


Abstract
Multidrug resistance (MDR) is one of the major obstacles to long term successful cancer chemotherapy. The use of MDR reversal (MDRR) agents is a promising approach to overcome the undesired MDR phenotype. To design more effective MDRR agents that are urgently needed for clinical use, a data set of 609 diverse compounds tested for MDRR activity against P388/ADR-resistant cell lines was submitted to the MULTICASE computer program for structure-activity analysis. Some substructural features related to MDRR activity were identified. For example, the CH2–CH2–N–CH2–CH2group was found in most of the active compounds, and the activity was further enhanced by the presence of (di)methoxylphenyl groups, whereas the presence of a stable quaternary ammonium salt, a carboxylic, a phenol, or an aniline group was found to be detrimental to activity. Possible explanations for these observations are proposed. Some physicochemical properties, e.g., the partition coefficient (logP) and the graph index (which in some sense measures the “complexity” of a molecule) were also found to be relevant to activity. Their role in MDRR was also rationalized. Based on our quantitative structure-activity relationship study of MDRR agents, some compounds with desired substructural features and activity were identified from the MACCS-II and National Cancer Institute DIS databases and tested experimentally. Our study may also help the rational design of anti-cancer drugs. Based on this study and on observations by other researchers, we postulate that P-glycoprotein-mediated resistance to paclitaxel could probably be eliminated by proper substitution of its benzamido and phenyl groups. Several novel compounds with the paclitaxel skeleton are proposed, which may lead to a new generation of paclitaxel anti-cancer drugs with less MDR potential.
The major difficulty with current cancer chemotherapeutic drugs is the clinically acquired or nonintrinsic resistance of cancer cells to these drugs (1). The most common drug resistance in cancer chemotherapy, now termed MDR, is defined as the ability of cancer cells exposed to a single drug to develop resistance to a diverse range of structurally and functionally unrelated drugs, including the anthracyclines (doxorubicin and daunomycin), the Vinca alkaloids (vincristine and vinblastine), podophyllotoxins, and actinomycin D (2, 3). The degree of cross-resistance displayed by MDR cells to individual drugs varies among cells lines (4). However, the similarity in the pattern of resistance to this set of chemotherapeutic agents suggests that there may exist a common underlying mechanism responsible for MDR. It is now generally believed that the MDR phenotype is mainly related to an increased production of a high molecular mass (ca. 170 kDa, depending on the degree of its glycosylation and/or phosphorylation) cell surface glycoprotein (Pgp or P170) (5, 6), expressed by a MDR gene, termed mdr1 (7, 8). The Pgp acts as an (ATP) energy-dependent drug efflux pump. In MDR cell lines, drugs entering the cells through passive diffusion bind to Pgp and are actively pumped outward from the cells. There is some evidence that Pgp can also decrease the influx of cytotoxic drugs into the cell (8). These two factors result in a decreased intracellular accumulation of the anti-cancer drug and reduced cellular cytotoxicity (6, 9). Pgp has also been found in some normal cells of such tissues as the kidney, liver, intestine, and adrenal gland. It is believed to perform certain biological function, such as the transport of peptides and other substrates (e.g., toxic metabolites) across the cellular membrane.
The use of MDRR agents is a promising approach to overcome the undesirable phenotype. It is observed (10, 11) that most MDRR agents share some common chemical properties, such as lipophilicity, cationic charge at physiological pH, and at least two planar aromatic rings. However, the extremely broad diversity of MDRR agents remains puzzling.
Current MDRR agents in clinical trials are far from perfect, mainly because of their inherent toxicity (e.g., dose-related cardiovascular toxicity, transient hyperbilirubinemia) and insufficient MDRR potency at the tolerated dosage. Therefore, development of new or so-called “second generation” reversal agents, with fewer side effects, that are more potent, and whose specific potency is in modulating MDR, is a high priority of research on drug resistance (12,13). More potent and less toxic MDRR agents are urgently needed for clinical use (14-16). However, the solution to this problem is rather difficult, because the mechanisms of MDR and MDRR are still not well understood.
Obviously, any attempt at identifying the relationships between the structure and the activity of MDRR agents is useful. Indeed, the relationship could help in our understanding of the mechanisms of MDR and MDRR and in the designing of more effective MDRR agents and/or anti-cancer drugs for clinical use. Several attempts at studying the structure-reversal activity relationships have been reported in the literature. The results of these studies are summarized in Table1. It should be noted that most of these studies were qualitative observations based on the examination of relatively small experimental data sets.
