


Abstract
There are conflicting reports about the role of nitric oxide in the down-regulation of cytochrome P450 that occurs when animals or cultured hepatocytes are exposed to inflammatory stimuli. Here, we investigated the participation of NO in the down-regulation of CYP2B1 by bacterial endotoxin (LPS) in rat hepatocytes cultured on Matrigel. LPS caused the down-regulation of CYP2B1 mRNA to 20% of control values within 12 h of treatment, and this was not reversed by concentrations of NO synthase inhibitors that completely blocked NO production. LPS was several orders of magnitude more potent in the down-regulation of CYP2B1 mRNA than in induction of NO production. In contrast, concentrations of LPS in the 1 to 100 ng/ml range induced NO production and produced a rapid down-regulation of CYP2B1 protein to 30% and <5% of control at 6 and 24 h, respectively, that could be completely prevented both by inhibitors of NO synthase and by LY83583, which prevents NO synthase-2 induction. The blockade of CYP2B1 down-regulation by NO synthase inhibitors was reversed by arginine, and the NO donors S-nitrosoglutathione andS-nitroso-N-acetylpenicillamine mimicked CYP2B1 protein suppression. Taken together, these experiments demonstrate two independent mechanisms of CYP2B1 down-regulation by LPS: a rapid, NO-dependent suppression of the protein occurring at high concentrations of LPS and a slower, NO-independent pretranslational suppression occurring at low concentrations of LPS.
Exposure of animals or cultured hepatocytes to inflammatory stimuli or cytokines causes decreases in total microsomal cytochrome P450 (P450), P450-catalyzed enzyme activities, and levels of P450 proteins and mRNAs in the hepatocyte (Morgan, 1997). These conditions also induce hepatic inducible nitric-oxide synthase [NOS2 (or iNOS)] activity and production of NO in both hepatocytes and Kupffer cells (Billiar et al., 1992). Because NO reacts with hemoproteins and is produced in the same cells in which P450 enzymes become inhibited or down-regulated, it has been hypothesized that the stimulation of NO production in the liver during inflammatory responses causes the observed changes in P450 activities and expression. As will be discerned from the following discussion, this has been a topic of extensive study and has yielded conflicting results.
Nitric oxide, peroxynitrite, and NO donors are capable of inhibiting the catalytic activities of hepatic microsomal P450s (Wink et al., 1993; Kim et al., 1995; Minamiyama et al., 1997) purified P450s (Roberts et al., 1998), and CYP8A1 (prostacyclin synthase) (Zou et al., 1998). This occurs by at least three mechanisms: reversible ligation of NO to (primarily ferrous) P450 heme (Wink et al., 1993), oxidation of P450 protein thiols (Takemura et al., 1999); and nitrosylation of specific tyrosine residues on the enzyme (Roberts et al., 1998). Inhibitors of NOS have been demonstrated to block or attenuate decreases in spectrally measurable microsomal P450 and P450-dependent catalytic activities associated with NOS2 induction by inflammatory stimuli in cultured rat (Stadler et al., 1994; Osawa et al., 1995) and human (Donato et al., 1997) hepatocytes, rat mesangial cells (Zou et al., 1998), and whole animals (Khatsenko et al., 1993; Muller et al., 1996; Khatsenko and Kikkawa, 1997; Takemura et al., 1999).
In contrast to its well-documented effects on P450 catalytic activities and spectral properties, the role of NO in the down-regulation of P450 mRNAs and proteins during an inflammatory response is more controversial. Studies in different laboratories using NOS inhibitors have produced conflicting results. For instance, Khatsenko and Kikkawa (1997) reported that NOS inhibitors could protect CYP2C11 and CYP3A2 protein and mRNAs from suppression by bacterial lipopolysaccharide (LPS) treatment of Sprague-Dawley rats. In contrast, we (Sewer and Morgan, 1998) and others (Takemura et al., 1999) found that the decreases in expression of these P450s were not affected by inhibition of NOS activity. Moreover, we found that LPS treatment suppresses the expression of CYP2C and 3A mRNA and proteins equally well in wild-type and NOS2-null mouse livers (Sewer et al., 1998), indicating that the suppression is NO-independent. Similarly conflicting results have been obtained from studies on the effects of NOS inhibitors on P450 down-regulation in cultured hepatocytes (Stadler et al., 1994; Carlson and Billings, 1996; Monshouwer et al., 1996). For example, our laboratory found no effect of NOS inhibition on the down-regulation of CYP2C11 mRNA and protein by LPS or interleukin (IL)-1 (Sewer and Morgan, 1997). In contrast, it was reported that NOS inhibition could block down-regulation in hepatocytes of CYP1A1 mRNA and protein, and of CYP2B, -2C11, -1A2, and -3A2 proteins (Carlson and Billings, 1996), caused by cytokine combinations.
