


Abstract
The decline in bone mineral density that occurs after long-term treatment with some antiepileptic drugs is thought to be mediated by increased vitamin D3 metabolism. In this study, we show that the inducible enzyme CYP3A4 is a major source of oxidative metabolism of 1α,25-dihydroxyvitamin D3 [1,25(OH)2D3] in human liver and small intestine and could contribute to this adverse effect. Heterologously-expressed CYP3A4 catalyzed the 23- and 24-hydroxylation of 1,25(OH)2D3. No human microsomal cytochrome P450 enzyme tested, other than CYP3A5, supported these reactions. CYP3A4 exhibited opposite product stereochemical preference compared with that of CYP24A1, a known 1,25(OH)2D3 hydroxylase. The three major metabolites generated by CYP3A4 were 1,23R,25(OH)3D3, 1,24S,25(OH)3D3, and 1,23S,25(OH)3D3. Although the metabolic clearance of CYP3A4 was less than that of CYP24A1, comparison of metabolite profiles and experiments using CYP3A-specific inhibitors indicated that CYP3A4 was the dominant source of 1,25(OH)2D3 23- and 24-hydroxylase activity in both human small intestine and liver. Consistent with this observation, analysis of mRNA isolated from human intestine and liver (including samples from donors treated with phenytoin) revealed a general absence of CYP24A1 mRNA. In addition, expression of CYP3A4 mRNA in a panel of duodenal samples was significantly correlated with the mRNA level of a known vitamin D receptor gene target, calbindin-D9K. These and other data suggest that induction of CYP3A4-dependent 1,25(OH)2D3 metabolism by antiepileptic drugs and other PXR ligands may diminish intestinal effects of the hormone and contribute to osteomalacia.
The activated form of vitamin D, 1α,25-dihydroxyvitamin D3 [1,25(OH)2D3], exerts profound effects on gene transcription and cell function in multiple tissues (Issa et al., 1998). Most of these effects are mediated through the binding of hormone to the nuclear vitamin D receptor (VDR) and enhanced transcription of its target genes. For example, in the small intestine, 1,25(OH)2D3 stimulates the production of a calcium binding protein (calbindin-D9K) that facilitates the transcellular movement of calcium from the intestinal lumen into the blood (Kumar, 1990; Walters et al., 1999). This phenomenon constitutes an important component of a highly regulated and dynamic system to maintain blood and cellular calcium homeostasis (Issa et al., 1998). 1,25(OH)2D3 activates the transcription of another human intestinal gene, CYP3A4 (Schmiedlin-Ren et al., 1997; Thummel et al., 2001; Makishima et al., 2002), in addition to the calcium binding protein gene. Induction of CYP3A4 by 1,25(OH)2D3 is VDR-dependent and requires binding of a VDR-RXR (retinoid X receptor) heterodimer to an ER-6 response element–152/–169 bp upstream of the CYP3A4 transcription start site, as well as a canonical DR-3 response element, found in a distal (–7719/–7733 bp) regulatory region of the gene.
The biological effects of 1,25(OH)2D3 are controlled in part by P450-catalyzed metabolism of the hormone to a series of oxidation products that generally exhibit reduced binding to VDR (Bouillon et al., 1995). The initial hydroxylation of 1,25(OH)2D3 occurs at the C-24, C-23, and C-26 positions. Formation of 1,24R,25(OH)3D3 is followed by sequential metabolism to yield a terminal product, calcitroic acid (Bouillon et al., 1995; Inouye and Sakaki, 2001). Sequential oxidation of the 23- and 26-hydroxy metabolites also occurs, resulting in the formation of the terminal metabolite, 1α,25R(OH)2-26,23S-lactone-D3 [1,25(OH)2D3-lactone]. CYP24A1 catalyzes the 24-hydroxylation reaction and is thought to catalyze C-23 hydroxylation as well (Endres and DeLuca, 2001). Indeed, enhanced CYP24A1 synthesis by 1,25(OH)2D3 is thought to provide negative-feedback control of hormonal effects in target tissues through the formation of inactive metabolite(s) (Sutton and MacDonald, 2003). Moreover, induction of oxidative metabolism of vitamin D3 molecular species by phenytoin, carbamazepine, and phenobarbital is thought to contribute to osteomalacia that often occurs in patients undergoing long-term treatment with these drugs (Pack and Morrell, 2004). This serious adverse effect has been attributed to an induction of CYP24A1 mediated by the pregnane X receptor (PXR), a nuclear hormone receptor (Pascussi et al., 2005).
Given that CYP3A4 gene transcription is activated by PXR and that CYP3A4 is also a 1,25(OH)2D3/VDR gene target, we tested the possibility that the inducible enzyme CYP3A4 catalyzes the hydroxylation of 1,25(OH)2D3, which could contribute to the termination of its biological effects and lead to drug-induced osteomalacia. Because CYP3A4 catalyzes the 24- and 25-hydroxylation of 1α-hydroxyvitamin D3 (Gupta et al., 2004, 2005), we reasoned that the enzyme might also hydroxylate the fully active form of the hormone. We found that CYP3A4, and not CYP24A1, played the dominant role in 23- and 24-hydroxylation of 1,25(OH)2D3 under constitutive and induced conditions in human small intestine and liver.
Materials and Methods
Materials. NADPH, troleandomycin, sodium periodate, n-butylboronic acid, 1α,25-dihydroxyvitamin D2, and N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) containing 1% trimethylsilylchlorosilane (TMCS) were purchased from Sigma-Aldrich (St. Louis, MO), and 1,25(OH)2D3 was purchased from Tetrionics (Madison, WI). Authentic 1α,24R,25-trihydroxyvitamin D3 and 1α,23S,25-trihydroxyvitamin D3 standards were prepared metabolically, as described previously (Sakaki et al., 2000; Uchida et al., 2004). Supersomes from baculovirus-transfected insect cells that coexpressed individual cytochrome P450 enzymes with P450 reductase (some with cytochrome b5) were purchased from BD Gentest (Woburn, MA). Cytochrome b5 was purchased from PanVera Corp. (Madison, WI). CYP24A1 was expressed in Escherichia coli and was purified as described previously (Sakaki et al., 2000). Caco-2 cells (American Type Culture Collection, Manassas, VA) were subcloned by limiting dilution as described previously (Schmiedlin-Ren et al., 1997). Dulbecco's modified Eagle's medium, nonessential amino acids, penicillin, streptomycin, and Hanks' balanced salt solution were obtained from Invitrogen (Carlsbad, CA). FBS was purchased from Hyclone Laboratories, Inc. (Logan, UT). Uncoated track-etched polyethylene terephthalate inserts and mouse laminin were obtained from Collaborative Biomedical Products (Bedford, MA).
Tissue Samples. Samples of human liver (n = 30) and jejunal mucosa (n = 20) from white donors were obtained from the University of Washington School of Pharmacy Human Tissue Bank (Seattle, WA). Among them, nine liver donors and one jejunum donor had taken phenytoin for various lengths of period (from a few days to long-term usage) before organ donation. Liver microsomes and jejunal homogenate were prepared according to previously published protocols (Paine et al., 1997). Protein concentrations were determined by the method of Lowry et al. (1951). Basic demographic information and detailed characterization of CYP3A4/5 expression has been published (Paine et al., 1997; Lin et al., 2002).