Summary of previous SAR studies of MDRR agents
Based on their SAR study on phenothiazines and thioxanthenes, Haitet al. (17) proposed an ideal structure of thioxanthenes for reversing MDR. They also proposed a model for the binding of modulators to Pgp and speculated on the structure of the drug-binding domain. In their model, the aromatic residues of the transmembrane α-helices of Pgp were thought to “sandwich” the tricyclic ring of thioxanthene MDRR agents through the overlapping of π electrons.
Priebe and Perez-Soler (18) explored a “double-advantage approach” for designing more effective anti-cancer anthracyclines able to overcome MDR. When the amine group, which is a common structural feature of many Pgp substrates, was substituted in ADR with a hydroxyl group to form hydroxyrubicin, the cross-resistance to ADR decreased substantially. This study revealed the importance of information gained from the study of the mechanisms of MDR and MDRR on the rational design of non-cross-resistant anti-cancer drugs.
One recent study carried out at the National Cancer Institute may also be useful in identifying anti-cancer drugs with Pgp-mediated MDR (19). In this report, Lee and his colleagues studied the ability of 58 cell lines to efflux the fluorescent dye rhodamine 123 as a functional assay for Pgp. There was a significant correlation between MDR1 expression and rhodamine efflux in the 58 cell lines (r = 0.788,p = 0.0001). The rhodamine efflux values, defined as the difference between the efflux values in the presence and absence of Pgp antagonist cyclosporin A, was used as a seed for the COMPARE program (20) analysis of compounds with a cytotoxicity profile similar to the rhodamine efflux profile. Hundreds of compounds with high correlation coefficients were identified from an National Cancer Institute database of >30,000 tested compounds. A high degree of reversal activity, up to 10,000-fold, for some of the compounds was noted in the presence of Pgp antagonist PSC 833. This finding suggested that compounds with predominately Pgp-mediated resistance were identified.
Dellinger et al. (21) carried out another interesting study. They synthesized a series of simple aromatic (alkylpyridiniums) and nonaromatic (alkylguanidiniums) organic cations differing in their lipophilicity by the stepwise addition of single alkyl carbons, and measured their cell growth inhibitory ability against both sensitive and MDR cells. They demonstrated that MDR cells were not cross-resistant to the nonaromatic guanidiniums but did show cross-resistance to those aromatic pyridiniums with chain lengths >4. They also found that a critical degree of lipophilicity (logP > −1) was required for the MDR cells to recognize these simple aromatic cations. Furthermore, resistance to pyridinium analogs in MDR cells was reversed by co-treatment with nontoxic doses of verapamil. These results provided convincing evidence that Pgp-mediated MDR and MDRR were observed.
Although much work strongly supports the hypothesis that the reversal of MDR is mediated by direct interaction of chemosensitizers with Pgp, Wadkins et al. (22) demonstrated that drug-lipid interactions resulting in modification of membrane properties may also play an important role in the mechanism by which these compounds reverse MDR. This complicated situation undoubtedly increases the complexity of the problem and the difficulty of finding a satisfactory SAR for MDRR agents.
As the exact mechanisms of MDR and MDRR are still to be uncovered (23) and the three-dimensional structure of Pgp is unknown, it is not practical to utilize (receptor) structure-based drug design methods (24) to design MDRR agents at this moment. On the other hand, a systematic and statistical analysis of the available experimental data with the help of a suitable computer program could help gain insight into the problem. In a previous communication, we have reported the (quantitative) structure-activity relationship (SAR/QSAR) study of a series of MDRR agents using the MULTICASE (MultipleComputer Automated StructureEvaluation; MULTICASE, Beachwood, OH) program (25) developed in our laboratory and obtained encouraging results (26). After identifying certain substructural features that were statistically significant for the observed activity, from a training data set of 137 diverse compounds, the program identified seven compounds that possessed some of these substructural features from the Fine Chemical Dictionary in MACCS-II. These compounds were obtained and tested experimentally. Four of them showed potent reversal activity in vitro. One of them was as potent as verapamil and had a quite different structure from the MDRR agents currently known to be under development.
With our initial success of the SAR study of MDRR agents, and with much more experimental data regarding the MDRR activity of diverse compounds becoming available, we set out to reinvestigate the SAR of MDRR agents in the hope that it may help us gain a better understanding of the mechanisms of MDR and MDRR and help in the identification of even more potent MDRR agents.