Some of the most convincing evidence for participation of NO in down-regulation of P450 mRNAs and proteins has been obtained with phenobarbital (PB)-inducible CYP2B enzymes. Administration ofN ω-nitro-l-arginine methyl ester to rats treated with PB and LPS blocked the suppression of CYP2B1/2 activity, mRNA and protein (Khatsenko et al., 1993, 1997). Inhibitors of NOS also blocked the suppression of CYP2B1/2 proteins by a cytokine combination in short-term cultures of rat hepatocytes (Carlson and Billings, 1996), although the regulation of CYP2B1/2 mRNAs in this system was not reported. However, Milosevic et al. (1999)reported recently that LPS caused suppression of CYP2B1 mRNA and protein only when hepatocytes were cocultured with Kupffer cells. This down-regulation was dependent on tumor necrosis factor-α (TNFα) release from the Kupffer cells and independent of NO production (Milosevic et al., 1999).
In the present study, we have used rat hepatocytes cultured on Matrigel to investigate the participation of endogenous NO in the down-regulation of CYP2B mRNAs and proteins by LPS and IL-1 in hepatocytes. Our results show for the first time that the down-regulation of a single P450 in hepatocytes in response to an inflammatory agent can occur by two independent mechanisms in a concentration-dependent manner. Down-regulation of CYP2B1 mRNA in this system occurs at low LPS concentrations and is independent of NO, whereas the suppression of CYP2B proteins occurs at high LPS concentrations and at early time points, and is caused by NO production.
Experimental Procedures
Materials.
Male Fischer 344 rats (175–200 g) from Harlan Sprague-Dawley (Indianapolis, IN) or Charles River Laboratories (Wilmington, MA) were used for hepatocyte isolation. Cell culture medium (Waymouth's MB 752/1), insulin, antibiotics, and murine recombinant IL-1β were purchased from Invitrogen (Carlsbad, CA). Chromatographically purified Escherichia coli LPS, serotype 0127:B8, N-monomethyl-l-arginine (NMA), aminoguanidine (AG), sulfanilamide,N-(1-naphthyl)ethylenediamine, glutathione, resorufin, pentoxyresorufin, NADPH:nitrate reductase, and Type IV collagenase were purchased from Sigma Chemical Co. (St. Louis, MO).S-nitroso-N-acetylpenicillamine (SNAP) andS-nitrosoglutathione (GSNO) were obtained from BIOMOL Research Laboratories Inc (Plymouth Meeting, PA). LY83583 was from Calbiochem (La Jolla, CA). Horseradish peroxidase-coupled goat anti-rabbit IgG was purchased from Jackson Immunoresearch Laboratories (West Grove, PA). The plasmid pAstNOS-4, containing the 3′ end of the NOS2 cDNA, was a generous gift from Dr. D.L. Feinstein (Cornell University, Ithaca, NY).
Hepatocyte Isolation, Culture, and Treatments.
Matrigel, prepared according to the method of Schuetz et al. (1988), was diluted to 6 to 7 mg/ml with Waymouth's medium, applied to 60-mm plastic culture dishes (Nunc; Fisher Scientific, Pittsburgh, PA), and allowed to gel at 37°C for 1 to 2 h. Isolation of rat hepatocytes was performed by in situ collagenase perfusion as described previously (Sewer and Morgan, 1997). Cells (3.0 × 106/60 mm plate) were plated in 3 ml of Waymouth's medium containing 0.15 μM insulin. Cultures were maintained for 5 to 7 days at 37°C in 5% CO2atmosphere. The medium was replaced every 48 h, commencing 4 h after plating.
Hepatocytes were treated with 1 mM PB to induce CYP2B1 expression, beginning 4 days after cell isolation, and PB was present at all times thereafter. Using this protocol, the levels of CYP2B1 mRNA and protein in the cultures were approximately 76 and 23%, respectively, of those in the livers of rats treated with PB in vivo (results not shown). Unless otherwise noted, cells were treated with LPS or cytokines beginning 48 h after PB addition to the medium. These treatments do not significantly affect cell viability measured by trypan blue exclusion (Sewer and Morgan, 1997).
Isolation of Total RNA and Slot-Blot Assays.
