


Abstract
It has recently been reported that rifampicin activates the glucocorticoid receptor and acts as an immunosuppressive drug. Because rifampicin constitutes an essential part of pulmonary tuberculosis therapy, we have examined whether it triggers glucocorticoid-like effects in alveolar cells. We have used reporter gene assays to measure the trans-activating and trans-repressing capacity of the glucocorticoid receptor after treating A549 human alveolar cells with rifampicin. The data show that rifampicin neither activated transcription from a promoter containing a glucocorticoid response element nor repressed the activity of activator protein 1 and nuclear factor κB, which are transcription factors involved in the immune response. In addition, rifampicin was also unable to inhibit the expression of an endogenous gene that contains activator protein 1 and nuclear factor κB response elements and encodes the proinflammatory cytokine RANTES (regulated upon activation normal T expressed and secreted protein). Finally, nuclear translocation of the glucocorticoid receptor, which occurs after ligand binding, was not triggered by rifampicin. In contrast, the glucocorticoid dexamethasone scored positive in all corresponding control experiments. In conclusion, rifampicin is not an activator of the glucocorticoid receptor in A549 alveolar cells. Our results support the clinical observation that rifampicin is not an immunosuppressive drug and suggest that the current medical practice concerning this antibiotic should not be changed.
Rifampicin (RIF) is a widely used antibiotic because of its clinical efficacy against a variety of organisms, including Streptococcus pneumoniae, Staphylococcus aureus,Hemophilus influenzae, Legionella pneumophila. Moreover, RIF is a major drug for mycobacterial infections (Houston and Fanning, 1994). Recently, Calleja et al. (1998) have reported that RIF may act as a ligand and an activator of the human glucocorticoid (GC) receptor (GR) in HepG2 and Jurkat cells. Using reporter gene assays, they demonstrated that RIF acts as a GC-like compound: not only could it induce the transcription of a gene controlled by a GC response element (GRE), but it also repressed the transcriptional activation from the IL-2 promoter, suggesting an interaction of the RIF-activated GR with activator protein 1 (AP-1) or nuclear factor κB (NF-κB) transcription factors.
GREs are found in genes involved in the control of neoglucogenesis, arterial pressure, and intraocular tension (Jantzen et al., 1987; Imai et al., 1993; Guo et al., 1995; Ming et al., 1996; Nguyen et al., 1998). Thus, trans-activation may account for some GC side effects (diabetes, arterial hypertension, hydrosodic retention, hypokaliemia, glaucoma). AP-1 and NF-κB DNA-binding sites, called 12-O-tetradecanoyl-phorbol-13-acetate response elements (TRE) and NF-κB response elements (NF-κBRE), respectively, are present in many genes coding for proinflammatory mediators and are necessary for the stimulation of their transcription (Cato and Wade, 1996). trans-Repression of AP-1 and NF-κB is considered the major mechanism whereby GCs exert their anti-inflammatory actions (Cato and Wade, 1996). If the ability of RIF to act as a GC at the molecular level is confirmed, RIF would have to be considered a potential immunosuppressive drug.
Nevertheless, the report of Calleja et al. (1998) is surprising to the physician because: 1) no GC-like adverse effects (arterial hypertension, hydrosodic retention, hypokaliemia, diabetes, osteoporosis, adrenal suppression, open angle glaucoma, immunosuppression, etc.) have been reported despite the long-term (at least 6 months) administration of RIF to millions of patients suffering from tuberculosis (Raviglione et al., 1995); 2) clinical benefits are gained when GC are added to RIF in the treatment of certain forms of tuberculosis (e.g., neurologic, pericarditis, pleural) (Senderovitz and Viskum, 1994; Dooley et al., 1997); and 3) RIF has proven to be effective in treating or preventing tuberculosis in patients with AIDS (Halsey et al., 1998). This discrepancy between clinical observations and the in vitro GC-like effects of RIF reported by Calleja et al. (1998) emphasizes the importance of verifying their findings. In this regard, Ray et al. (1998) were unable to demonstrate any effect of RIF on a GRE-dependent reporter gene in AtT20 and COS-7 cells. In addition, they showed that, as opposed to a GC, RIF did not suppress adrenocorticotropic hormone release from the pituitary corticotroph cell line AtT20. The molecular hypotheses evoked to explain these discrepant data were the action of cell-type specific RIF metabolites, a lower RIF intracellular concentration caused by the P-glycoprotein efflux pump action, or a possible contamination of RIF by other compounds (Ray et al., 1998).