Duodenal samples came from two sources. Tissue collection protocols were approved by the governing Institutional Review Board (University of Washington, Seattle, WA, and University of North Carolina, Chapel Hill, NC). The first set of samples were pinch biopsies collected from 19 patients undergoing diagnostic endoscopy at Harborview Medical Center (Seattle, WA) for suspected gastroesophageal reflux disease. Homogenates of the biopsy tissue were prepared and used for catalytic activity measurements. The second set of duodenal pinch biopsy samples were collected from 20 healthy volunteers at University of North Carolina Medical Center (Chapel Hill, NC). Total mRNA was isolated from each sample in this set and used for gene-specific, real-time quantitative PCR assays. Two human kidney RNA samples were obtained from BD Biosciences Clontech (Palo Alto, CA). One sample originated from a single person, and the other was pooled from several kidney tissues.
Treatment of Caco-2 Cells with 1,25(OH)2D3. Caco-2 cells, passage number 19, were seeded onto semipermeable laminin-coated inserts at 105 cells/cm2 and cultured as described previously (Schmiedlin-Ren et al., 1997; Fisher et al., 1999). After reaching confluence, cells were fed for 3 days with Dulbecco's modified Eagle's medium containing 0.1 mM nonessential amino acids, 100 U/ml sodium penicillin, 100 μg/ml streptomycin, 0.1 M sodium selenite, 3 μM zinc sulfate, 45 nM racemic α-tocopherol, 5% heat-inactivated FBS, and 0.5 μM 1,25(OH)2D3. Medium was collected before and after 3 days of 1,25(OH)2D3 treatment for analysis of 1,25(OH)2D3 metabolites. The cells were harvested and used for total RNA extraction.
Isolation of Total RNA and Real-Time Quantitative PCR. Total RNA was isolated from human duodenal, jejunal, and liver samples and Caco-2 cells using TRIzol reagent (Invitrogen, Carlsbad, CA). Analysis of each product revealed no evidence of significant mRNA degradation. Reverse transcription was performed using random hexamers, according to the manufacturer's instructions for the SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen). The real-time quantitative PCR was performed with the ABI 7900 system (Applied Biosystems, Foster City, CA). The primers and probe sequences for CYP3A4 were as described previously (Lin et al., 2002). For all other gene targets (villin, calbindin-D9K, and CYP24A1), Assays-on-Demand products from Applied Biosystems (containing appropriate gene-specific primers and probe) were used. For CYP24A1, we confirmed results from the Applied Biosystems assay using the following two sets of primers for PCR: set 1, CTCATGCTAAATACCCAGGTG (forward), TCGCTGGCAAAACGCGATGGG (reverse); set 2, GATTCCTTTATGGCATTAGGG (forward), AAACTTTGAAACATGCCCTG (reverse).
Characterization of 1,25(OH)2D3 Metabolism. All 1,25(OH)2D3 metabolism experiments with recombinant cytochrome P450 (P450) enzymes, human liver microsomes, and intestinal homogenate were carried out in 0.1 M potassium phosphate buffer at 37°C, pH 7.4, with a final volume of 0.5 ml. After preincubation of the source enzyme with 1,25(OH)2D3 for 5 min, the reaction was initiated by the addition of NADPH (final concentration, 1 mM). 1,25(OH)2D3 was dissolved in methanol. For all incubations, the total percentage of organic solvent was ≤1%.
When screening the various recombinant P450 enzymes for relative catalytic activity, 20 pmol of enzyme was incubated with 10 μM 1,25(OH)2D3 for 20 min. Reaction kinetic parameters for product formation catalyzed by recombinant CYP3A4 (coexpressed with cytochrome P450 reductase and cytochrome b5) was determined using 8 pmol of enzyme and 0.02 to 50 μM 1,25(OH)2D3 incubated for 4 min. Comparison of metabolic activity from different human tissue donors was performed by incubating 0.05 mg of liver microsomal or 0.15 mg of intestinal homogenate protein with 6 μM 1,25(OH)2D3 for 4 and 45 min, respectively. For inhibition experiments, troleandomycin (TAO, 20 μM) was preincubated with the source enzyme and NADPH for 15 min before a 20-μl aliquot (containing the appropriate amount of source enzyme) was then transferred to the final incubation tube. Otherwise, ketoconazole (100 nM; dissolved in dimethyl sulfoxide) was added directly to the incubation mixture before addition of NADPH.
Incubation reactions were terminated with the addition of 1.2 ml of MeOH. To this mixture, 30 ng of 1,25(OH)2D2 (internal standard) was added and centrifuged. The resulting supernatant was subjected to reversed-phase solid phase extraction (SPE) chromatography for partial purification of vitamin D molecules using the following procedures. A solid-phase extraction column (Strata C18-M: 55 μm, 140A; 500 mg/3 ml) (Phenomenex, Torrance, CA) was preconditioned with hexane, 2-propanol, and methanol/water (70:30). The methanolic extract was then loaded onto the head of the conditioned SPE column and the column was washed with methanol/water (70:30), and then hexane. The desired vitamin D products were eluted with hexane/2-propanol (80:20) and dried under N2. The residue was reconstituted in 0.1 ml of methanol:water (70:30) and subjected to LC/MS analysis (described below). Using this protocol, the substrate, products, and internal standard had a recovery of approximately 100%. Methods used for characterizing CYP24A1-catalyzed 1,25(OH)2D3 metabolism have been published previously (Sakaki et al., 2000). Conditions for testing the effects of CYP3A inhibitors on CYP24A1 catalytic activity were the same as described for CYP3A4.
Purification of Monohydroxylated 1,25(OH)2D3 Metabolites and Identification by GC/MS. Two milligrams of human liver microsomes were incubated with 1,25(OH)2D3 (75 μM) and 1 mM NADPH for 40 min to produce hydroxylated metabolites. Metabolites were separated by liquid chromatography and detected by UV absorbance (264 nm). Eluting fractions containing three metabolite peaks (M1, M2, and M3) were isolated and the solvent removed under a stream of N2. Each peak residue was dissolved in 40 μl of acetonitrile and transferred to a 2-ml glass vial containing 30 μl of N,O-bis(trimethylsilyl)trifluoroacetamide plus 1% TMCS. After heating at 42°C for ∼15 h, 2 μl were injected onto a 5% phenylmethylpolysiloxane GC column (30 m × 0.25 mm). Gas chromatography/mass spectrometric analysis of the HPLC-purified metabolites was accomplished on a QP2010 GC/MS (Shimadzu, Columbia, MD). The oven was held at 200°C for 1.5 min, followed by a 20°C/min ramp to a hold at 280°C. Under these GC conditions, the trimethylsilyl (TMS) derivatives of putative 1,24R,25-trihydroxyvitamin D3, 1,23S,25-trihydroxyvitamin D3 metabolites, and 1,25(OH)2D3 standard eluted at 19.0, 20.9, and 16.8 min, respectively. Eluting peaks were introduced into the mass spectrometer and ionized by electron impact (70 eV); data were acquired over m/z 50 to 800.
Two experiments (periodate cleavage and n-butylboronic acid esterification) were conducted to further confirm the regiochemistry of M2, the putative 24-hydroxylated 1,25(OH)2D3 metabolite. For the periodate cleavage experiment, human liver microsomes were incubated with 1,25(OH)2D3 and the biological reaction products were partially purified by MeOH treatment and SPE chromatography, as described above. The reaction products or 200 ng of 24,25(OH)2D3 was treated with aqueous 5% NaIO4 at room temperature for 40 min. Products of the chemical reaction were then extracted with chloroform, dried under nitrogen, reconstituted in MeOH/H2O (7:3), and subjected to LC/MS analysis. For the n-butylboronic acid esterification experiment, 80 μl of 0.5% n-butylboronic acid (dissolved in ethyl acetate) was added to HPLC-purified M2 or 1,25(OH)2D3, 24,25(OH)2D3, and 25,26(OH)2D3, and incubated for 30 min at 40°C. At the end of the esterification reaction, organic solvent was removed under nitrogen and 50 μl of BSTFA plus 1% TMCS was added to the residue for derivatization and GC/MS analysis.