Total hepatocyte RNA was prepared by the acid phenol extraction method (Chomczynski and Sacchi, 1987). Relative levels of CYP2B1 and NOS2 mRNAs in the samples were measured by a slot blot assay using the CYP2B1-specific oligonucleotide described by Omiecinski et al. (1985)or a cDNA to the 3′ end of the NOS2 mRNA (Galea et al., 1994), respectively. Total RNA was denatured using formaldehyde and loaded onto Nytran filters (Schleicher & Schuell, Keene, NH) in the slot blot manifold. The RNA was immobilized by both UV irradiation and baking at 80°C. The CYP2B1 oligonucleotide probe was labeled using T4 polynucleotide kinase and [γ-32P]ATP and was hybridized with the immobilized RNA overnight at 45°C in 1× standard saline/phosphate/EDTA buffer (0.18 M NaCl and 1 mM EDTA in 10 mM sodium phosphate buffer, pH 7.7) containing 5× Denhardt's solution, 0.5% SDS, 0.1 mg/ml yeast tRNA, and 1 mM EDTA. Blots were washed twice for 30 min with 1× SSC (0.15 M NaCl in 15 mM sodium citrate, pH 7.0) containing 0.5% SDS, and twice for 30 min with 0.2× SSC, 1.0% SDS at room temperature. The final wash was for 30 min with 0.1× SSC, 0.5% SDS at 38°C. The NOS2 cDNA was labeled using the Megaprime labeling kit (Amersham Pharmacia Biotech, Piscataway, NJ) and [α-32P]dCTP. Hybridization and washing conditions for this probe have been described previously (Sewer et al., 1997). Bound 32P-labeled probes were detected and quantified on a PhosphorImager (Molecular Dynamics, Santa Clara, CA). All slot-blot results were normalized to the content of poly(A+) RNA, measured by probing slot blots with an oligo(dT)30 probe (Sewer and Morgan, 1997).
Microsomal Protein Isolation, Western Blotting, and Enzyme Assays.
Microsomes were isolated by differential ultracentrifugation, using a Beckman TLK tabletop ultracentrifuge (Beckman Coulter, Inc., Fullerton, CA) as described previously (Iber et al., 1997). Microsomal protein concentration was determined by the method of Lowry et al. (1951) using bovine serum albumin as a standard. Relative levels of CYP2B1 protein in the microsomes were measured by Western blotting, under conditions of linearity with respect to the amount of protein applied. Blots were incubated for 1 h at room temperature in phosphate-buffered saline containing a 1:12,500 dilution of rabbit CYB2B1 antiserum (Duignan et al., 1987). After washing three times with phosphate-buffered saline, they were incubated for 1 h with a 1:2000 dilution of horseradish peroxidase-coupled goat anti-rabbit IgG. Bound antibody complexes were measured by enhanced chemiluminescence detection (ECL; Amersham Pharmacia Biotech), followed by autoradiography and video densitometry (Lynx system; Applied Imaging, Santa Clara, CA).
Pentoxyresorufin O-dealkylase activity (PROD) of the microsomes was determined by measuring the rate of formation of the fluorescent product resorufin (Lubet et al., 1985) using a fluorescence microplate reader (SpectraMax Gemini; Molecular Dynamics). Reactions were performed under conditions of linearity with respect to protein concentration and time in amber plastic microcentrifuge tubes containing 50 to 200 μg of microsomes, 5 mM HEPES buffer, pH 7.6, 20 μM pentoxyresorufin, and 1 mM NADPH in a total volume of 500 μl. After 5 min of incubation at 37°, reactions were terminated by the addition of 500 μl of acetonitrile. The tubes were centrifuged at 13,000g for 2 min, and 150 μl of supernatant was transferred to a light-shielded 96-well microplate. The amount of resorufin formed was determined by measuring the fluorescence of the samples at excitation and emission wavelengths of 535 nm and 590 nm, respectively, and by comparison with a resorufin standard curve.
Analysis of Nitrite and Nitrate Concentration.
The stable products of NO synthesis, nitrate and nitrite (NOx), were measured in the culture media using a colorimetric method based on the Griess reaction as described previously (Sewer and Morgan, 1997). Nitrate was reduced to nitrite with NADPH:nitrate reductase (98% conversion). Nitrite concentrations in the reduced samples were measured at 540 nm with a Thermomax microplate reader (Molecular Devices) using the Griess reagent and a sodium nitrite standard curve.
Statistical Analysis.
One-way analysis of variance and the Neumann-Keuls test were used to determine differences among treatment groups. Data from slot-blot and Western blot assays were expressed in arbitrary units or as a percentage of the mean of an appropriate control group in each experiment.
Results
In initial experiments, we optimized the conditions for maximal induction and stable expression of CYP2B1 mRNA in the presence of PB. As reported by others using different culture conditions (Sidhu and Omiecinski, 1995), the concentration dependence of induction over the period of 0 to 72 h in our cultures was biphasic (not shown), and the optimal concentration of 1 mM PB was selected for all other experiments.
Concentration-Dependent Suppression of CYP2B1 by LPS Treatment.