Rifampicin constitutes an essential part of drug regimens administered for pulmonary tuberculosis therapy. It is, therefore, important to examine whether RIF triggers GC-like effects in alveolar cells. A549 human pulmonary cells seem to be appropriate for such a study because they express the GR and respond to GC (Kwon et al., 1995;Mathieu et al., 1999). Moreover, they retain important functional and morphological characteristics of normal human alveolar cells, which are one of the targets of Mycobacterium tuberculosis (Mehta et al., 1996). To evaluate the possible effects of RIF on GRE-mediatedtrans-activation and on AP-1 and NF-κB activities, A549 cells were transiently transfected with reporter genes controlled by GRE, TRE, or NF-κBRE. To examine a physiologically relevant target gene, we measured the possible effect of RIF on the expression of an endogenous gene that contains TRE and NF-κBRE and encodes the proinflammatory cytokine ‘regulated upon activation normal T expressed and secreted protein’ (RANTES). This mediator was quantified because it is involved in many inflammatory diseases, including AIDS, autoimmune encephalomyelitis, Crohn’s disease, arthritis, glomerulonephritis, and asthma (Cocchi et al., 1995; Venge et al., 1996; Berrebi et al., 1997; Scarlatti et al., 1997; Barnes et al., 1998; Chen et al., 1998; Glabinski et al., 1998; Gonzalo et al., 1998). RANTES is also one of the major HIV-suppressive factors (Cocchi et al., 1995). Moreover, it has recently been reported that RANTES is induced by infection of mononuclear phagocytes with mycobacteria, is present in lung alveoli during active pulmonary tuberculosis, and involved in the recruitment of cells for tuberculosis granuloma formation (Sadek et al., 1998). Finally, we tested whether RIF was able, as are other GR ligands, to trigger nuclear translocation of the GR.
Experimental Procedures
Materials.
Transferrin-polylysine, poly-l-lysine (P2636), spermine, dexamethasone (DEX), and RIF (R3501, R7382) were purchased from Sigma (St. Louis, MO). Luciferin and dithiothreitol (DTT) were purchased from Promega (Madison, WI). Tumor necrosis factor α (TNF-α) was purchased from Pharmingen (San Diego, CA). CoA was purchased from Boehringer Mannheim (Mannheim, Germany). Geneticin was purchased from Gibco/BRL (Gaithersburg, MD). To prepare DEX and RIF solutions, dilutions with medium were made freshly each day from original stocks. The DEX stock was at a concentration of 10−3 M in absolute ethanol, whereas RIF was at 10−2M in dimethyl sulfoxide.
Plasmids.
The plasmid pHHLuc contains sequences from −223 to +105 of the mouse mammary tumor virus long terminal repeat promoter (including one functional GRE) fused to the luciferase gene and was purchased from the American Type Culture Collection (Rockville, MD). The luciferase reporter construct 5xTRE TATA Luc, containing five copies of the AP-1-binding site (TRE) from the collagenase gene upstream of a TATA element, was a gift from Peter Herrlich (Institute of Genetics, Karlsruhe, Germany) (Jonat et al., 1990). The 3xIgκ Cona Luc plasmid, which contains three tandem repeats of the NF-κBRE from the Igκ chain linked to the conalbumin minimal promoter and the luciferase gene, was obtained from Alain Israël (Institut Pasteur, Paris, France). The plasmid CMVβ-gal, consisting of the cytomegalovirus early promoter linked to the β-galactosidase gene, was used as a control vector to correct for variations in transfection efficiency. The expression vector for the NF-κB subunit p65 (pECEp65) and control vector (pECE) were donated by Carl Scheidereit (Max Delbrueck Center for Molecular Medicine, Berlin, Germany) (Naumann et al., 1993). The expression vector for the human c-Fos (pCIFos) (Roux et al., 1991) was provided by Dany Chalbos (Institut National de la Santé et de la Recherche Médicale U148, Montpellier, France) and the control vector pCI was purchased from Promega.
Preparation of AdCMVnull Adenovirus.
AdCMVnull adenovirus, a replication-defective strain of human adenovirus type 5 in whichE1A and E1B sequences were replaced by the CMV early promoter, was propagated on the complementing 293 cell line as described previously (Graham et al., 1977).
Cell Culture.
A549 human lung carcinoma cells were maintained in Ham’s F12 medium containing 10% fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM glutamine. They were seeded into 48-well cluster plates (50,000 cells/well) for reporter and RANTES assays and into 24-well cluster plates (50,000 cells/well) containing coverslips for immunofluorescence analysis.
Transient Transfection.
One day after seeding the cells, medium was replaced by 100 μl/well of serum-free medium. For reporter assays, the DNA to be transfected included 25 ng/well of the CMVβ-gal plasmid for normalization of transfection efficiency, 60 ng/well of the reporter plasmids pHHLuc, 5xTRE TATA Luc, or 3xIgκ Cona Luc, and (where indicated in the figure legends) 20 ng/well of the expression vectors for c-Fos (pCIFos) and for the NF-κB subunit p65 (pECEp65). The corresponding empty vector (pCI or pECE) was added so that each well contained the same total amount of DNA (600 ng).