After identification of the two major metabolites, M2 and M3, an incubation of 1,25(OH)2D3 (75 μM) with liver microsomes (2.0 mg) for 40 min was repeated. An aliquot of the reconstituted residue was subjected to HPLC chromatography for separation and isolation of M2 and M3. The concentration of these two metabolite standards was calculated using their UV absorbance at 264 nm and the molar extinction coefficient (1.80 × 104 M–1 · cm–1) of 1,25(OH)2D3. These solutions were used for construction of standard curves and quantification of unknown metabolite concentrations.
LC/MS Assay for 1α,25-Dihydroxyvitamin D3 Monohydroxylation Products. The quantification of metabolites of 1,25(OH)2D3 was achieved with an HPLC/MS system consisting of a Quattro II tandem quadrupole mass spectrometer (Micromass, Manchester, UK) and 10-AD HPLC (Shimadzu). Separation of the isobaric monohydroxy-metabolites was achieved using a Symmetry C8, 2.1 × 150-mm, 3.5-μm column (Waters, Milford, MA). The column was heated to 45°C to reduce internal pressure. Analysis was carried out under gradient conditions of two mobile phases: methanol with 0.25 mM sodium acetate and water with 0.25 mM sodium acetate. The flow rate was 0.25 ml/min. An aliquot (20 μl) of reconstituted residue was injected for LC/MS analysis. Hydroxy metabolites of 1,25(OH)2D3 eluted under the following conditions: a linear increase in the methanol phase from 70 to 90% over 14 min, maintained at 90% until 17 min, returned to initial conditions over 1 min, and then maintained under that condition for 5 min before a new injection. Atmospheric pressure electrospray ionization in the positive mode was employed with the cone voltage of 40 eV. The product (M+Na)+ ions of 1,25(OH)2D3, its monohydroxylated metabolites, and 1α,25-dihydroxyvitamin D2 (internal standard) were monitored at m/z 439, 455, and 451, respectively. Data acquisition and analysis were carried out using MassLynx-NT 3.4 (Micromass).
Hepatic and Intestinal Metabolism of Midazolam. Incubation of midazolam, an established CYP3A probe (Wrighton et al., 2000), with human liver microsomes or intestinal homogenate was performed according to published procedures (Lin et al., 2002). The 1′-OH midazolam metabolite was analyzed by an LC/MS method from Thummel et al. (2001), except that selective ion monitoring at m/z 342 and 347 [for 1′-OH midazolam and 2H-labeled 1′-OH midazolam (internal standard), respectively] was employed.
Data Analysis. Linear regression analysis was performed by SPSS version 8.0 (SPSS Inc., Chicago, IL). Enzyme kinetic parameters were estimated using KaleidaGraph software (Abelbeck/Synergy, Reading, PA).
Results
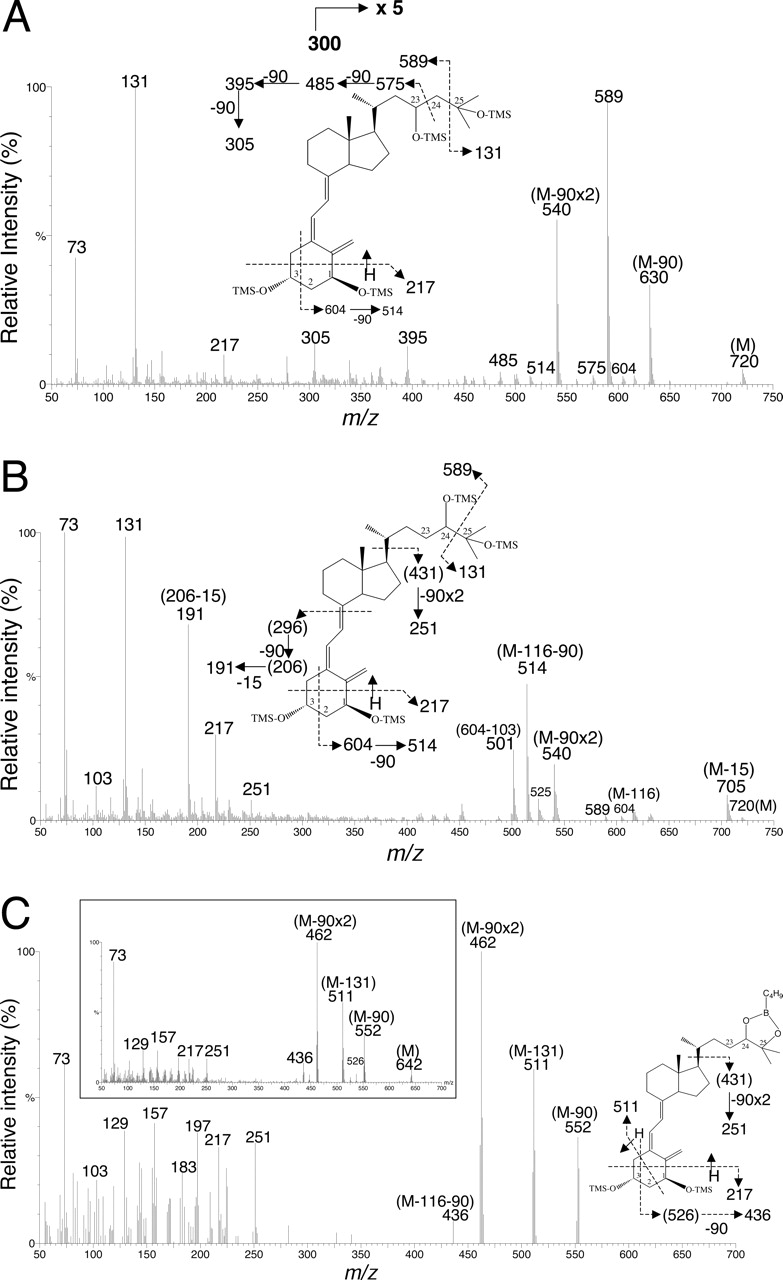
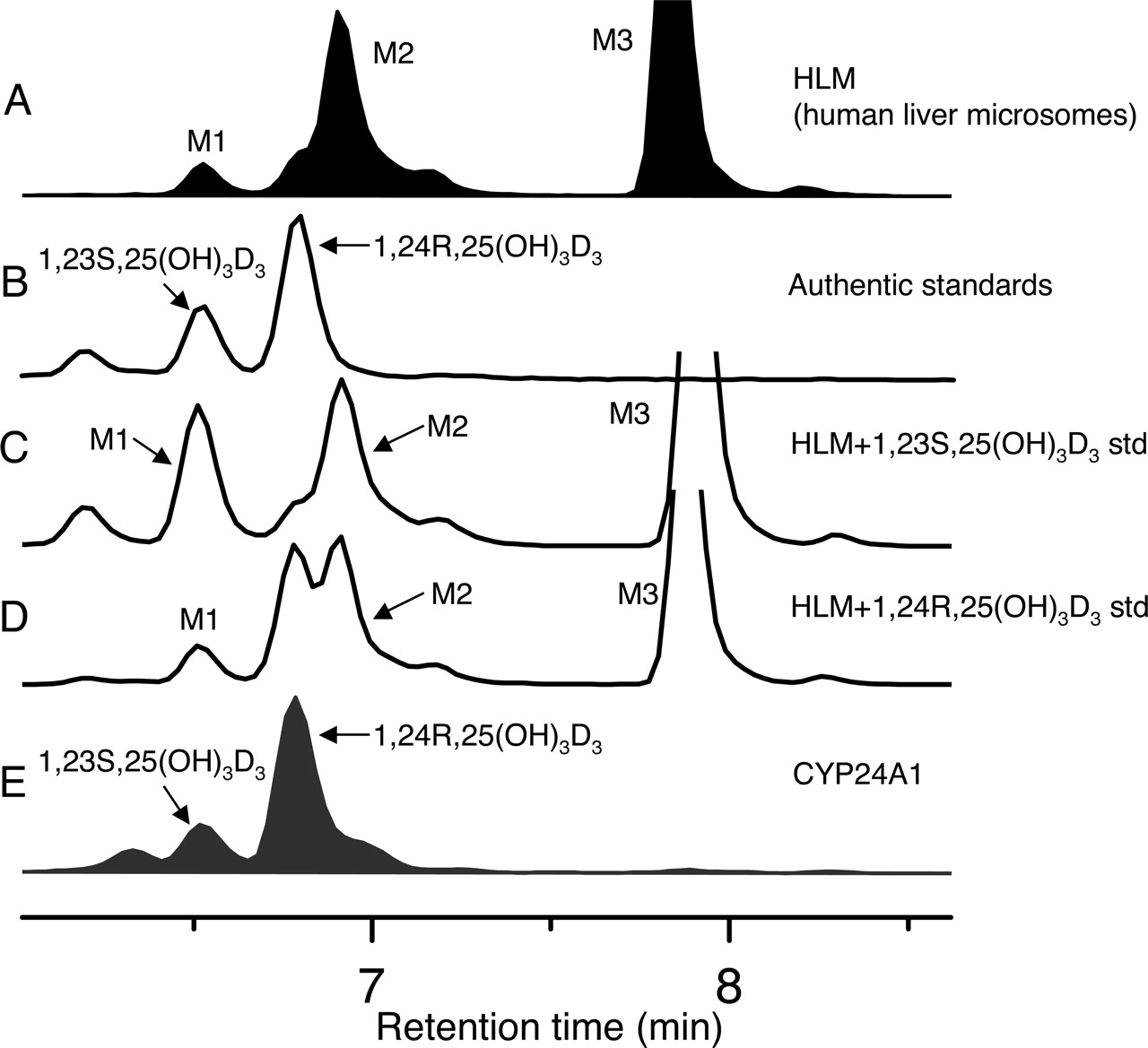
Metabolism of 1,25(OH)2D3 by Human Liver and Small Intestine. A novel LC/MS assay (see Materials and Methods) was developed to fully characterize the metabolism of 1,25(OH)2D3. Three monohydroxylated metabolites (M1, M2, and M3) were detected after incubation of human liver microsomes and duodenal and jejunal homogenate with 1,25(OH)2D3 (Fig. 1). To identify the metabolites, approximately 500 ng of M2 and M3 generated from human liver microsomal incubation was purified and subjected to TMS derivatization and analysis by GC/MS. The resulting mass spectrum of M3 (Fig. 2A) was fully consistent with the structural assignment of the product as 1,23,25(OH)3D3 (Napoli and Horst, 1983). An m/z of 720 represents the molecular ion for the TMS derivative. The presence of the major fragment ion at m/z 131 demonstrated that the C(26)H3-C(25)OH-C(27)H3 structure of the original 1,25(OH)2D3 was intact in this metabolite. The ion at m/z 575 was produced by TMS-derivatized C(23)-C(24) bond cleavage and is unique to 23-hydroxylated 1,25(OH)2D3. Sequential losses of 90 Da [(CH3)3SiOH] from 575 gave m/z 485, 395, and 305, respectively, also confirming the presence of a 23-hydroxyl group in the parent molecule. To determine the stereochemistry of M3, we compared its reverse phase elution time with that of authentic standards (Fig. 3). M1 but not M3 coeluted with 1,23S,25(OH)3D3. Therefore, it was concluded that M3 was the other diastereomer, 1,23R,25(OH)3D3.