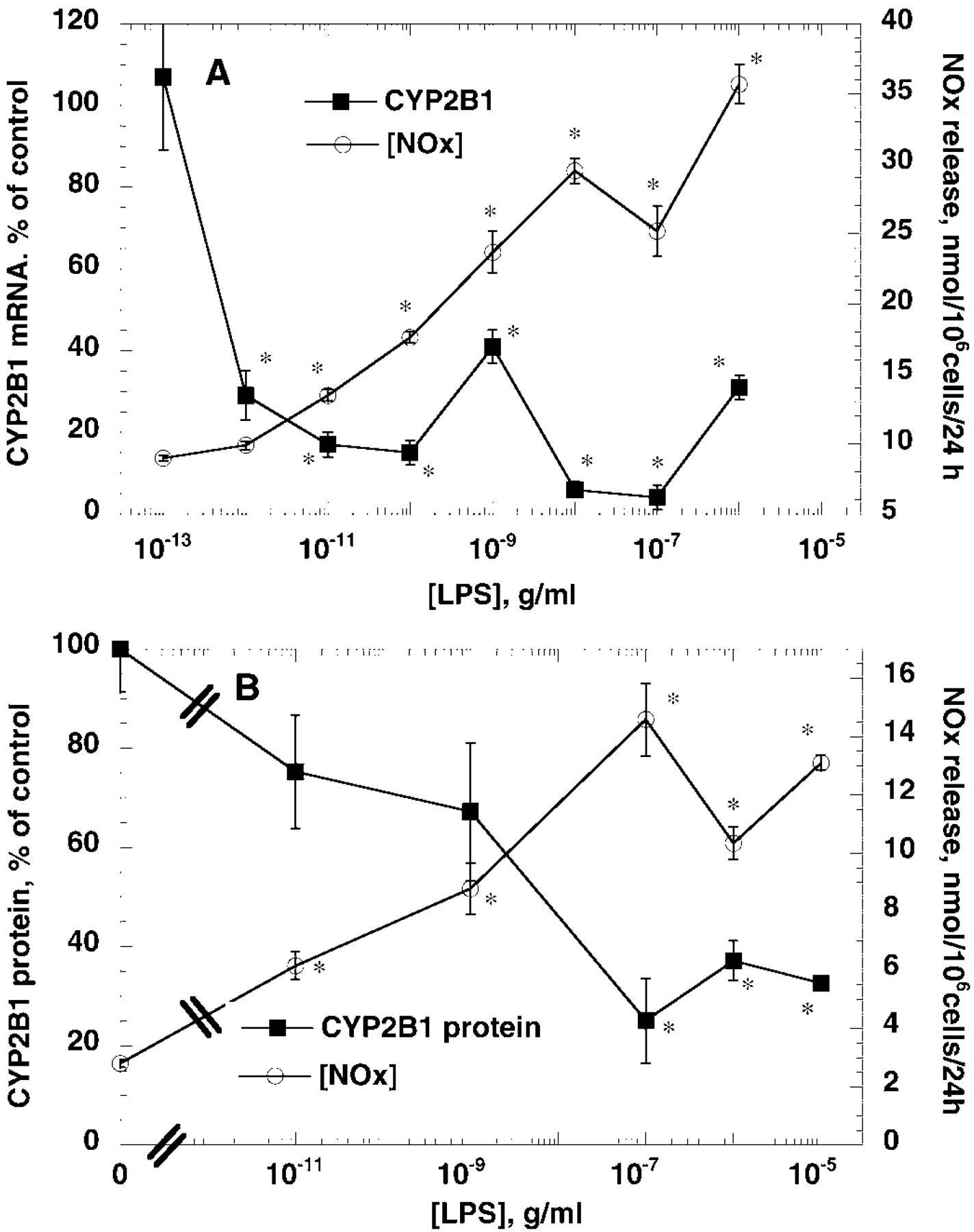
Treatment of the cells with LPS for 24 h caused a suppression of PB-induced CYP2B1 mRNA (Figs.1A and 2A), and protein expression (Figs. 1B and 2B). However, LPS was more potent in suppression of CYP2B1 mRNA (EC50 < 1 pg/ml) than in induction of NO production (EC50 >10 ng/ml) (Fig. 2A). On the other hand, LPS treatment suppressed CYP2B protein expression by LPS with potency similar to that of NOS2 induction (Fig. 2B). Western blots of microsomes from PB-treated hepatocyte yielded one major band and one minor band with a slightly lower mobility. These bands had identical mobilities to purified CYP2B1 and 2B2, respectively, and were sufficiently resolved to be measured independently by video densitometry (data not shown). Although these bands tended to be affected similarly by the various treatments, only the CYP2B1 band was measured in these studies. Treatment of cells for 6 h or 24 h with 10−5 or 10−10 g/ml LPS or 5 ng/ml IL-1 had no effect on the expression of glyceraldehyde 3-phosphate dehydrogenase mRNA measured by Northern blotting (data not shown).