The DNA was diluted in 20 mM HEPES, pH 7.4, 150 mM NaCl, complexed with AdCMVnull adenovirus, transferrin-polylysine, and poly-l-lysine and added to the cells as described previously (Kohout et al., 1996). Using this technique, up to 50% of the cells could be transfected. After transfection, the cells were treated or not with 10−7M DEX and/or 10−6M RIF for 20 h.
Luciferase and β-Galactosidase Assays.
Transfected cells were lysed in 100 μl of 15 mM Tris·HCl, pH 7.8, 60 mM KCl, 15 mM NaCl, 2 mM EDTA, 0.15 mM spermine, and 1 mM DTT. After a brief centrifugation, the supernatant was recovered for the reporter assays. Luciferase activity in 40 μl of cell extract was measured in a luminometer after injection of 100 μl of luciferin mix (25 mM Tris-acetate, pH 7.8, 41 mM DTT, 0.125 mM EDTA, 4.67 mM MgSO4, 0.34 mM coenzyme A, 0.66 mM ATP, and 0.59 mM luciferin). For β-galactosidase assay, 10 μl of cell extract was mixed with 67 μl of the chemiluminescent substrate Galacton-Plus and processed according to the manufacturer’s recommendations (Tropix, Bedford, MA). β-galactosidase activity was measured to correct for variations in transfection efficiency.
RANTES Immunoassay.
A549 cells were left untreated or stimulated with 10 ng/ml of TNF-α alone or in combination with 10−7M DEX, 10−6M RIF, or 10−7M DEX and 10−6M RIF. Concentration of RANTES in supernatants collected at 20 h was determined using a quantitative sandwich enzyme immunoassay technique and following the manufacturer’s recommendations (R&D Systems, Oxon, UK).
Subcellular Distribution of the GR.