The electron impact MS spectrum of M2 is shown in Fig. 2B. It possessed a molecular ion of m/z 720 with major fragments at 705 (M-15), 604 (M-116), 589 (M-131), 540 (M-90 × 2), 514 (M-116–90), 251 (431–90 × 2), 191 (296–90-15), and 131. The major fragment at m/z 131 corresponds to C(24)-C(25) cleavage and loss of C3H7OTMS, indicating no substitution at C(26) and C(27). This fragmentation pattern also indicated that the hydroxylation site was at the side chain and was consistent with 1,24,25(OH)3D3 but not with 1,23,25(OH)3D3 (Napoli and Horst, 1983) or 1,25,26(OH)3D3 (Reinhardt et al., 1981). When we compared the LC/MS elution time of M2 with that of authentic standards (Fig. 3), we found that it did not coelute with 1,24R,25(OH)3D3 and hypothesized that it must be the other diastereomer, 1,24S,25(OH)3D3.
To confirm the presence of vicinal hydroxyl groups on M2, we conducted two additional experiments. In the first experiment, we treated the isolated incubation mixture of human liver microsomes and 1,25(OH)2D3 with sodium periodate, which selectively cleaves C–C bonds with vicinal hydroxyl (or vicinal hydroxyl-ketone) groups (Beckman et al., 1996). Upon LC/MS analysis (data not shown), we observed the disappearance of M2, but not M1 or M3 (both 23-hydroxylated) or the substrate, 1,25(OH)2D3. In the second experiment, purified M2 was esterified with n-butylboronic acid, which produces a cyclic boronate from a molecule that possesses vicinal hydroxyl groups (Masuda et al., 1996) and was then derivatized with BSTFA plus 1% TMCS. Its mass spectrum after GC/MS (Fig. 2C) showed the fragments of 552 (M-90), 511 (M-131), 462 (M-90 × 2), 436 (M-116–90), 251, 217, and 73. The fragmentation pattern of derivatized M2 was essentially identical (except for the absence of the molecular ion) to that of the cyclic boronate derivative of 1,24R,25(OH)3D3 standard (Fig. 2C, inset). Together, these data strongly indicated the presence of vicinal hydroxyl groups at the side chain of M2, and thus the structure was assigned as 1,24S,25(OH)3D3.

Representative mass spectrometric ion chromatograms ((M+Na)+, m/z = 455) for mono-hydroxylated 1,25(OH)2D3 products from incubations of 1,25(OH)2D3 with: pooled human liver microsomes in the absence of NADPH (0.2 mg of microsomes (A); 20 μM 1,25(OH)2D3; 15 min incubation); pooled human liver microsomes in the presence of NADPH (0.2 mg of microsomes; 20 μM 1,25(OH)2D3; 15 min incubation) (B); pooled human intestinal homogenate in the presence of NADPH (0.4 mg of homogenate; 20 μM 1,25(OH)2D3; 15 min incubation) (C); and recombinant CYP3A4 coexpressed with P450 reductase and b5 in the presence of NADPH (8 pmol enzyme; 14 μM 1,25(OH)2D3; 4-min incubation) (D). The inset shows an expanded view of M1 and M2 peaks.
Hydroxylation at the 23R-position was favored over hydroxylation at the 24S-position, resulting in 23R-/24S-product ratios of 2.8 ± 0.1 and 2.6 ± 0.2 from liver and intestinal tissues, respectively (Fig. 1). These product ratios were independent of 1,25(OH)2D3 concentration (0.02–50 μM) in the incubation (data not shown).

Mass spectral analysis of the trimethylsilyl derivative of: M3, identified as 1,23,25(OH)3D3 (the ion signals between m/z 300 and 750 were amplified 5-fold) (A); M2, identified as 1,24,25(OH)3D3 (B); and M2, esterified with n-butylboronic acid (B-C4H9) (C). The inset shows the mass spectrum of the trimethylsilyl derivative of 1,24R,25(OH)3D3 standard esterified with n-butylboronic acid.
