberan group (81%) than in the placebo group (92%) (table 1).
Summary of patient characteristics
A high level of compliance with study treatment was observed, and >90% of patients in each treatment group were ≥75% compliant. Compliance with twice-daily (morning and evening) eDiary symptom recording was also high; the mean level of recording compliance was 91% in lesogaberan- and 88% in placebo-treated patients.
Treatment response
In the efficacy analysis set, significantly more lesogaberan-treated patients than placebo-treated patients responded to treatment (primary variable: 16% vs 8% responders; p=0.026); the difference between treatment groups was 9% (95% CI 0 to 17) (figure 3A). A sensitivity analysis including all randomised patients with known responder status (lesogaberan n=110; placebo n=109) showed similar treatment response (lesogaberan 15% vs placebo 8%; difference between treatment groups 7% (95% CI −1 to 16).

Proportion of (A) patients with treatment response (at most one 24 h period with heartburn or regurgitation of not more than mild intensity during the last 7 days of treatment); (B) symptom-free days during the 4 week treatment period for heartburn and regurgitation.
There was a lower mean maximum symptom intensity in the lesogaberan treatment group during the last treatment week as compared with placebo. However, when taking baseline intensity into consideration, the reduction of symptom intensity was similar in both treatment groups (table 2).
Mean maximum symptom intensity at baseline and last week of treatment for the primary Reflux Disease Questionnaire (RDQ) items
Only treatment (lesogaberan vs placebo OR 4.42 (95% CI 1.20 to 15.16), p=0.01) and maximum intensity of heartburn and regurgitation at screening (OR 0.14 (95% CI 0.05 to 0.42), p<0.001) were significant covariates in the explorative multiple logistic regression model of treatment response.
In a post hoc analysis of the efficacy analysis set, applying the responder definition on daytime symptoms and night-time symptoms separately (ie, at most one night with at most mild symptoms during the last seven nights on treatment, and analogously for daytime symptoms), treatment comparisons of daytime were similar to night-time. The analysis of morning recordings (response during the previous night) showed that 29% of lesogaberan- and 15% of placebo-treated patients met the criteria for response (between-group difference 14%; 95% CI 3% to 25%); 21% of lesogaberan- and 10% of placebo-treated patients met response criteria when bedtime recordings (response during the daytime) were analysed (between-group difference 12%; 95% CI 2% to 21%).
Time to treatment response and the sustained absence of symptoms
Over the 4 week treatment period, treatment with lesogaberan, compared with placebo, resulted in a significant treatment effect on the cumulative proportion of responders over time (log-rank p=0.0195; figure 4). This was the case for both heartburn (log-rank p=0.0011) and regurgitation (log-rank p=0.0077) (figure 5A,B). Furthermore, treatment with lesogaberan, compared with placebo, resulted in a significantly greater treatment effect with respect to time to the sustained absence of symptoms (heartburn and regurgitation; log-rank p=0.0408).