Cells were fixed by 4% paraformaldehyde and permeabilized for 4 min at 0°C by 0.5% (v/v) Triton X-100 in PBS. After blocking in PBS containing 3% BSA and 0.1% (v/v) Tween 20, the cells were incubated for 1 h at room temperature with the anti-GR monoclonal antibody NCL-GCR (Novocastra, Newcastle upon Tyne, UK) diluted 1:20 in the same buffer. This antibody is directed against an epitope at the amino terminus of the GR. A 1:50 dilution of goat serum was then added for 15 min, removed and replaced by tetramethylrhodamine isothiocyanate-coupled goat anti-mouse IgG diluted 1:100 for 30 min. Extensive washes were made after fixation, permeabilization, and incubation with antibodies. Cells were mounted and observed with a Nikon Optiphot-2 fluorescence microscope (Nikon, Tokyo, Japan) equipped with a Bio-Rad 1024 laser scanning confocal imaging system (Bio-Rad, Hercules, CA) and a 60× (1.4 aperture) objective.
Statistical Analysis.
All data represent the mean ± S.E.M. Data were analyzed with the Instat software (GraphPad Software, San Diego, CA) using the Mann-Whitney U test. Statistical significance was set at p < .05.
Results
Rifampicin Does Not Induce trans-Activation through a GRE.
trans-Activation through the endogenous GR was assessed after transfection of A549 human alveolar cells with the GC-inducible reporter plasmid pHHLuc (Fig.1). Rifampicin did not alter transcriptional activity from pHHLuc, whereas treatment with the synthetic GC DEX resulted in a 5-fold induction of this activity. Combination of RIF and DEX produced the same stimulating effect as DEX alone. The inability of RIF to induce trans-activation through a GRE was confirmed on the human HeLa cell line (data not shown).
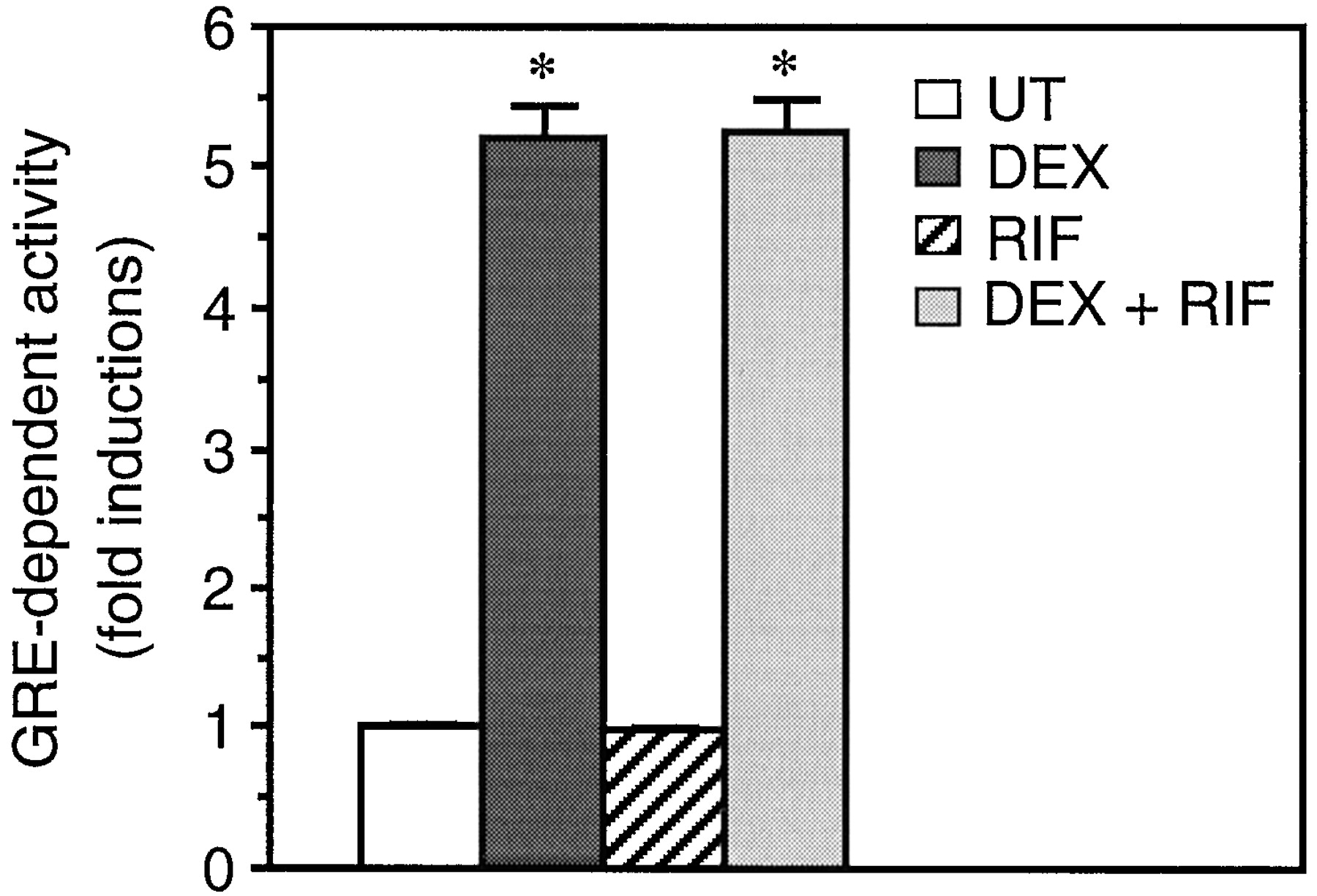
RIF does not induce trans-activation through a GRE. After transfection with the GRE-dependent reporter plasmid pHHLUC, A549 cells were left untreated (UT) or stimulated with 10−6M RIF and/or 10−7M DEX for 20 h, as indicated. Transcriptional activity was then measured by luciferase and β-galactosidase assays as described in Experimental Procedures. Data are shown as fold inductions over basal activity (activity in untreated cells) and represent the means ± S.E.M. of three independent experiments performed in triplicate. *p < 0.001 versus basal activity.