Metabolism of 1,25(OH)2D3 by CYP3A4 and CYP24A1. When 1,25(OH)2D3 (14 μM) was incubated with recombinant CYP3A4 for 4 min, we observed the formation of 1,23R,25(OH)3D3 and 1,24S,25(OH)3D3 as well as a minor metabolite 1,23S,25(OH)3D3 (Fig. 1). Moreover, the 23R-/24S-product ratio produced by CYP3A4 was similar in stereochemical preference but slightly larger (5.4 ± 1.2) than that generated by human liver and intestine (Fig. 1). When 1,25(OH)2D3 was incubated with recombinant CYP24A1, only two monohydroxylated products were detected, with formation of 1,24R,25(OH)3D3 favored 4:1 over that of 1,23S,25(OH)3D3 (Fig. 3). Therefore, although CYP3A4 and CYP24A1 monohydroxylate 1,25(OH)2D3 at the same carbon atoms, they confer opposite product stereoselectivity.
Screen of 1,25(OH)2D3 Hydroxylation Activity by Various Recombinant P450 Isoforms. We screened 11 other recombinant human P450 enzymes for 1,25(OH)2D3 hydroxylation activity and found that only CYP3A5 (supplemented with cytochrome b5, 3:1 molar ratio) generated the monohydroxyl products and that the reaction occurred at a rate ∼4% that of CYP3A4 under similar cytochrome b5 reconstitution conditions (Table 1). When cytochrome b5 was not added to either enzyme preparation, CYP3A5 had ∼10% the activity of CYP3A4. CYP3A4-mediated 1,25(OH)2D3 hydroxylation activity was also cytochrome-b5–dependent. The rate was 3.5-fold higher when enzyme was supplemented with exogenous b5 and 6-fold higher when b5 was coexpressed with CYP3A4 compared with the absence of b5 (Table 1).
1,25(OH)2D3 23R-hydroxylase activity for various recombinant human microsomal cytochrome P450 enzymes
Incubations were carried out with 20 pmol of enzymes for 20 min. Substrate concentration was 10 μM.

Mass spectrometric ion chromatograms ((M+Na)+, m/z = 455) for mono-hydroxylated 1,25(OH)2D3 products from: incubation of 1,25(OH)2D3 with pooled human liver microsomes in the presence of NADPH (0.2 mg of microsomes; 20 μM 1,25(OH)2D3; 15-min incubation) (A); authentic standards of 1,23S,25(OH)3D3 and 1,24R,25(OH)3D3 (B); 1,23S,25(OH)3D3 standard spiked into the sample for A (C);1,24R,25(OH)3D3 standard spiked into the sample for A (D); and incubation of 1,25(OH)2D3 with recombinant CYP24A1 reconstituted with adrenodoxin and adrenodoxin reductase in the presence of NADPH (Sakaki et al., 2000) (E).
Kinetic Parameters for CYP3A4-Catalyzed 1,25-(OH)2-D3 Metabolism. To compare the metabolic efficiency of recombinant CYP3A4 with that published for CYP24A1, we measured product formation rates over a range of substrate concentrations (0.02–50 μM). Incubation times were limited to 4 min to maintain initial rate kinetics. In addition, higher substrate concentrations were not used to avoid substrate precipitation. Formation of 23R- and 24S-hydroxy-1,25(OH)2D3 exhibited hyperbolic kinetics; thus, the Michaelis-Menten equation was fit to the data. The resulting kinetic parameters are listed in Table 2 along with published data for CYP24A1 (Kasudo et al., 2003). The total intrinsic clearance (Vmax/Km; sum of 23- and 24-hydroxy metabolites) of CYP3A4 was approximately 6% that of CYP24A1.
Kinetic parameters of recombinant CYP3A4, human liver microsomes, human intestinal homogenate, and recombinant CYP24A1 toward 1,25(OH)2D3
Values reported represent mean ± S.D. for two replicate incubations (P450) or four pooled liver or jejunal samples. Total intrinsic clearance is the sum of Vmax/Km for all products observed in the reaction.
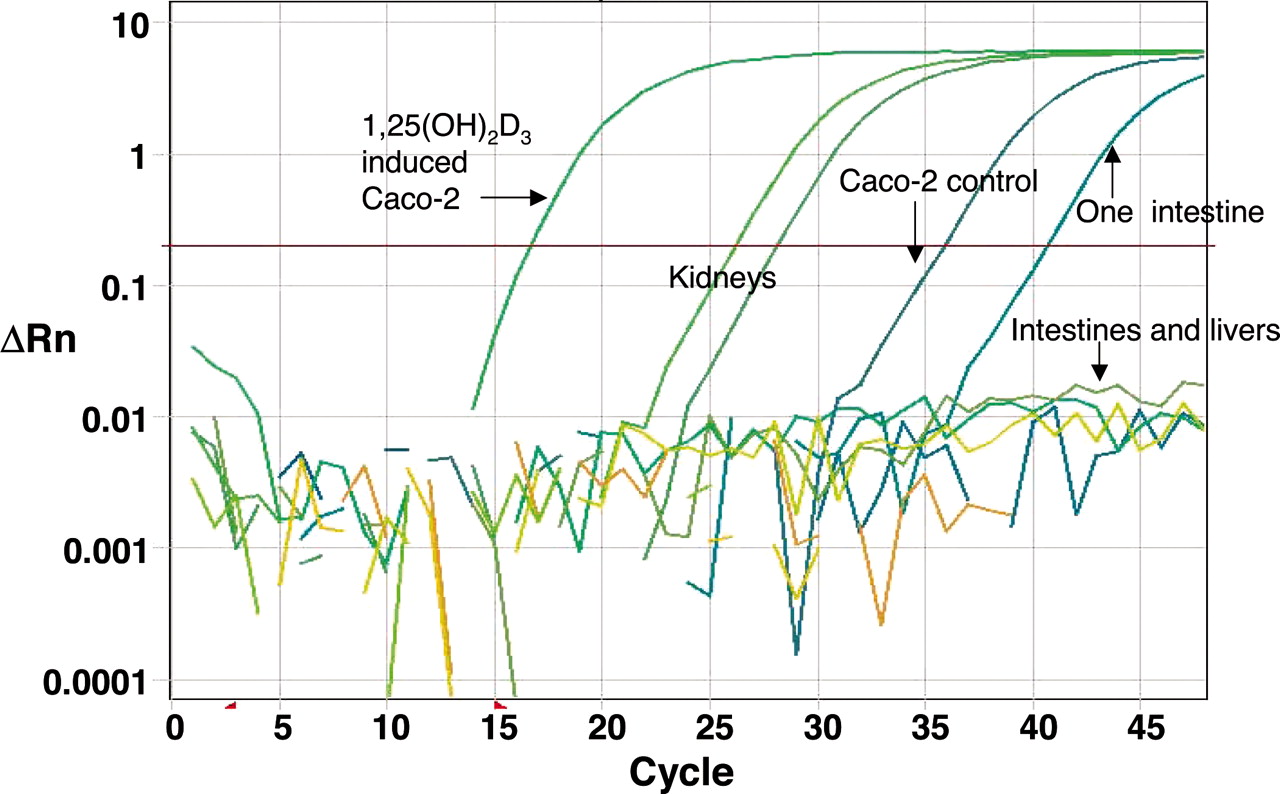
General Absence of CYP24A1 Expression in Human Small Intestine and Liver. Because CYP24A1 is a much more efficient 1,25(OH)2D3 hydroxylase than CYP3A4, we sought to determine why CYP24A1 did not dominate 1,25(OH)2D3 metabolism in human liver and small intestine [based on 1,25(OH)2D3 metabolite profiles]. When we carefully examined the constitutive expression of CYP24A1 mRNA in human small intestine and liver using both traditional PCR-gel and quantitative real-time PCR methods, we did not detect a signal corresponding to CYP24A1 mRNA in 28 human liver, 24 human jejunal, and 19 human duodenal biopsy samples (Fig. 4). Only a single duodenal biopsy sample produced a CYP24A1 mRNA amplification signal, but this signal was much weaker in comparison to that detected for human kidney. We also examined the expression of CYP24A1 mRNA in nine livers and one jejunal mucosa whose donors had taken phenytoin. Three of the livers had CYP3A4 protein levels (125–220 pmol/mg of protein) much higher than the average of all other “nonexposed” livers (60 ± 41 pmol/mg of protein, n = 21). We found that CYP24A1 signals from two of these nine livers were very weak (threshold cycle = 41 and 43, respectively), and that no signals were observed from the other phenytoin-exposed livers or the jejunal mucosa. In addition, the 1,25(OH)2D3 metabolic profiles produced by these “induced” liver microsomal samples were similar to those of the other livers but different from that of CYP24A1.