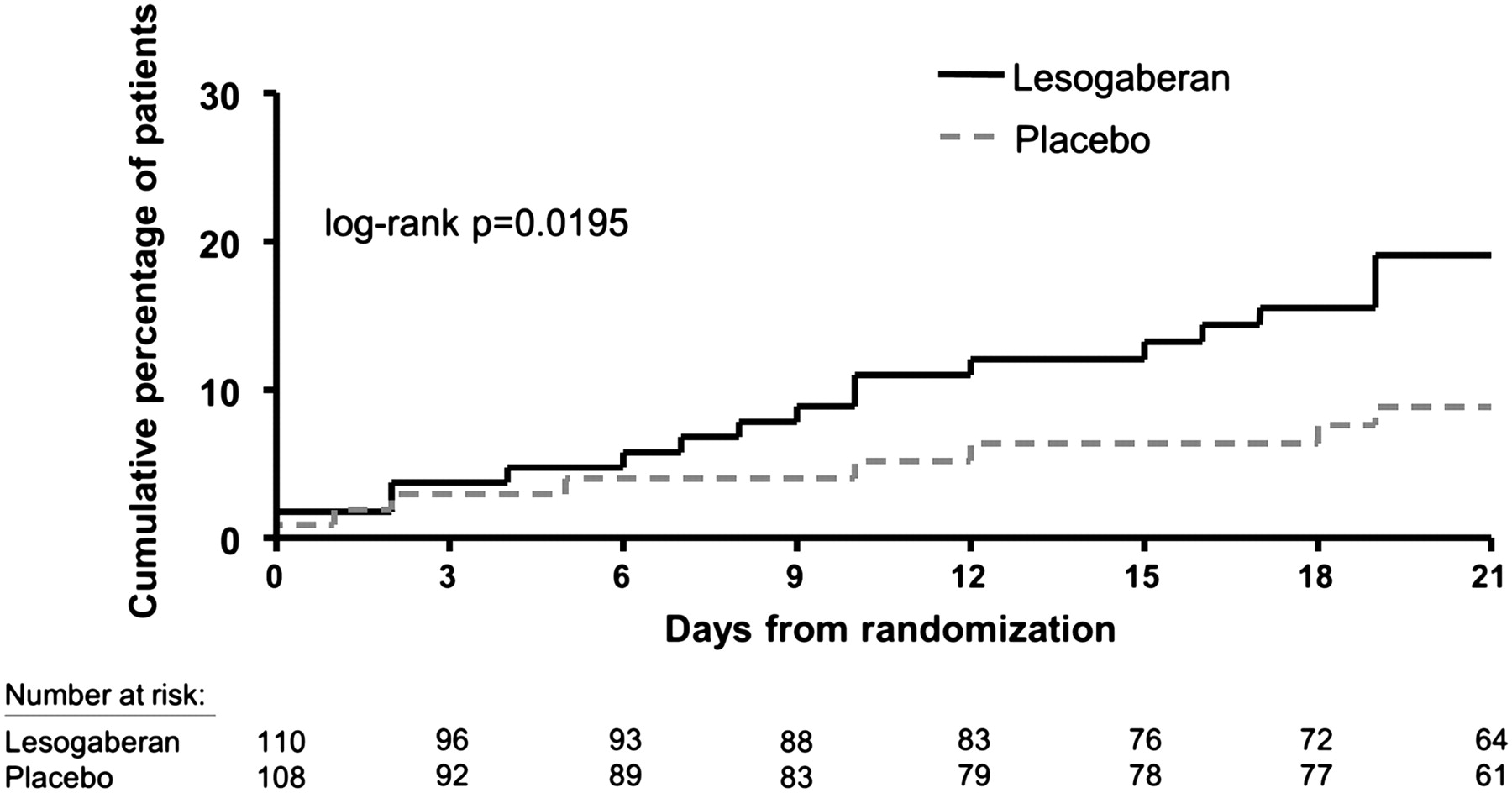
Kaplan–Meier curve of time to treatment response (the first of seven consecutive days with at most 1 day of not more than mild heartburn and regurgitation); day 0 corresponds to the first day of treatment.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Kaplan–Meier curve of time to treatment response (the first of seven consecutive days with at most 1 day of not more than mild heartburn (A) or regurgitation (B)); day 0 corresponds to the first day of treatment.
In accordance with the findings for treatment response, lesogaberan-treated patients had a higher proportion of days free of both heartburn and regurgitation symptoms over the 4 week treatment period compared with placebo (20% and 10%, respectively; between-group difference 10%; 95% CI 4% to 15%; figure 3B). The difference between lesogaberan- and placebo-treated patients in terms of heartburn-free days was 14% (95% CI 6% to 23%) and 13% with regard to regurgitation-free days (95% CI 4% to 22%) (figure 3B).
In the exploratory analysis of Gastrointestinal Symptom Rating Scale dimension scores, no clinically relevant differences in the mean change from baseline were observed between patients treated with lesogaberan and those who received placebo after the last week of treatment. The mean number of rescue antacid tablets consumed during each treatment day was similar in both treatment groups.
Tolerability
Lesogaberan was generally well tolerated. No deaths or treatment-related serious AEs were reported. The most commonly reported AEs were diarrhoea, transient episodes of paraesthesia, and nausea (table 3). Most AEs were mild or moderate in intensity.
Summary of adverse events*
Four lesogaberan- and two placebo-treated patients discontinued due to AEs. The reasons for discontinuation in the lesogaberan-treated patients were dizziness, headache, facial hypoaesthesia and nausea (female patient); headache, hypertension and vomiting (male patient); diarrhoea, dysphonia and nausea (female patient); and pruritus (male patient). The reasons for discontinuations among the placebo-treated patients were chest pain (female patient) and cough (male patient).
No clinically relevant changes in vital signs, electrocardiography recordings or laboratory tests were observed in either treatment group.
Discussion
This was the first randomised, placebo-controlled study to investigate the efficacy on symptom relief and tolerability of lesogaberan as add-on treatment in patients with GORD with persistent reflux symptoms despite PPI therapy. Patients presenting with persistent GORD symptoms despite PPI therapy have become a frequent occurrence in GI practice,17 imposing a considerable burden on healthcare resources. The mechanisms of symptom generation in these patients have not yet been fully elucidated and treatment options are currently limited.