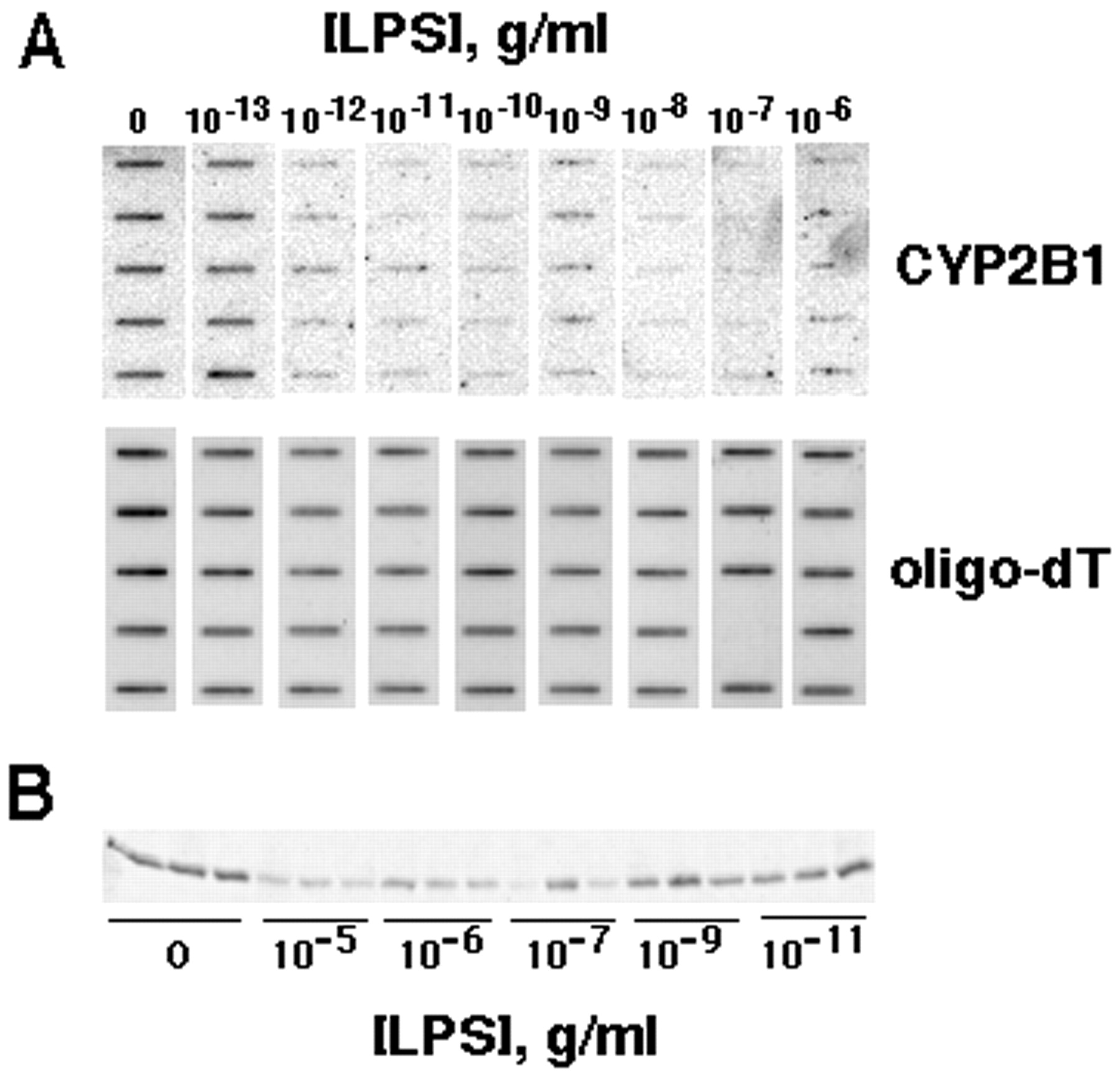
Concentration dependence of CYP2B1 mRNA and protein suppression by LPS in cultured hepatocytes. Hepatocytes were treated with 1 mM PB for 48 h; then, LPS was added at the indicated concentrations in fresh medium containing PB. Hepatocytes were harvested and total RNA and microsomal protein were prepared 24 h after LPS addition. A, top, a PhosphorImage of a slot blot loaded with equal amounts (3.33 μg) of total hepatocyte mRNA and probed with the CYP2B1-specific oligonucleotide. Bottom, the poly(A+) RNA contents of the samples measured by probing a blot loaded with one fifth of the amount of the identical samples with an oligo-dT probe. In the lower panel, one slot is blank because of sample loss. B, Western blot of microsomal proteins probed with CYP2B1 antibody. Each lane contains 2 μg of microsomal protein from two pooled culture plates.