Materials and Methods
Experimental procedure for MDR and MDRR testing.
The experimental procedures used in this study followed the protocol previously described by Ramu and Ramu (27, 28, 32). In this test system, the P388 murine leukemia cells and an ADR-resistant subline P388/ADR were maintained in RPMI 1640 medium supplemented with 10% fetal calf serum, 10 μm 2-mercaptoethanol, penicillin base (50 IU/ml), and streptomycin (50 μg/ml). An inoculum of cells was transferred to fresh medium once every 4 days to maintain exponential growth. The sensitivity of both cell lines to a given drug was assessed as follows: 1 × 106 cells were cultured in 10 ml of medium in the presence of various drug concentrations. Once a day for 4 days the density of the cells was measured with a Coulter counter. The cell growth rate was calculated from the slope of the log cell density versus time curve by linear regression analysis. The growth rate at each drug concentration was expressed as a percentage of the control rate (no drug). Dose-response curves were thus produced and used to determine the drug concentration effective in inhibiting the growth rate by 50% (ED50). In repeated experiments the standard deviation of this parameter was consistently <10% of the ED50 values obtained. The ability of a compound to ameliorate MDR (RF) was evaluated by dividing the ED50values obtained in the P388/ADR cells incubated in the absence of ADR by those in the presence of 200 nm ADR. This ADR dose was just below the concentration that produced a detectable growth-inhibitory effect on P388/ADR cells, but was rather toxic for the parent P388 cells. (The ED50 values of ADR obtained in P388 and P388/ADR cells were 35 and 900 nm, respectively. The fold of resistance of P388/ADR cells to ADR is 25.7.)
Modeling methodology.
The MULTICASE program is based on a hierarchical statistical analysis of a database (called the training set) composed of various compounds along with their biological activity data. The fundamental assumption of the MULTICASE methodology is that, if a substructure is not relevant to the observed activity, it will appear with an even distribution in both active and inactive compounds; if it is responsible for the observed activity, it will be found predominantly in active compounds. The program can automatically identify those fragments that are related to the observed activity or inactivity of a compound based on the binomial distribution criteria and bayesian probability rules. By proper combination of fragments that are favorable for activity we may obtain some new compounds with the desired activity, and in some cases, they may possess novel structural skeletons. The knowledge that the program gained during the training process can then be used to predict the biological activity of novel compounds that were not included in the training set. Therefore, the MULTICASE program may help us identify new lead compounds and design more potent agents with desired activity. Details of the methodology of MULTICASE can be found elsewhere (25).
Database.
During the past decade, many compounds have been tested for their MDRR ability under diverse experimental conditions. For example, the cytotoxins and sensitive cell lines used in the selection of the MDR cell lines are often different from one another (4), as shown in Table 1. The methods and procedures used to select MDR cell lines were shown to cause extreme diversity in the pattern of cross-resistance (29). Furthermore, the experimental procedures and expression of experimental results may also be different (26). MDRR agents that are active in one MDR cell line may or may not have similar activity in another cell line (30). The reported differences in MDRR activity may be due either to differences in the properties of the molecules tested, or to differences in the methodology used (31). Therefore, the comparison of the reversal activity of compounds tested under diverse experimental conditions may present a serious obstacle to a good SAR. Recent studies by Ramu and Ramu (27, 28, 32) reported experimental results on the MDRR activity of several hundreds of compounds (see experimental section for their experimental procedures), including many clinical drugs, under the same experimental conditions. Because of the self-consistence of the experimentally tested biological activity data and the large number of compounds under consideration, one may expect to obtain more reliable SAR results from these data, if a relationship between chemical structures and MDRR activity does indeed exist.
We collected the chemical structures of 609 compounds and their corresponding MDRR activity data as reported by Ramu and Ramu. Among them the activity data of 385 compounds were directly from the reports of Ramu and Ramu (27, 28, 32) and those of the remaining 224 compounds had since been determined in Dr. Ramu’s Laboratory.1 The chemical structures were entered into the MULTICASE program using the KLN code (33, 34) along with their MDRR activity (expressed as the RF). Among the 609 compounds, 301 were designated as active (RF > 4.2), 77 as marginally active (2.0 < RF ≤ 4.2), and 231 as inactive (RF ≤ 2.0). Because of the large diversity of chemical structures in the database, it is practically impossible to analyze the relationship between structure and activity using traditional QSAR methods, which can only deal with congeneric data sets. The database was therefore submitted to the MULTICASE program that was designed specifically for the SAR analysis of noncongeneric data sets.