Rifampicin Does Not Repress AP-1 Activity.
To study the effect of RIF on AP-1 activity, A549 cells were transfected with the AP-1-dependent reporter plasmid 5xTRE TATA Luc (Fig.2). Overexpression of the AP-1 component c-Fos triggered a 4.5-fold induction of transcriptional activity. RIF did not repress c-Fos-induced AP-1 activity, whereas DEX treatment resulted in a 30% inhibition. Combination of RIF and DEX produced the same repressing effect as DEX alone.
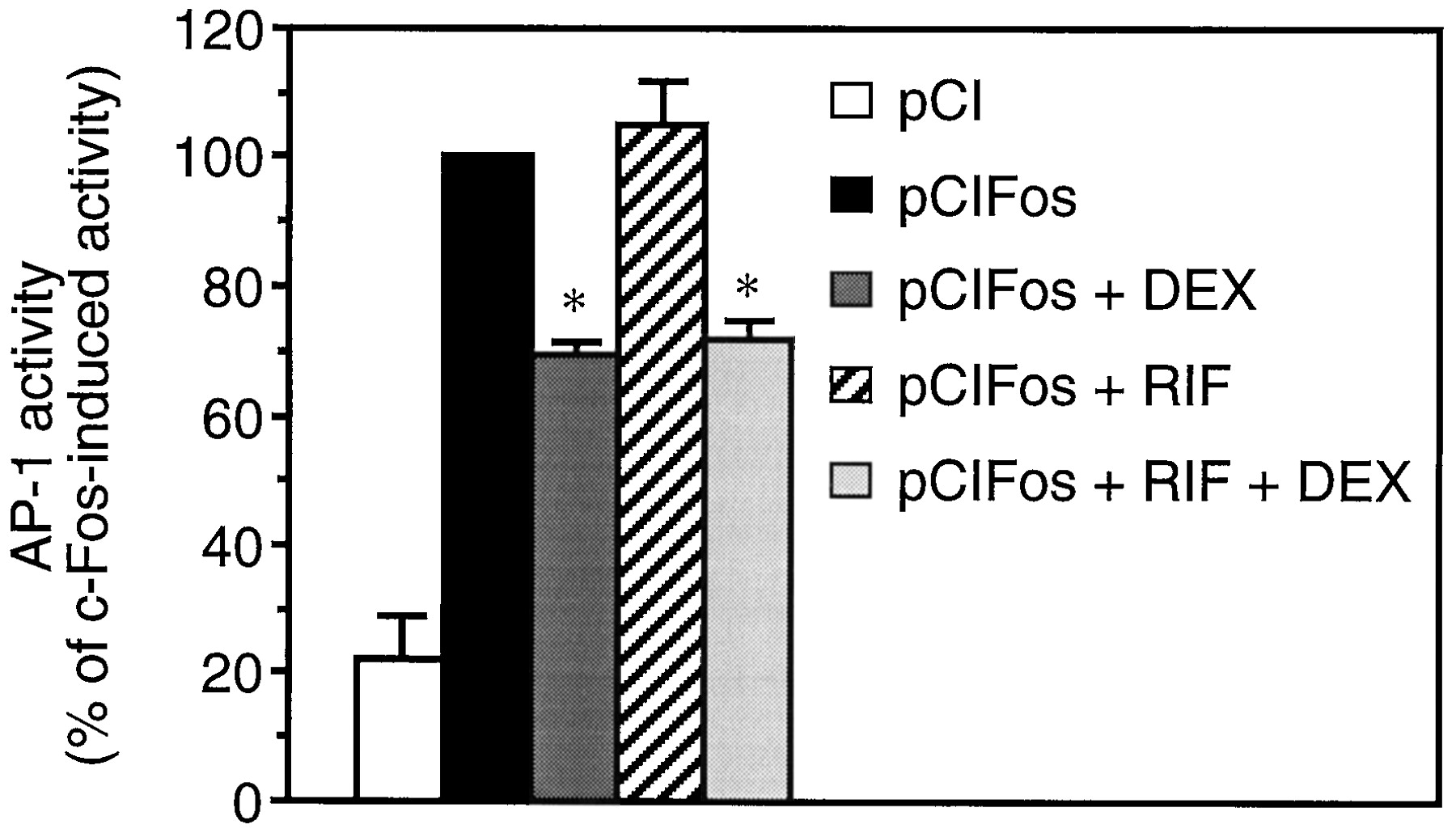
RIF does not repress AP-1 activity. After transfection with the AP-1-dependent reporter plasmid 5xTRE TATA Luc and the empty vector pCI or the c-Fos expression vector pCIFos, A549 cells were stimulated with 10−6M RIF and/or 10−7M DEX for 20 h, as indicated. Transcriptional activity was measured by luciferase and β-galactosidase assays as described in Experimental Procedures. Data are shown as a percentage of the c-Fos induced activity and represent the means ± S.E.M. of three independent experiments performed in triplicate. **p < 0.001 versus c-Fos-induced activity.
Rifampicin Does Not Repress NF-κB Activity.
The effect of RIF on NF-κB activity in A549 cells was assessed after transfection with the NF-κB-responsive reporter plasmid 3xIgκ Cona Luc (Fig.3). Overexpression of the NF-κB p65 subunit resulted in a 6-fold induction of transcriptional activity. RIF did not repress p65-induced NF-κB activity, whereas DEX treatment produced a 26% inhibition. Combination of RIF and DEX produced the same repressing effect as DEX alone.