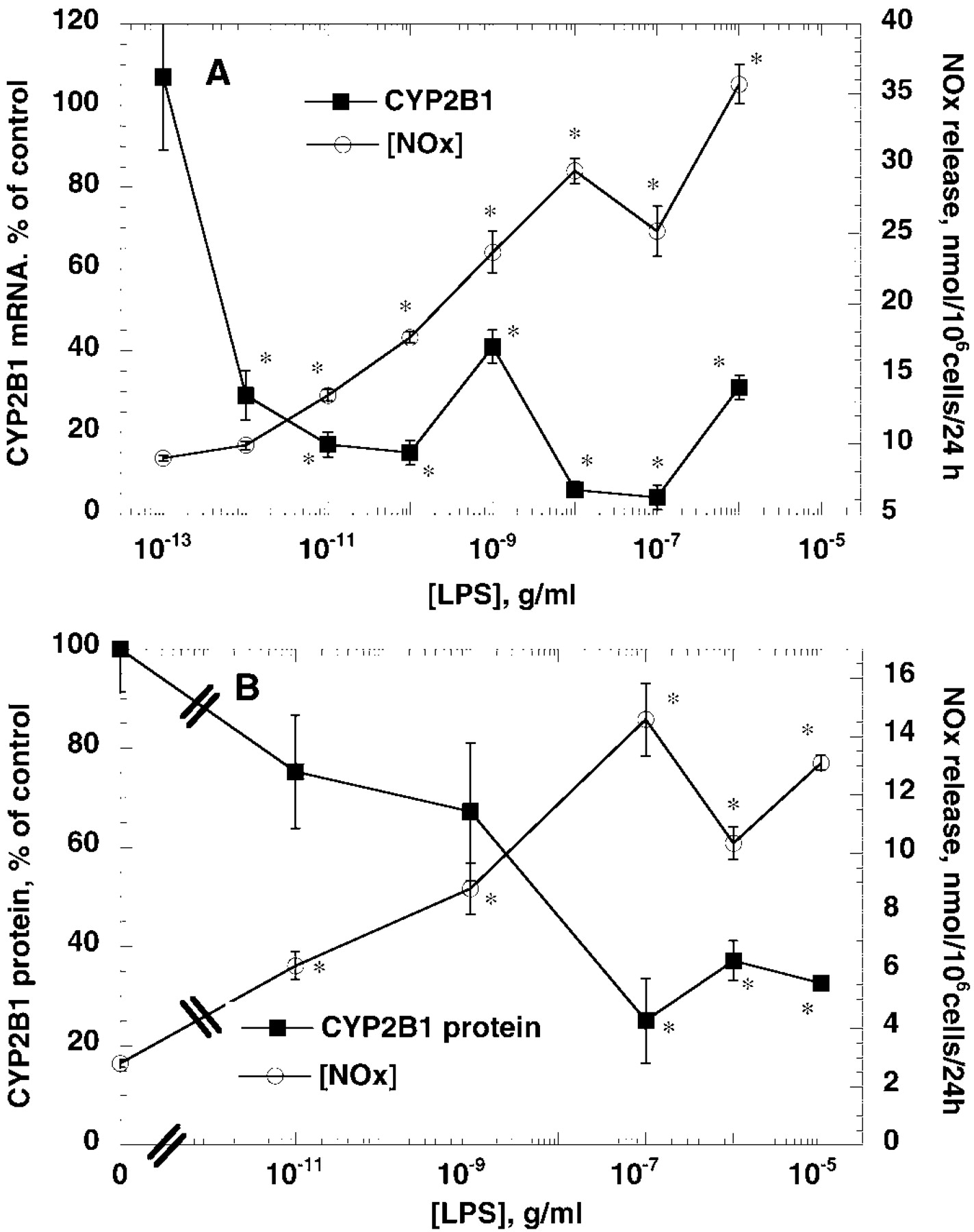
Concentration dependence of suppression of CYP2B1 mRNA and protein by LPS compared with induction of NO synthesis. Hepatocytes were treated as described in the legend to Fig. 1. Medium was collected for assay of NOx formation, and cells were harvested for preparation of total RNA or microsomal protein in separate experiments. A, CYP2B1 mRNA suppression compared with NOx release. Data represent the means ± S.E. of five independent samples for each group, and values for CYP2B1 mRNA are expressed as a percentage of the mean of the group treated with medium (PB, no LPS). B, CYP2B1 protein suppression compared with NOx release. CYP2B1 data represent the means ± S.E. of three independent samples for each group and are expressed as a percentage of the mean of the group treated with medium (PB, no LPS). NOx data are the means of ± S.E. of four to five independent samples for each group.
NO-Independent Suppression of CYP2B1 mRNA by LPS.
The earliest detectable suppression of CYP2B1 mRNA by either high or low concentrations of LPS occurred 12 h after addition of LPS to the medium (Fig. 3). CYP2B1 mRNA levels remained low 24 h (Fig. 2) and 48 h (Fig. 3) after treatment. Interestingly, low concentrations of LPS produced a transient induction of CYP2B1 mRNA 6 h (but not 3 h) after addition to the medium (Fig. 3). Suppression of CYP2B1 mRNA was independent of NO production by the hepatocytes, because concentrations of the NOS inhibitor NMA that completely blocked NO production failed to affect down-regulation of CYP2B1 mRNA by LPS (Fig. 4). A similar lack of effect of 300 μM AG, which also blocked NO production stimulated by LPS, on CYP2B1 down-regulation was observed (not shown).