Results
Biophores.
The program identified 35 different biophores for this database. The most statistically significant ones, based on their binary probability of being relevant to activity (25), are listed in Table 2.
Most significant biophors for MDRR activity
The most significant biophore can be expressed in the generic form of C–C–X–C–C, where X = N, NH, or O (preferably a tertiary nitrogen), linked to two unsubstituted alkyl groups. There are 260 compounds with this substructural feature, of which 193 are active, 21 are marginally active, and 46 are inactive. This result is consistent with our previous results (26) and possibly related to other researchers’ observations that most MDRR agents carry a positively charged nitrogen at physiological pH. It is to be noted, however, that our results point toward a partially positively charged tertiary nitrogen atom but not toward a stable quaternary nitrogen atom. This may indicate that although a partial positive charge enhances intrinsic MDRR activity, it may also be that a highly positive charge such as found on quaternary nitrogen atoms prevents the molecule from entering the cell and thereby negates the agents potential MDRR activity.
Another new observation is that the configuration of this biophore seems to be important in the present study, which was not recognized in our previous study. Indeed it is now found that compounds in which the biophore exists in a trans-conformation (i.e., at least one branch is in a linear chain) are more often active than those containing the biophore in a cis-configuration (where the biophore is part of a single ring). Indeed, 72% of the molecules containing the trans biophore are active, whereas only half of those with the cis biophore, constrained within a ring, show activity 
Trans Cis
Generally speaking, the biophore describes a substructure in which a nitrogen atom is attached to two lipophilic moieties. The preferential trans configuration may indicate that it is the nitrogen atom that interacts with the target, possibly Pgp, and that a more favorable interaction results from a larger separation between the attached lipophilic groups.
A QSAR analysis was performed on the 260 compounds containing biophore 1. The analysis helped us to identify 17 parameters (modulators) significant in modulating the MDRR activity of compounds containing biophore 1 (see Table 3). A correlation coefficient ofr = 0.743 was obtained.
QSAR values for biophore 1
Although the correlation may not be impressive, one has to keep in mind that it encompasses very diverse molecules. Nevertheless, the low correlation coefficient indicates that, although a qualitative discrimination between actives and inactives is well established, quantitative potency predictions are not to be expected. This may be due to the fact that the activity is either modulated by a great variety of undetermined factors, or that we have not been able to find the modulators, if they exist, that would rationalize the potency of these 260 compounds.
The square of the logarithm of the partition coefficient, (logP)2, was identified as the most significant modulator as it explains much of the variance. This is not surprising as the effect of MDRR agents depends on their partitioning into (across) the lipid domain of the cells (8). In fact, Zamora et al. (10) previously found that log P was an important feature for a set of 25 MDRR agents, and we also reported that logP modulates the MDRR activity (26). Furthermore, modulator number 14, water solubility of the drug, was found to have a negative effect on the reversal activity. This effect is consistent with that of the partition coefficient, because, generally speaking, the more water-soluble, the more difficult it will be for a compound to partition into the lipid phase to exhibit its biological effect. The second modulator, the o-dimethoxylphenyl group (e.g., present in the verapamil series), is an significant activating fragment. It may play a role during the MDRR agent/Pgp binding, in which the interaction between the partially positively charged hydrogens of the aromatic residues on Pgp and the partially negatively charged oxygens (of o-dimethoxylphenyl group) on MDRR agents may be enthalpically favorable for the binding (35).
The second biophore exists in 19 compounds, of which 17 are active, one is marginally active, and one is inactive. The possible reason for the inactivity of imoxiterol which possesses this biophore may be attributed to its relatively low log P value and high water solubility. Two modulators were found for the 19 compounds containing biophore 2. Again, water solubility was found to be a deactivating parameter for activity.
Some of the other biophores (e.g., biophores 4, 5, and 6) simply define lipophilic centers. Biophore 7 is associated with the dihydropyridine calcium channel blockers, e.g., nicardipine.
Deactivating fragments.
The MULTICASE program also found some substructures that are predominantly (exclusively) present in inactive molecules and may actually be responsible for this inactivity. Some of them are listed in Table 4.