The eDiary data collected at screening and during the run-in period indicated that the patients in the trial had a high GORD symptom load at baseline and thus confirmed that there is substantial need to improve symptom control in patients with GORD who do not respond sufficiently to PPI therapy. In this study a responder definition was applied that did not involve complete resolution of symptoms, as this may resemble what is achievable in these patients in clinical practice.
Patients' characteristics were generally well matched between the treatment groups. However, fewer male patients were included in the lesogaberan treatment group as an artefact of randomisation. The higher proportion of women in the lesogaberan treatment group may account for a somewhat higher mean age of this treatment group; women were required to not be of childbearing potential and thus were, on average, older than the male participants. Although gender and age were not significant covariates of treatment response, the safety-based exclusion of women of childbearing potential in this study may limit the generalisability of these study findings.
Overall, lesogaberan 65 mg twice daily, administered as add-on treatment to a PPI, significantly improved heartburn and regurgitation symptoms in this patient population (16% of lesogaberan- and 8% of placebo-treated patients). In addition to the primary efficacy variable, a consistent pattern of effects of lesogaberan on other efficacy variables versus placebo was noted. For example, lesogaberan-treated patients also had higher proportions of days free from heartburn and/or regurgitation symptoms compared with placebo over the whole treatment period, and lesogaberan treatment was beneficial in terms of the time to treatment response. Although these data provide proof of the concept that lesogaberan reduces GORD symptoms, it has to be acknowledged that the number of responders was low, with a therapeutic gain of 9%. However, to put this into context, the patients had a high baseline symptom burden and it should be noted that a substantial reduction in symptom frequency and intensity was required to fulfil the responder definition. By chance the placebo group turned out to have higher symptom intensity before treatment. Although this may have influenced the outcome in the study, symptom intensity was included in an explorative model adjusting for baseline covariates thought potentially to affect the outcome, which showed that study treatment remained a significant factor for response at the 5% level. The criterion for treatment response in this study included total symptom freedom on six of the seven last treatment days for both heartburn and regurgitation, allowing mild intensity on only 1 day. By including both heartburn and regurgitation the responder definition was more stringent than responder definitions commonly used in studies in patients with untreated GORD including only heartburn. Moreover, not all symptoms in patients with GORD with persistent symptoms despite PPI treatment can be expected to be due to reflux and, as such, these symptoms would not be likely to be influenced by reflux inhibition.17 In particular, the population of patients with GORD with persistent reflux symptoms despite PPI treatment is known to overlap significantly with groups of patients with functional heartburn who are characterised by poor responsiveness to PPIs.17 It would therefore be important to find criteria to identify and exclude these patients in future studies.
It should also be noted that no dose finding was done in this study, and only one dose of lesogaberan was tested in the intended patient population. Dose-ranging studies in dogs have shown that at least 50% mean reduction of TLOSRs can be achieved in dogs,12 while a mean reduction of 25% of TLOSRs was observed on lesogaberan 65 mg twice daily in a pharmacodynamic study in patients with GORD and persistent symptoms despite PPI treatment.14 This might indicate that improved pharmacodynamic effects might also be achieved in man on higher doses than given in the current and previous study in patients with GORD. However, whether greater efficacy against symptoms can also be achieved with higher doses of lesogaberan has to be explored further in a dose-finding study. Furthermore, the degree of response that is ‘clinically meaningful’ in this patient population remains to be fully determined.
The improvement in regurgitation is particularly noteworthy, and is in accordance with the reported mechanism of action of lesogaberan in humans in terms of decreasing TLOSRs and decreasing reflux episodes.13 14 The post hoc analysis revealed that the treatment differences were similar when daytime and night-time symptoms were analysed separately. The apparently higher response rates noted in the post hoc analysis of morning and bedtime recordings may be due to it being relatively easier to achieve a response (defined as at most one daytime or night-time period with at most mild symptom intensity) during seven consecutive 12 h day or night periods than during seven consecutive 24 h periods. The fact that TLOSRs occur primarily postprandially during the daytime, and in this study lesogaberan had a similar degree of efficacy during the night-time and the daytime suggest that some degree of efficacy may be related to lesogaberan-induced increases in lower oesophageal sphincter pressure14 or other, as yet unknown, mechanisms.