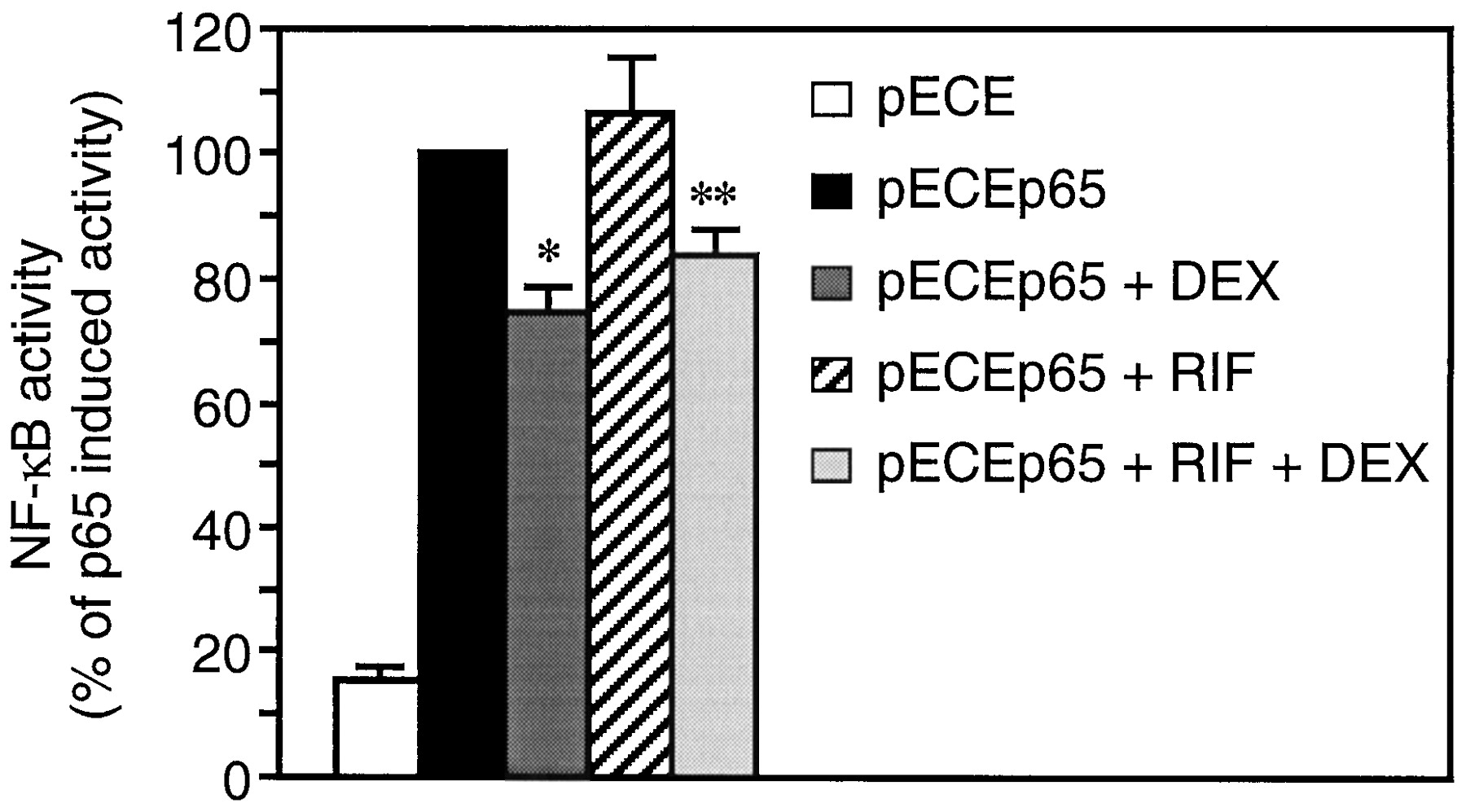
RIF does not repress NF-κB activity. After transfection with the NF-κB-dependent reporter plasmid 3xIgκ Cona Luc and the empty vector pECE or the p65 expression vector pECEp65, A549 cells were stimulated with 10−6M RIF and/or 10−7M DEX for 20 h, as indicated. Transcriptional activity was measured by luciferase and β-galactosidase assays as described in Experimental Procedures. Data are shown as a percentage of the p65-induced activity and represent the mean ± S.E.M. of three independent experiments performed in triplicates. *p < 0.001 versus p65-induced activity; **p < 0.01 versus p65-induced activity.
Rifampicin Does Not Affect RANTES Production.
Because our previous experiments were made using transfected promoter-gene constructs, we investigated whether the expression of an endogenous gene containing AP-1 and NF-κB sites would be affected by RIF. One such gene codes for the proinflammatory cytokine RANTES (Nelson et al., 1993), which is positively regulated by TNF-α in A549 cells (Kwon et al., 1995). Our data show that upon TNF-α stimulation, concentration of RANTES in cell supernatants rose from 1.24 ± 0.43 pg/ml to 919 ± 65 pg/ml (Fig. 4). Concomitant RIF treatment did not modify the concentration of RANTES. In contrast, DEX treatment strongly antagonized (by 73%) the stimulatory effect of TNF-α on RANTES production. Moreover, combination of RIF and DEX produced the same repressing effect as DEX alone.