Deactivating fragments (biophobes) for MDRR activity
There are 19 compounds which contain the carboxylic group, –COOH, of which 18 were completely inactive, and only one such compound, difloxacin, showed marginal activity. Seven compounds with a permanent positive charge (quaternary ammonium), were all found to be inactive. This may be due to the fact that they are unable to delocalize the negative (for carboxylic acids, which exist mainly in the dissociated form at physiological pH) or the positive (quaternary ammonium) charge and penetrate into the cell membrane to interact with the active size(s) on Pgp. The presence of a primary amine group or a hydroxyl group on an aromatic ring (aniline or phenol) is detrimental to MDRR activity, as is a nitrogen atom in an aromatic ring. The fewer detrimental effects of the phenol, aniline, and pyridine groups compared with those of the −COOH and quaternary ammonium groups may be attributed to the fact that their negative (for phenol) or positive (for aniline and pyridine) charge can be partially delocalized within the aromatic system. Therefore, in the design of new MDRR agents, one should avoid the presence of these deactivating substructural features.
Design of potential MDRR agents.
Before a QSAR model can be used in drug design, it is imperative to check its ability to predict the activity of “unknown” compounds. For this purpose, we selected the 351 molecules that show unequivocal activity or lack thereof and randomly divided them into two subsets, one subset with 175 compounds (I/M/A = 75/0/100) and another subset with 176 compounds (I/M/A = 80/0/96). Each time we used one subset as the training set for MULTICASE analysis and the other subset as the testing set. The observed correct prediction rates were 81 and 82%, respectively. These results are highly acceptable, which confirms the predicting ability of the MULTICASE methodology.
Although it is still difficult to postulate a general mechanism for MDRR from the results shown above, we may be able to develop MDRR agents with some of the biophores and/or activating modulator features. The MACCS-II Fine Chemicals Directory, which contains 64,132 commercially available compounds, was searched for compounds containing the top biophore. Several hundreds of compounds were identified and seven of them were predicted to be (very) active on the basis of the QSARs obtained from the analysis of the database. The structures of these compounds are shown in Fig. 1. Compound 8, manidipine (CV-4093), a newly synthesized dihydropyridine calcium channel blocker, was predicted to be an extremely active MDRR agent. The probability for this compound to show MDRR activity is very high (99%), owing to the presence of several biophores. The National Cancer Institute database of about 230,000 compounds was further searched using the constraints listed in Table 5.

MULTICASE-identified MDRR agents from MACCS-II.
Constraints for the NCI DIS database search
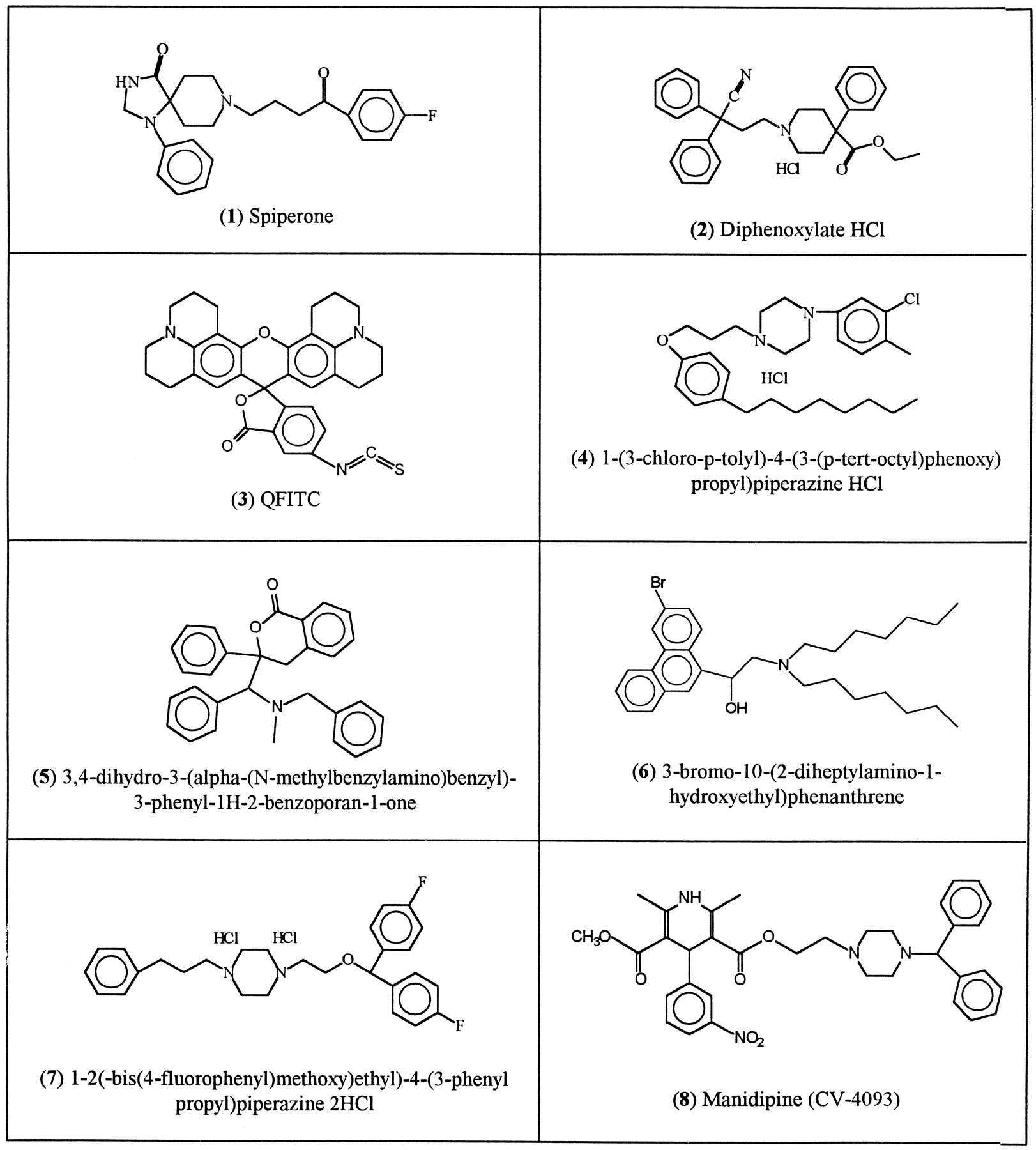