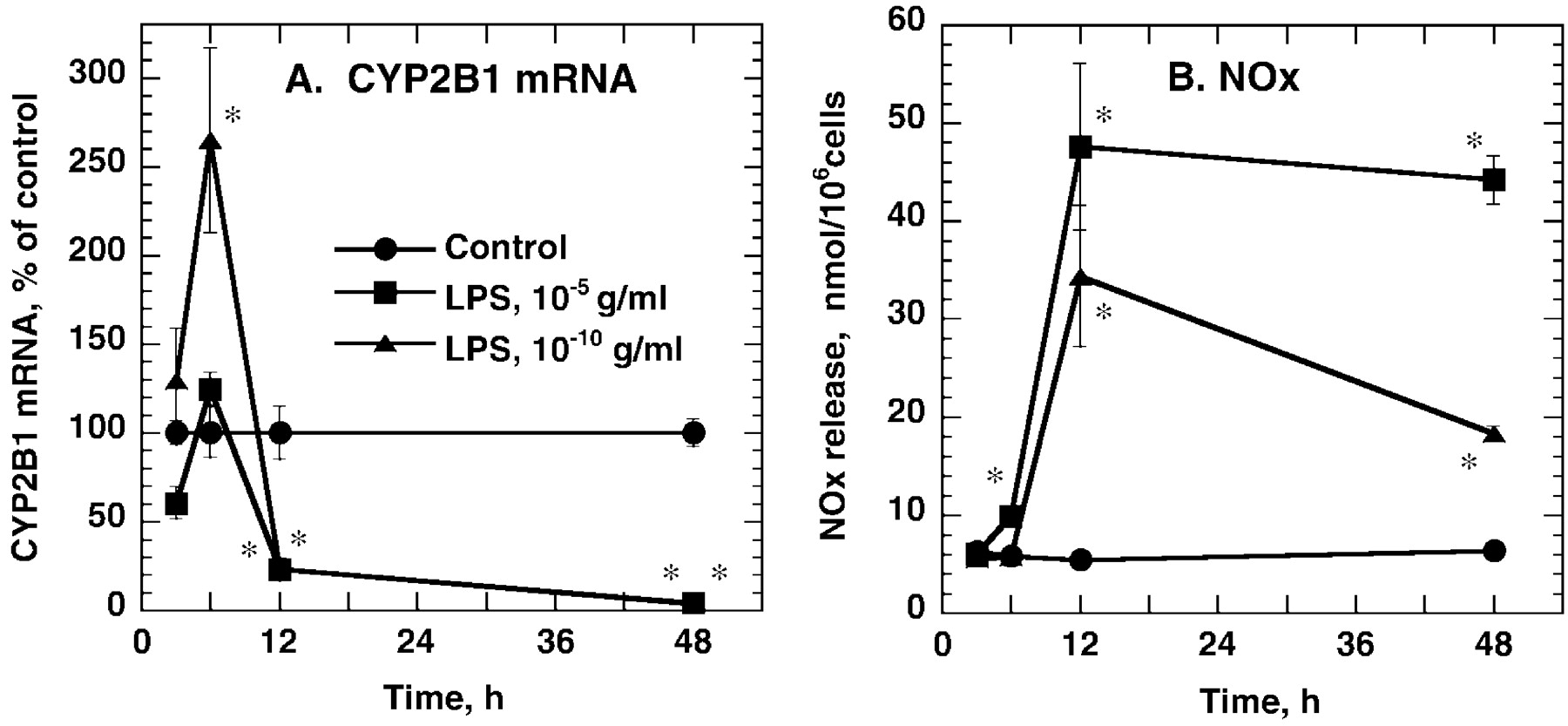
Time course of CYP2B1 mRNA suppression by different concentrations of LPS. Hepatocytes were treated with PB as described in the text, and then exposed to medium or the indicated concentrations of LPS for 3, 6, 12, or 48 h. Cells were harvested for preparation of microsomes and total cellular RNA. A, CYP2B1 mRNA determined by slot blotting. Values are expressed as a percentage of the group mean at each time point. B, NOx release. Data represent the means ± S.E. of three to four independent samples. Some samples were omitted from analyses because their values deviated more than 2 S.D. from the group mean. *p < 0.05, significantly different from control at appropriate time point.

Lack of effect of NMA on suppression of CYP2B1 mRNA by LPS. After 48-h treatment with 1 mM PB, hepatocytes were treated for 24 h with fresh medium containing PB ± 10 μg/ml LPS and the indicated concentrations of NMA for 24 h. Relative CYP2B1 and NOS2 mRNA levels determined by slot blotting were normalized to the poly(A+) contents of the samples and are expressed as arbitrary units. Values are the means ± S.E. of three to six independent samples for each group. a, significantly different from control; b, significantly different from LPS-treated group,p < 0.05.