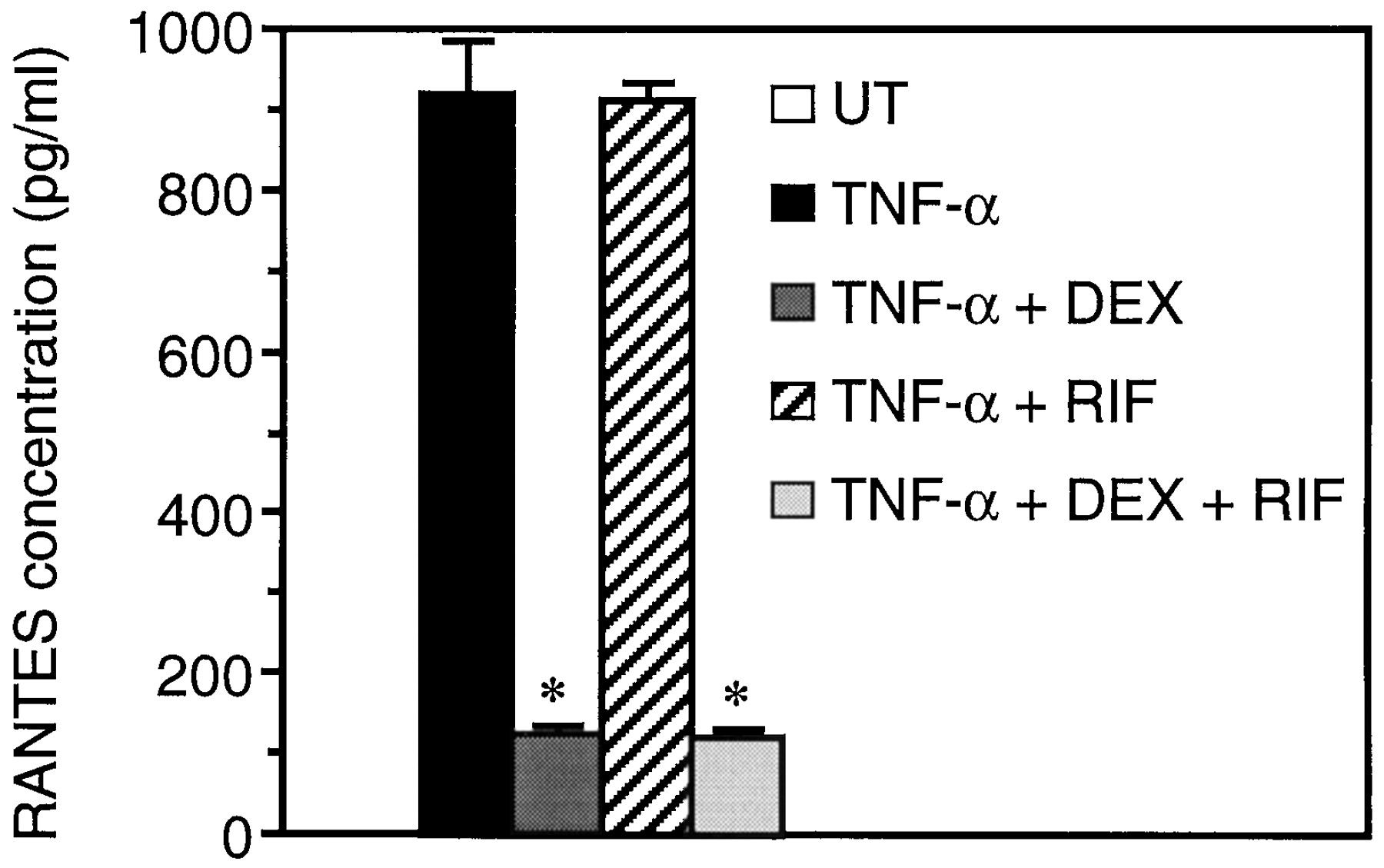
RIF does not affect RANTES production. A549 cells were left untreated (UT) or stimulated with 10 ng/ml TNF-α in the absence or presence of 10−7M DEX and/or 10−6M RIF for 20 h, as indicated. Concentration of RANTES in the supernatant was determined using a sandwich enzyme immunoassay as described in Experimental Procedures. Data are shown as absolute values ± S.E.M. *p < 0.05 versus concentration after stimulation with TNF-α.
Rifampicin Does Not Affect the Subcellular Localization of the GR.
Nuclear translocation of the GR occurs after ligand binding. Therefore, the effect of RIF on subcellular localization of the GR was assessed using immunofluorescence microscopy. In untreated cells, the endogenous GR was detected in both the cytoplasm and nucleus of cells, but expression was predominant in the latter compartment (Fig.5a). After DEX treatment, cytoplasmic expression of GR could no longer be detected, which indicates the occurrence of a hormone-driven nuclear translocation of the GR (Fig.5b). In contrast, subcellular localization of the endogenous GR was not influenced by RIF (Fig. 5c). Similar data were obtained on the subcellular localization of overexpressed GR (data not shown).

RIF does not affect the subcellular localization of the GR. A549 cells were left untreated (UT) or stimulated with 10−7M DEX or 10−6MRIF, as indicated. Twenty hours later, cells were processed for anti-GR immunofluorescence as described in Experimental Procedures. Scale bar, 50 μm.
Discussion
In this study, we report that RIF has no GC-like effects in a human alveolar cell line. The ability of RIF to activate the GR has recently been both reported and controverted. Whereas Calleja et al. have shown that RIF bound and activated the GR (Calleja et al., 1998), others were unable to demonstrate any effect of RIF on a GRE-dependent reporter gene or on adrenocorticotropic hormone synthesis in AtT20 cells (Ray et al., 1998). To explain this discrepancy, it was speculated that AtT20 cells may overexpress the P-glycoprotein efflux pump, which would cause resistance to RIF (Ray et al., 1998). This possibility was ruled out in our study, because A549 cells do not express the P-glycoprotein (Kavallaris et al., 1997).