Based on the PPI doses used in this study, it is possible that PPI treatment and thus the acid suppression could have been suboptimal in some patients. Whether symptomatic improvement may have been achieved with twice-daily (double-dose) PPI treatment, which is often tried in patients with persistent GORD symptoms despite PPI treatment, remains unknown. However, there is no regulatory-approved indication for twice-daily PPI treatment in patients with GORD and there is a lack of supportive clinical data for a beneficial effect on GORD symptoms in this regard. Moreover, a substantial proportion of symptoms in patients with partial response to PPI therapy is related to weakly acidic reflux.17 18 Since PPIs seem to have very little effect on the total number of reflux episodes, the benefit with regards to reducing symptoms by PPI dose escalation in order to produce a more pronounced acid suppression would be limited in these patients.19
The eDiary approach in this study led to a high actual level of compliance of patients' symptom recordings, with recording at the correct time rather than retrospectively or in advance, while minimising the potential for clinician underestimation of symptom severity relative to patient ratings that may occur particularly before treatment and for more severe symptoms.20 The use of twice-daily symptom recording with the eDiary compared with, for example, 1 week symptom recall may have increased the likelihood of symptoms being recorded and, taken together, these factors have contributed to the production of a high-quality data set and robust analysis with regard to symptom response.
The potential difficulty in maintaining blindness in randomised clinical trials of GABAB agonists due to the occurrence of paraesthesia and potentially CNS-related AEs did not seem to be a problem in this study since the occurrence of such AEs was similar in both treatment groups. Moreover, few patients were withdrawn as a result of problems related to tolerability, and there were no safety concerns raised for lesogaberan in this study.
Conclusions
Lesogaberan, administered as an add-on treatment to a PPI, significantly improved heartburn and regurgitation symptoms, and was well tolerated, in patients with persistent GORD symptoms despite PPI therapy. Although the proportion of responders was small, these findings suggest that reflux inhibition can potentially be a new therapeutic approach to address the unmet medical need in patients with GORD. However, further studies are necessary to determine which patients would derive benefit from reflux inhibition and to define what is a clinically meaningful response in these patients.
Acknowledgments
The authors acknowledge the following investigators and their support teams for participation within the study: (Australia) Professor Gerald Holtmann, Adelaide; (Belgium) Dr Hubert Piessevaux, Brussels; Dr Luc Botelberge, Wilrijk; Dr Vinciane Muls, Brussels; and Dr Christoph Wagner, Eupen; (France) Professor Jean-Paul Galmiche, Nantes; Professor Frank Zerbib, Bordeaux; Professor François Mion, Lyon; Dr Gérard Leothaud, Ales; Dr Jean Paul Vove, Bordeaux; and Dr Jacques Tondut, Angers; (Germany) Dr Alexander Meining, Munich; Dr Albin Lütke, Koblenz; Dr Andreas Hackelsberger, Wiesbaden; Dr Peter Uebel, Ludwigshafen; Dr Gerald Tangerding and Dr Joachim Sauter, Wangen; Dr Wolfgang Brandt, Potsdam; Dr Elke Bästlein, Köln; and Dr.Harvey Juergens, Oelde; (Hungary) Dr István Rácz, Györ; Dr Zoltán Döbrönte, Szombathely; Dr Ágnes Király, Pécs; Dr Róbert Schnabeland and Dr Zsolt Tulassay, Budapest; Dr Tibor Szalóki, Vác; Dr János Lonovics, Szeged; Dr János Kersák, Siófok; and Dr László Bárány, Nagykanizsa; (Norway) Dr Charlotte Jacobsen and Dr Ingunn Haakerud, Oslo; Dr Christen Bang, Bergen; Dr Jon Florholmen, Tromsø; Dr Harald Hart, Stavanger; Dr Dag Arne Lihaug Hoff, Ålesund; Dr Gjermund Johnsen, Trondheim; Dr Ulf Tønnes Braut Fjøsne, Levanger; and Dr Thomas De Lange, Rud; (Romania) Dr Tudor Nicolaie and Professor Rado Voiosu, Bucharest; Dr Ioan Brandeu, Satu-Mare; Assoiate Professor Simona Bataga, Mures Targu; Dr Felicia Coman, Brasov; and Associate Professor Dobru Daniela, Mures.In addition, the authors would like to acknowledge members of the AstraZeneca study team (Ann Larkö, Marianne Brunander, Karin Arneklev, John Adler, Göran Hasselgren and Michelle Anderson; Mölndal, Sweden) and the study committee Data Safety Monitoring Board (Lars Wilhelmsen, Anders Odén and Rolf Olsson; Gothenburg, Sweden).
References
Footnotes
Funding This study was supported by AstraZeneca.
Competing interests GEB was supported by a grant (Odysseus programme, G.0905.07) of the Flemish ‘Fonds voor Wetenschappelijk Onderzoek’ (FWO) and has received financial support in the form of consultation fees and grant/research funding from AstraZeneca, and in the form of consultation fees from Movetis. HB has no competing interests to declare. JGH has received financial support in the form of speaker's fees and grant/research funding from AstraZeneca. DGS, KB, MK and HD are employees of AstraZeneca.
Ethics approval This study was conducted with the approval of an independent Institutional Review Board/Research Ethics Committee within each country.
Provenance and peer review Not commissioned; externally peer reviewed.