In contrast to human intestine, CYP24A1 mRNA was readily detected in cultured Caco-2 cells before and after treatment with 1,25(OH)2D3 (Fig. 4), although at a relatively low level before 1,25(OH)2D3 treatment. CYP24A1 mRNA was induced ∼40,000-fold by 3 days of 0.5 μM 1,25(OH)2D3 treatment. In the same experiment, CYP3A4 mRNA was induced 7000-fold (data not shown). It is noteworthy that analysis of the medium from cells treated with 1,25(OH)2D3 revealed that the metabolic profile was more similar to that produced by CYP24A1 (Fig. 5), not CYP3A4 and human liver and intestinal tissues.
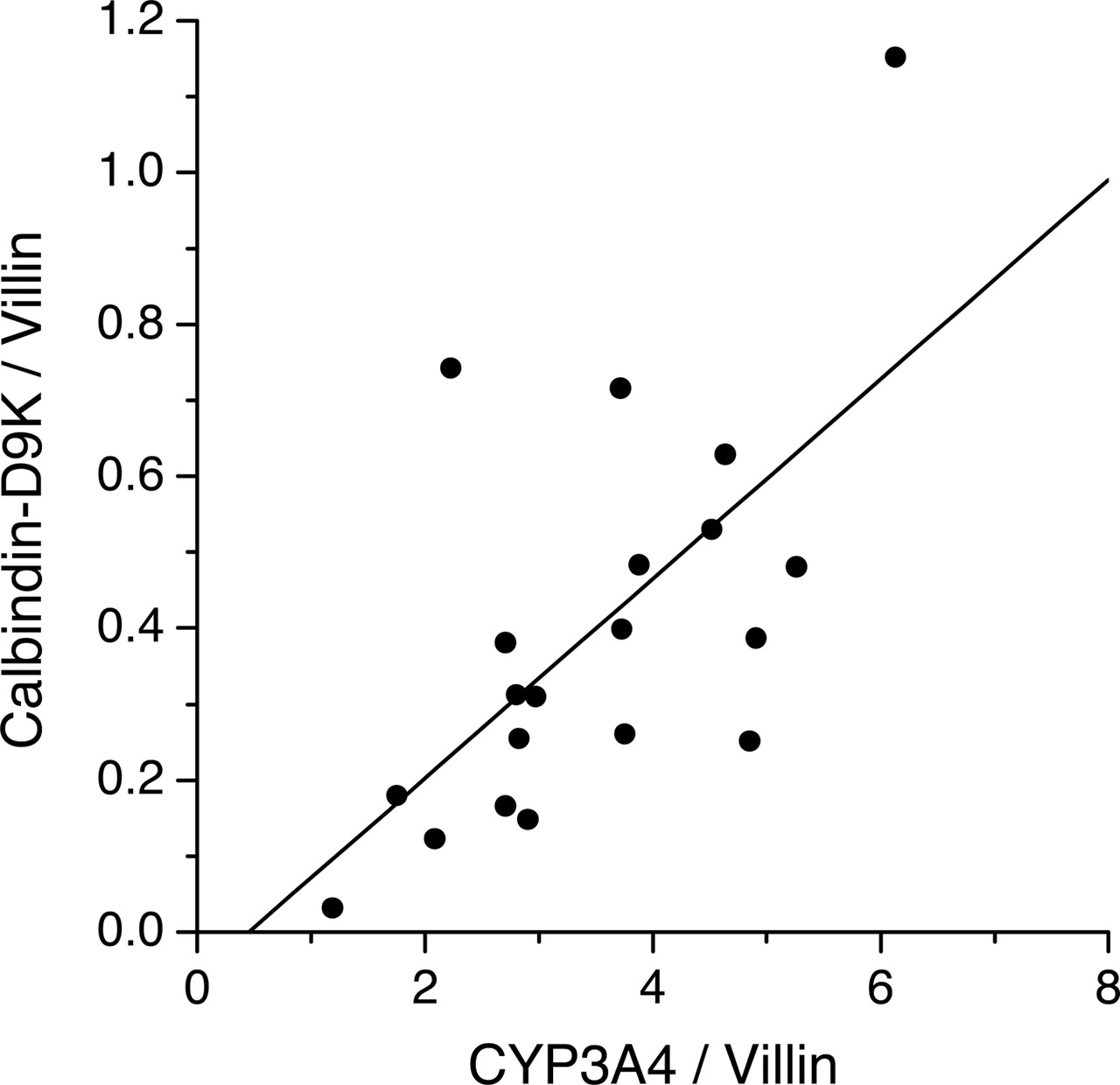
Association between Calbindin-D9K and CYP3A4 Expression in Human Duodenal Samples. Our inability to detect CYP24A1 mRNA in human intestinal tissues raised the question of whether this was part of a general pattern of reduced 1,25(OH)2D3 signaling in these tissues as a result of an altered nutritional (fasting) or physiological status (brain death) of some of the tissue donors. However, this did not seem to be the case, in that there was abundant expression of another known VDR target gene, calbindin-D9K, in the duodenal pinch biopsies collected from healthy volunteers and in duodenal and proximal jejunal mucosa collected from organ donors. Indeed, there was a positive and significant association between CYP3A4 and calbindin-D9K expression among the 20 duodenal biopsy samples (r = 0.64; p < 0.01) (Fig. 6).
Inhibition of 1,25(OH)2D3 Monohydroxylation by TAO and Ketoconazole. To confirm that CYP3A4, and not CYP24A1, is the dominant enzyme catalyzing 1,25(OH)2D3 monohydroxylation in both human small intestine and liver tissues, we preincubated each tissue subfraction with the CYP3A-specific mechanism-based inhibitor TAO. We found that for CYP3A4, human intestinal homogenate, and liver microsomes, preincubation of 20 μM TAO for 15 min resulted in ≥84% loss of both 23- and 24-hydroxylation activities (Table 3). Although ketoconazole is not a specific P450 inhibitor, it does show some selectivity and is a potent CYP3A4 inhibitor at nanomolar concentrations. Indeed, at a concentration of 100 nM, ketoconazole inhibited CYP3A4-catalyzed 1,25(OH)2D3 hydroxylation by 98%. The same degree of inhibition was observed when 100 nM ketoconazole was added to human intestinal and hepatic incubations with 6 μM 1,25(OH)2D3. In sharp contrast, cDNA-expressed CYP24A1 was not inhibited by TAO or ketoconazole (102 and 95% of control, respectively) when tested under the incubation conditions that essentially abolished CYP3A4 and human liver and intestinal activity.