About 200 compounds were identified. Eleven of them (Fig.2) were predicted to be active by the MULTICASE program and believed to be worthy of further investigation. Compounds 5, 6, 7, and 9 are variations of a common theme and were selected to test the hypothesis that a high positive charge on the biophore’s tertiary nitrogen atom is beneficial to activity. Compound 12 is manidipine, discussed above, and compounds 13 and 14 were selected as an afterthought to test the hypothesis that a large number of phenyl rings are beneficial to MDRR activity.

MULTICASE-identified MDRR agents from the National Cancer Institute DIS.
All the samples except no. 12 (manidipine) were obtained from the National Cancer Institute’s Anticancer Drug Discovery Program (via Dr. G. W. A. Milne). Manidipine, manufactured by Takeda Chemical Industries of Osaka, Japan, was provided to us by Dr. Shaoming Huang of the Medical School of Case Western Reserve University. Manipidine and the 13 compounds obtained from the National Cancer Institute were tested experimentally for MDRR activity using the protocol described above, and the results are listed in Table 6.
MDRR tests results (All ED50 values are in μM unit)6-a
Discussion
Our study shows that certain structure-activity relationships exist among MDRR agents. Some substructural features activating or deactivating MDRR activity have been identified. Furthermore, we found that there are significant differences in molecular weight, logP, water solubility, and global graph index between active and inactive compounds to allow for easy discrimination. It is also clear that the size of a molecule (in terms of molecular weight, global graph index) is an important determinant for MDRR activity, although the number of phenyl groups in the molecule does not seem to be critical. Furthermore, positively charged nitrogen atoms are not seen to be necessarily associated with high MDRR activity as our QSAR seems to suggest. However, we believe that one of the important factors is the existence of weak polar interactions, including aromatic-aromatic, oxygen-aromatic, amino-aromatic interactions between MDRR agents and Pgp. Weak polar interactions are believed to play an important role in stabilizing protein structures and drug/protein binding (35). It has already been noted that the transmembrane segments of Pgp are very rich in aromatic amino acid residues (17, 36), which are thought to play a functional role in drug/Pgp binding because of the overlapping of π electrons (17) and aromatic-aromatic interaction (36).
Based on the results of our analysis, we identified and tested 14 new compounds for potential MDRR activity (Table 6). Of these, two (13 and 14) were selected in spite of the fact that they were predicted to be inactive by our program. Four of the remaining molecules (5, 6, 7, and 9) were selected to test our hypothesis about quaternary nitrogen derivatives. As can be seen, seven out of the 14 molecules are indeed active. The inactives are the two that were predicted to be inactive (13 and 14), and the 4 molecules that were predicted to be active on the basis of their large nitrogen positive charge. Overall, we believe that this is a remarkable result (10 out of 14 molecules were predicted correctly), considering that none of the tested compounds belong to a class previously known to be endowed with MDRR activity. Of these molecules, 2, 3, 10, and 12 (manidipine) seem to be the most promising.