Regarding the current use of RIF to treat tuberculosis, particularly in patients with AIDS, the most important question to address is whether this compound has immunosuppressive properties. To this end, Calleja et al. (1998) reported that RIF decreased transcription from a transiently transfected IL-2 promoter-reporter construct in Jurkat cells. However, the effect of RIF on IL-2 protein production was not assessed. To gain insights into the molecular mechanism of this transcriptional repression, we first tested whether RIF repressed AP-1 and NF-κB activities, which control many genes encoding inflammatory mediators such as IL-2 and RANTES. We then evaluated the effect of RIF on the expression of RANTES at the protein level. Our data show that RIF neither inhibited AP-1 and NF-κB activities nor repressed the production of RANTES. Importantly, the lack of GC-like effects of RIF was not specific to A549 cells, because no trans-activating effect of RIF was found in the human cell line HeLa (data not shown) and in COS-7 monkey cells (Ray et al., 1998). Together these data suggest that RIF is unlikely to act as an immunosuppressor in vivo.
As in the previous in vitro studies (Calleja et al., 1998; Ray et al., 1998), we used RIF at 10−6M, which is 100 times higher than the reported Kd of RIF for the GR (Calleja et al., 1998). Moreover, 10−6M of RIF is a pharmacologically relevant concentration, because it is reached in human lung and serum after administration of this antibiotic at the regular dose of 10 mg/kg body weight (Boman and Malmborg, 1974; Kiss et al., 1976). Consequently, the absence of reported GC-like adverse effects of RIF in vivo is also not explained by a low concentration of RIF reached in target tissues.
In contrast to the putative immunosuppressive effect of RIF, its role as a powerful inducer of CYP3A is clearly established. These isoforms metabolize a vast array of drugs, including cyclosporin and oral contraceptives, in the liver. By increasing drug clearance through CYP3A induction, RIF can cause acute graft rejections in patients with transplants or can disrupt unplanned pregnancy in women. Such drastic effects are not produced by GC, which also induce CYP3A (Schuetz et al., 1996). Because GRE half-sites are present in aCYP3A gene, it was speculated that RIF could induce CYP3A through activation of the GR (Blanchard, 1998). Rather, it seems that the molecular mechanism by which RIF induces CYP3A involves the recently identified human nuclear receptor termed hPAR, which selectively induces human but not murine CYP3A expression through another regulatory sequence than GRE present in human but not murineCYP3A genes (Bertilsson et al., 1998).
Using indirect immunofluorescence microscopy, we show that RIF was unable to induce nuclear translocation of the GR, a process that occurs after binding of various GR ligands, including agonist and antagonist GC (Qi et al., 1990). We also report that most of the endogenous GR molecules resided constitutively in the nucleus of A549 cells. Thus, RIF did not elicit GC-like effects, most likely because of its inability to trigger nuclear translocation of cytoplasmic GR and to transcriptionally activate nuclear GR. Moreover, RIF did not behave as an antagonist, because it did not affect the transcriptional effects of DEX. Taken together, these data strongly suggest that RIF is not a ligand and activator of the GR in A549 cells.
In conclusion, as opposed to a representative GC, RIF did not activate the GR in a human alveolar cell line and did not repress the production of RANTES, a major player in the immune response in various diseases, particularly AIDS and tuberculosis. Our in vitro data support clinical experience: physicians prescribing RIF for antibiotic therapy are not confronted with the usual adverse effects of GC. Therefore, the current medical practice concerning this antibiotic should not be changed.
Acknowledgments
We are very grateful to P. Herrlich, A. Israël, C. Scheidereit, and D. Chalbos for providing plasmids. We thank P. Atger for help with Fig. 5. Confocal microscopy analysis was performed using the facilities at the Center Régional d’Imagerie Cellulaire de Montpellier.
Footnotes
-
Send reprint requests to: Dr. Pascal Demoly, Service des Maladies Respiratoires-INSERM U454, Hôpital Arnaud de Villeneuve, CHU de Montpellier, 34295 Montpellier Cedex 5, France. E-mail:demoly{at}montp.inserm.fr
-
G.M. is supported by a grant from the Conseil Régional du Languedoc-Roussillon and by a grant from the Ministère de la Recherche.
- Abbreviations:
- RIF
- rifampicin
- GC
- glucocorticoid
- GRE
- glucocorticoid response element
- GR
- glucocorticoid receptor
- DEX
- dexamethasone
- AP-1
- activator protein 1
- NF-κB
- nuclear factor-κB
- RANTES
- regulated upon activation normal T expressed and secreted protein
- DTT
- dithiothreitol
- TNF-α
- tumor necrosis factor α
- TRE
- 12-O-tetradecanoyl-phorbol-13-acetate response element
- NF-κBRE
- NF-κB response element
- Received November 30, 1998.
- Accepted January 25, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}