Inhibition of 1,25(OH)2D3 mono-hydroxylation activity of CYP3A4, liver microsomes, intestinal homogenate, and CYP24A1 by TAO and ketoconazole (KTZ)
TAO (20 μM) was preincubated with the source enzyme and NADPH for 15 min before a 20-μl aliquot (containing the appropriate amount of source enzyme) was then transferred to the final incubation tube. Ketoconazole (100 nM) was added directly to the incubation mixture before addition of NADPH.
Interindividual Differences in Human Hepatic and Duodenal 1,25(OH)2D3 Metabolism. The metabolism of 1,25(OH)2D3 by different liver and duodenal tissues was highly variable, consistent with differences in CYP3A4 expression. In addition, the rates of 23- and 24-hydroxylation were each highly correlated with the rate of metabolism of an established CYP3A probe reaction, midazolam 1′-hydroxylation. This was true for both liver microsomes and duodenal homogenate [between 1,25(OH)2D3 23R-hydroxylation activity and midazolam 1′-hydroxylation activity: r = 0.94, liver and r = 0.83, duodenum]. Not surprisingly, the rates of 23R- and 24S-hydroxylation were highly correlated with each other (r = 0.98, liver; r = 0.97, duodenum), suggesting that the metabolites were produced by the same enzyme. Moreover, the ratio of 1′-OH midazolam to 1,23R,25(OH)3D3 formation rate for human liver microsomes and duodenal homogenate was similar (30 and 55, respectively, compared on the basis of picomoles per minute per milligram of protein), again indicative of metabolites that are produced by the same enzyme in these two tissues. To evaluate the production of M1 (1,23S,25(OH)3D3) more accurately, 0.5 mg of liver microsomes was used to repeat the correlation study. We found that, among the panel of human liver microsomes tested, the peak area for M1 metabolite produced was highly correlated with that of 23R-hydroxy-1,25(OH)2D3 and 24S-hydroxy-1,25(OH)2D3 (r = 0.97 and 0.97, respectively).
Discussion
The oxidative metabolism of 1,25(OH)2D3 is generally regarded as a critical process in the termination of biological effects of the hormone in many of its target tissues and in its ultimate elimination from the body (Sutton and MacDonald, 2003). Two major pathways of systemic elimination that involve CYP24A1-catalyzed 24R- and 23S-monohydroxylation have been described previously (Bouillon et al., 1995). Our data suggest that CYP3A4 also contributes to the elimination of 1,25(OH)2D3 in humans, particularly within hepatocytes and enterocytes, through the formation of 23R- and 24S-hydroxy metabolites (Fig. 7).
Unlike CYP24A1, CYP3A4 exhibits relatively broad substrate specificity, metabolizing a wide array of structurally diverse xenobiotic and endobiotic molecules, including several different steroids and bile acids (Guengerich, 1999; Nebert and Russell, 2002). It also catalyzes the 24- and 25-hydroxylation of vitamin D2 (but not vitamin D3) and of 1α-hydroxyvitamin D2/D3 (Gupta et al., 2004, 2005). Our data, together with those of Gupta et al. (2004, 2005), demonstrate that CYP3A4 catalyzes the 24-hydroxylation of both 1α-hydroxyvitamin D2 and 1,25(OH)2D3 with the same stereospecificity (i.e., producing a 24S-stereoisomer). In addition, CYP3A4 has been shown to be a 23R-, 24S-, and 24R-hydroxylase of a bile acid intermediate, 5β-cholestane-3α,7α,12α,25-tetrol (Honda et al., 2001). Indeed, the ratio of 23R-/24S-metabolites of 5β-cholestane-3α,7α,12α,25-tetrol produced by recombinant CYP3A4 is ∼2.7, similar to that observed in the current study when 1,25(OH)2D3 was used as a substrate for CYP3A4. It is noteworthy that the 24-hydroxylation activity of CYP3A4 toward 1,25(OH)2D3, determined in this study, was much higher (>20-fold) than the activity found by Gupta et al. (2005) toward 1α-hydroxyvitamin D2/D3. However, carbon-23 of 1,25(OH)2D3 is the preferred hydroxylation site for CYP3A4, and it represents the dominant path of oxidative metabolism in human liver and small intestine.

Real-time PCR traces of CYP24A1 amplification from human intestine, liver, kidney, and Caco-2 cells. The samples tested include those (nine livers and one jejunal intestine) from the donors who had taken the P450 inducer phenytoin for various periods of time (1 day to prolonged usage) before organ donation. In all the liver (n = 30) and intestinal (n = 44) samples examined, only one duodenal sample and two liver samples gave an extremely low signal with a threshold cycle >40; others showed no amplification signal at all. In contrast, both kidney samples tested gave a robust signal.

Comparison of the formation of mono-hydroxylated 1,25(OH)2D3 products by mass spectrometric ion chromatograms ((M+Na)+, m/z = 455) from: cell culture medium from Caco-2 cells that were treated by 0.5 μM 1,25(OH)2D3 for 3 days (A) and incubation of 1,25(OH)2D3 with recombinant CYP24A1 reconstituted with adrenodoxin and adrenodoxin reductase in the presence of NADPH (Sakaki et al., 2000) (B).
This finding is not surprising given the near complete absence of CYP24A1 mRNA in the same human tissue preparations that we studied, including those from donors treated with phenytoin. The absence of basal intestinal and hepatic CYP24A1 mRNA expression in rodents (Komuro et al., 1999; Cheng et al., 2003) has been reported previously. Our metabolic data suggest that CYP24A1 is not present at a level sufficient to affect the 23-/24-hydroxy metabolite profile in those control and phenytoin-treated human tissues, even at a low (20 nM) substrate concentration that would be more likely to favor CYP24A1. In this regard, it is worth noting that cultured Caco-2 cells do not recapitulate the 1,25(OH)2D3 metabolism profile of the human intestinal mucosa, and this most likely reflects an altered state of gene (i.e., CYP24 and CYP3A4) regulation in the immortalized cell line.

Correlation between calbindin-D9K and CYP3A4 mRNA expression in 20 human duodenal biopsy samples. Villin was used as the internal control gene for normalization of the data. The correlation coefficient was 0.64 (p < 0.01).
We have only limited information on what medicines the organ donors received and the duration of exposure before organ harvest. Eight donors received phenytoin during their stays in the intensive care unit (ICU) before death. With respect to the three livers with an elevated CYP3A4 level, the donor received phenytoin for 6 days or longer before procurement. Regarding the rest of the livers with relatively moderate or low CYP3A4 expression, the donor had received phenytoin for 1∼3 days in the ICU. Thus, the duration of phenytoin exposure might have determined the “apparent” CYP3A4 inducibility. Unfortunately, plasma phenytoin levels at the time of procurement were not available for all subjects to permit a more rigorous evaluation of interliver differences on CYP3A4.
We used CYP3A-specific inhibitors (TAO and ketoconazole) to confirm that CYP3A4, not CYP24A1, is the dominant enzyme catalyzing 1,25(OH)2D3 monohydroxylation in both human small intestine and liver tissues. We also used the recombinant CYP24A1 (which served as a control) to show that neither inhibitor diminished the activity of this enzyme. TAO, a specific mechanism-based inhibitor of CYP3A4, did not affect the activity of the recombinant CYP24A1, as expected. With regard to ketoconazole, it has been shown previously (Ohyama and Okuda, 1991) to inhibit 24-hydroxylation activity toward 25(OH)D3 using CYP24A1 purified from rat kidney (substrate concentration at 20 μM). However, it was not a potent inhibitor of CYP24A1, with 22% and 64% inhibition of catalytic activity at 2 and 20 μM ketoconazole, respectively. Thus, our observations are consistent with these published results: no apparent inhibition was observed at concentrations of ketoconazole equal to 0.1 or 1 μM. In sharp contrast, ketoconazole is a potent inhibitor toward CYP3A4, with a Ki ∼ 0.02 to 0.1 μM.