Because MDRR agents probably compete with anti-cancer agents in binding to Pgp, it seems likely that for MDRR agents to exhibit potent activity they should in some way “resemble” the anti-cancer drug that is used to select the MDR cell lines. From the point of view of computer-aided drug design, it may be worthy to develop some “similarity indices” between MDRR agents and anti-cancer drugs, and employ them as independent variables in QSAR analysis in conjunction with those parameters already generated by MULTICASE at present.
It is generally believed that Pgp-mediated MDR is one of the most common mechanisms of drug resistance (8). The anti-cancer drug entering the cell by passive diffusion has strong binding affinity to Pgp. Thus it is pumped outward from the cell by Pgp, an energy-dependent drug-efflux pump. The function of MDRR agents is thought to compete with the anti-cancer drug in binding to Pgp, or blocking the drug-efflux function of Pgp, resulting in a decrease of drug efflux by Pgp. Therefore, results from the MULTICASE SAR analysis on MDRR agents should also have implications for the rational design of new anti-cancer drugs with no or less MDR. First, the biophore substructural features responsible for MDRR activity should be avoided in new anti-cancer drug candidates in case that they may be pumped outward from the cells by Pgp, resulting in decreased intracellular drug concentration and cytotoxicity. In fact, several clinical anti-cancer drugs (e.g., vincristine and vinblastine) with observed MDR are predicted to be good Pgp substrates by the MULTICASE program because of the presence of biophore(s). Second, proper combination of the biophobes (their presence prevents the binding to Pgp) for MDRR and biophores (pharmacophores) for anti-cancer activity may enable one to obtain compounds with desired anti-cancer activity and no undesired MDR phenotype. Therefore, we conclude that the results from this study cannot only be used directly as a guide in the development of new MDRR agents, but also may be helpful for the rational design of anti-cancer drugs with no or less MDR.
Keeping this hypothesis in mind, we noticed that the most promising anti-cancer drugs developed in the last decade were paclitaxel (Taxol; Bristol Myers-Squibb, New Brunswick, NJ) and its analog, docetaxel (Taxotere) (37). However, cross-resistance was also reported for paclitaxel (38). The main reason of resistance to paclitaxel was proposed to be due to the overexpression of Pgp (38) and can be fully reversed by several chemosensitizers in vitro (39). We postulate that if the nitrogen atom of the benzamido group and some of the three phenyl rings of paclitaxel are properly substituted, the resulting paclitaxel analogs should not bind effectively to Pgp but might still be effective anti-cancer drugs. Our postulate is based not only on our present investigation of the SAR of MDRR agents, but also on observations by other researchers: 1) Priebe and Perez-Soler (18) substituted the amine group of ADR with a hydroxyl group and the resulting analog, hydroxyrubicin, showed much less cross-resistance (Fig. 3); 2) Gros and his colleagues (40) found that, when the acetamido group of colchicine was eliminated, the resulting analog, deacetamidocolchicine, was not a Pgp substrate and did not show cross-resistant to colchicine (Fig. 4); and 3) Docetaxel seems to be a more effective anti-cancer drug than paclitaxel (Fig.5).

Hydroxyrubicin shows much less cross-resistance compared with doxorubicin.

Deacetamidocolchicine is not cross-resistant to colchicine.