The general absence of CYP24A1 mRNA in the small intestine is puzzling, in that it is clearly a VDR gene target. It is possible that CYP24A1 mRNA has a shorter half-life (in comparison to calbindin-D9K and CYP3A4 mRNA) and that it disappeared under the conditions preceding tissue collection (short- or long-term fasting, for example). It is also possible that the intestine and liver lack an essential coactivator protein or that a corepressor protein is present. However, the effect of these modifying transcription factors would have to be selective, given the observed expression of other VDR target genes, such as calbindin-D9K and CYP3A4.

Pathways of tissue-specific mono-hydroxylation of 1,25(OH)2D3 catalyzed by CYP3A4 and CYP24A1. The R group represents structure of 1,25(OH)2D3 minus the side chain.
The metabolism of 1,25(OH)2D3 by intestinal CYP3A4 suggests that the enzyme may exert negative feedback control of constitutive 1,25(OH)2D3 transcriptional effects in this tissue, much the same way that CYP24A1 does in the kidney and possibly other tissues. 1,25(OH)2D3 activates CYP3A4 expression in cell culture through a VDR-dependent mechanism (Schmiedlin-Ren et al., 1997; Thummel et al., 2001), and experiments with mice suggest that regulation of intestinal CYP3A by 1,25(OH)2D3 occurs in vivo (Makishima et al., 2002). Thus, increased transcription of intestinal VDR gene targets, including CYP3A4, in response to 1,25(OH)2D3 would result in enhanced metabolic elimination of 1,25(OH)2D3, tempering the magnitude and duration of the cellular effects of the hormone. Basal expression of CYP24A1 was not apparent in the pinch biopsies of healthy human duodenum that we examined, in contrast to the well correlated calbindin-D9K and CYP3A4 expression, suggesting that the transcription of CYP24A1 in the intestine is not activated by 1,25(OH)2D3 under normal physiological conditions.
Long-term treatment with some antiepileptic drugs has a negative effect on bone mineral density (Gascon-Barré, 2004; Pack and Morrell, 2004). Previous investigators attributed the occurrence of osteomalacia from phenytoin, phenobarbital, and carbamazepine to the induction of hepatic vitamin D3 metabolism (Hahn et al., 1972; Gascon-Barré et al., 1984; Gascon-Barré, 2004). However, it is not clear exactly which enzymatic system in the liver or which vitamin D3 metabolite(s) were involved. This adverse effect was recently attributed specifically to a PXR-mediated activation of CYP24A1 expression (Pascussi et al., 2005). However, induction of CYP3A4-dependent 23R- and 24S-hydroxylation of 1,25(OH)2D3 offers an equally, if not more, plausible explanation. In this regard, it is intriguing to speculate why patients receiving long-term antiepileptic therapy develop symptoms of osteomalacia despite normal or elevated blood levels of the active hormone 1,25(OH)2D3 (Eastwood et al., 1979; Gascon-Barré, 2004). Data from this study show that CYP24 expression is relatively high in human kidney. However, renal PXR expression is extremely low compared with liver and intestine (Lehmann et al., 1998), and this could restrict the inducibility of CYP24A1 by PXR ligands and limit systemic changes in 1,25(OH)2D3 after treatment with antiepileptic drugs (Eastwood et al., 1979; Gascon-Barré, 2004) if the kidney is the dominant organ for blood 1,25(OH)2D3 clearance. This line of reasoning is supported by the lack of effect of rifampin on renal digoxin clearance, a P-glycoprotein-mediated process from another PXR gene target (ABCB1), despite marked induction of hepatic and intestinal PXR gene targets (Greiner et al., 1999).
These observations suggest the possibility that a local intestinal, rather than systemic, 1,25(OH)2D3 deficiency leads to drug-induced osteomalacia. Induction of intestinal CYP3A4 (Kolars et al., 1992) could accelerate 1,25(OH)2D3 catabolism and, thus, reduce 1,25(OH)2D3 intracellular concentration leading directly to impaired calcium absorption. In addition, if the intestinal epithelia rely on biliary secretion of 1,25(OH)2D3-glucuronide as an important source of 1,25(OH)2D3 for gene regulation (after hydrolysis and reuptake of the active hormone) (Wiesner et al., 1980; Gascon-Barré and Gamache, 1991), induction of hepatic CYP3A4-dependent 1,25(OH)2D3 metabolism could also contribute to reduced intestinal calcium uptake and disease. This mechanism does not exclude the possibility that induction of hepatic and intestinal CYP24A1 by PXR activation also contributes to an increase in “local” 1,25(OH)2D3 metabolism (Pascussi et al., 2005). However, we observed an absence of CYP24A1 mRNA expression in the control and phenytoin-treated human intestine and liver samples. Therefore, unless it can be shown that these drugs induce hepatic and intestinal CYP24A1 expression and its metabolic contribution in vivo, induction of CYP3A4 may be the more clinically relevant event. Regardless of the ultimate mechanism, our data and those of Pascussi et al. (2005) should alert health care providers to monitor bone mineral density for patients receiving long-term treatment with any effective (in vivo) PXR ligand, not just antiepileptic drugs.
In sum, we have determined that CYP3A4 catabolizes the endogenous hormone 1,25(OH)2D3. Although a much less efficient hydroxylase compared with CYP24A1, CYP3A4 seems to be the dominant source of 1,25(OH)2D3 23- and 24-hydroxylase activity in both human small intestine and liver tissues (including samples from donors treated with phenytoin). Because the 1,25(OH)2D3/VDR signaling pathway regulates CYP3A4 expression, these data suggest that CYP3A4 provides negative feedback control of 1,25(OH)2D3 effects in the intestine. In addition, induction of intestinal and hepatic CYP3A4 activity provides a plausible mechanism for the decline in bone mineral density that occurs in patients receiving long-term therapy with effective PXR ligands (e.g., some antiepileptic drugs and rifampin).
Acknowledgments
We thank William N. Howald for expert advice with the GC/MS analysis. We thank Dr. Allan E. Rettie for critical review of the manuscript. We would also like to thank Ruihai Liu, Masako Hashizume, Suzanne Tay, and Hiromi Hamamoto (Kyoto University) for technical assistance.
Footnotes
- Received July 27, 2005.
- Accepted October 5, 2005.
Financial support for this work was provided in part by National Institutes of Health grants GM63666, GM32165, ES07033 (to K.E.T.), and GM38149 (to P.B.W.).
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.105.017392.
ABBREVIATIONS: 1,25(OH)2D3, 1α,25-dihydroxyvitamin D3; 1,23,25(OH)3D3, 1α,23,25-trihydroxyvitamin D3; 1,24,25(OH)3D3,1α,24,25-trihydroxyvitamin D3; VDR, vitamin D receptor; PXR, pregnane X receptor; BSTFA, N,O-bis(trimethylsilyl)trifluoroacetamide; TMCS, trimethylsilylchlorosilane; PCR, polymerase chain reaction; P450, cytochrome P450; TAO, troleandomycin; SPE, solid phase extraction; HPLC, high-performance liquid chromatography; GC, gas chromatography; MS, mass spectroscopy; TMS, trimethylsilyl; LC, liquid chromatography.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}