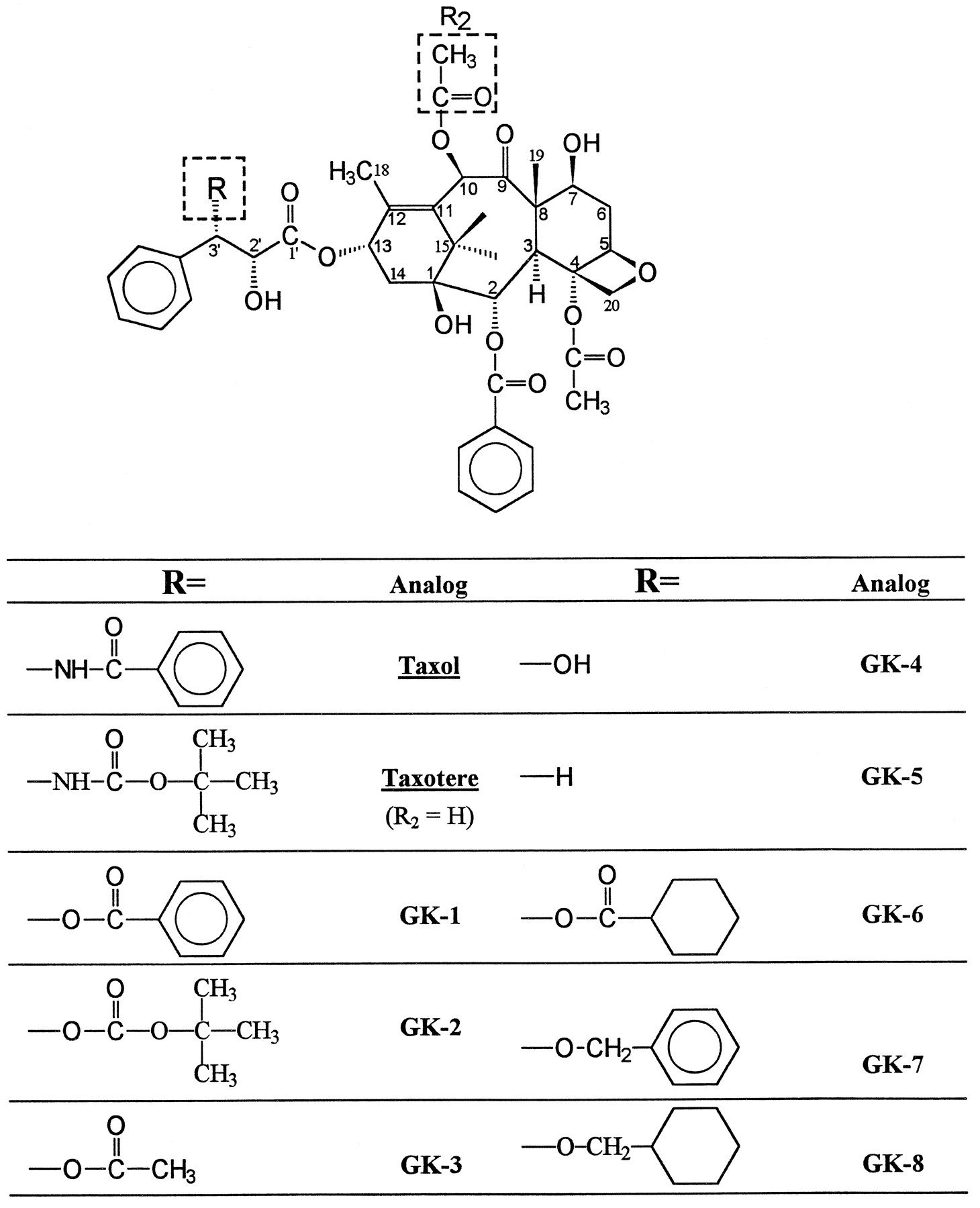
Structures of paclitaxel (Taxol)/docetaxel (Taxotere) and proposed new analogs (GK) with less MDR.
Although extensive substitutions have been made on the structure of paclitaxel (37), little attention has been paid on the replacement of the nitrogen atom and/or phenyl rings and the available activity data against MDR cells are quite limited. We propose the structures of several paclitaxel analogs (Fig. 5) that we believe should be able to show reduced Pgp-mediated MDR.
Acknowledgments
We are grateful to Dr. G. W. A. Milne and Dr. Shaomeng Wang of the National Cancer Institute for providing us samples and help with the National Cancer Institute DIS database searching.
Footnotes
- Received September 5, 1996.
- Accepted April 18, 1997.
-
Send reprint requests to: Dr. Gilles Klopman, Chemistry Department, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, OH 44106-7078. E-mail:gxk6{at}po.cwru.edu
-
↵1 A. Ramu and N. Ramu, unpublished observations.
Abbreviations
- ADR
- adriamycin (doxorubicin)
- I/M/A
- distribution of inactive, marginally active, and active compounds
- MDR
- multidrug resistance
- MDRR
- multidrug resistance reversal
- Pgp
- P-glycoprotein
- QSAR
- quantitative structure-activity relationship
- RF
- multidrug resistance reversal fold
- SAR
- structure-activity relationship
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}