NOS Inhibitors Block Suppression of CYP2B1 Protein Expression by High Concentrations of LPS.
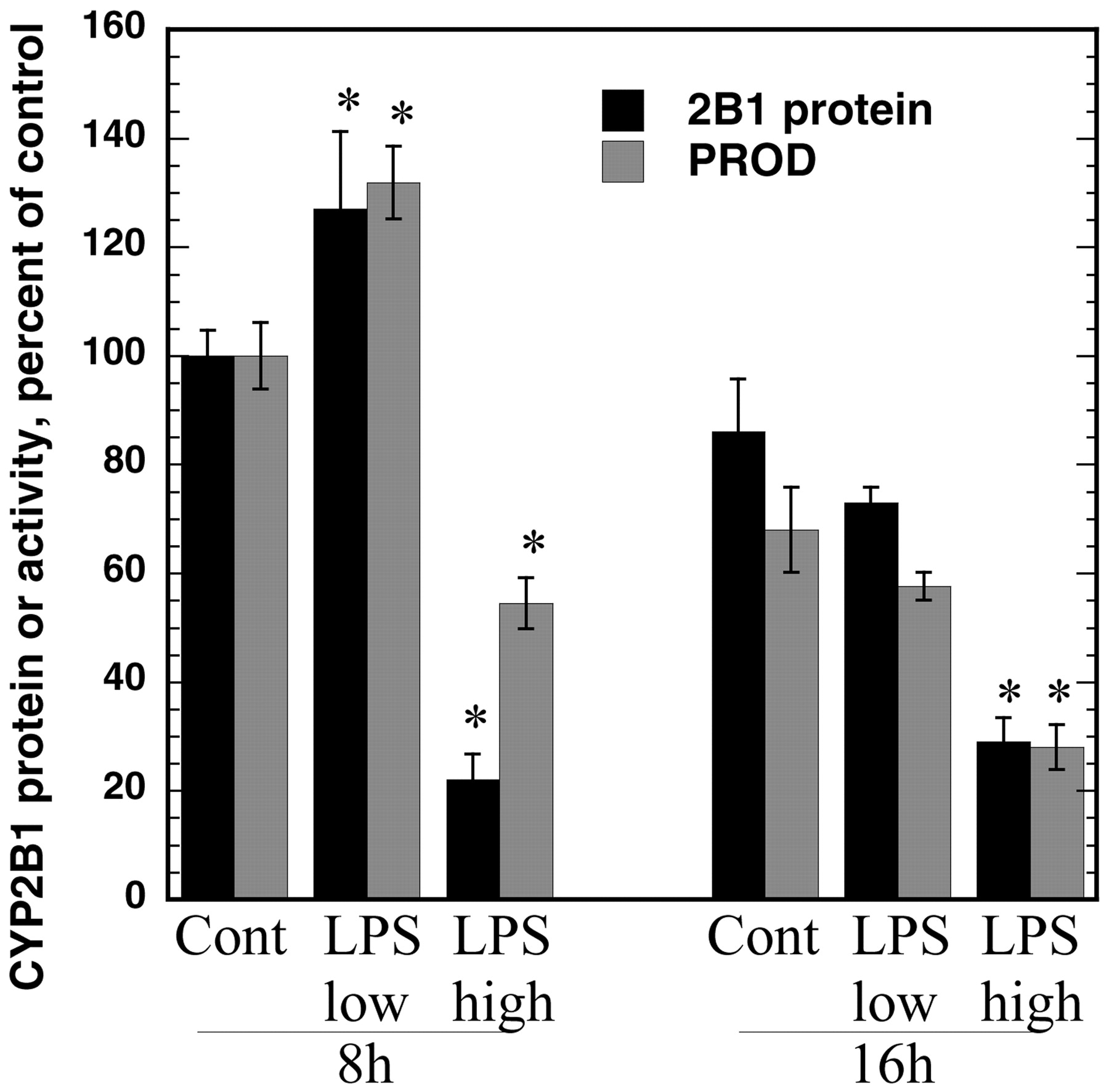
High concentrations of LPS suppressed CYP2B1 protein levels and PROD activities as early as 8 h after addition of LPS to the cultures (Fig. 5). This suppression persisted 16 h (Fig. 5) and 24 h (Fig. 2B) after treatment. In contrast, low concentrations of LPS that suppress CYP2B1 mRNA within 12 h (Fig. 3) failed to suppress CYP2B1 protein or PROD activity (Fig. 5) at 8 h and, in agreement with the induction of CYP2B1 mRNA at 6 h (Fig. 3), actually produced a small induction of CYP2B1 protein at 8 h (Fig. 5).
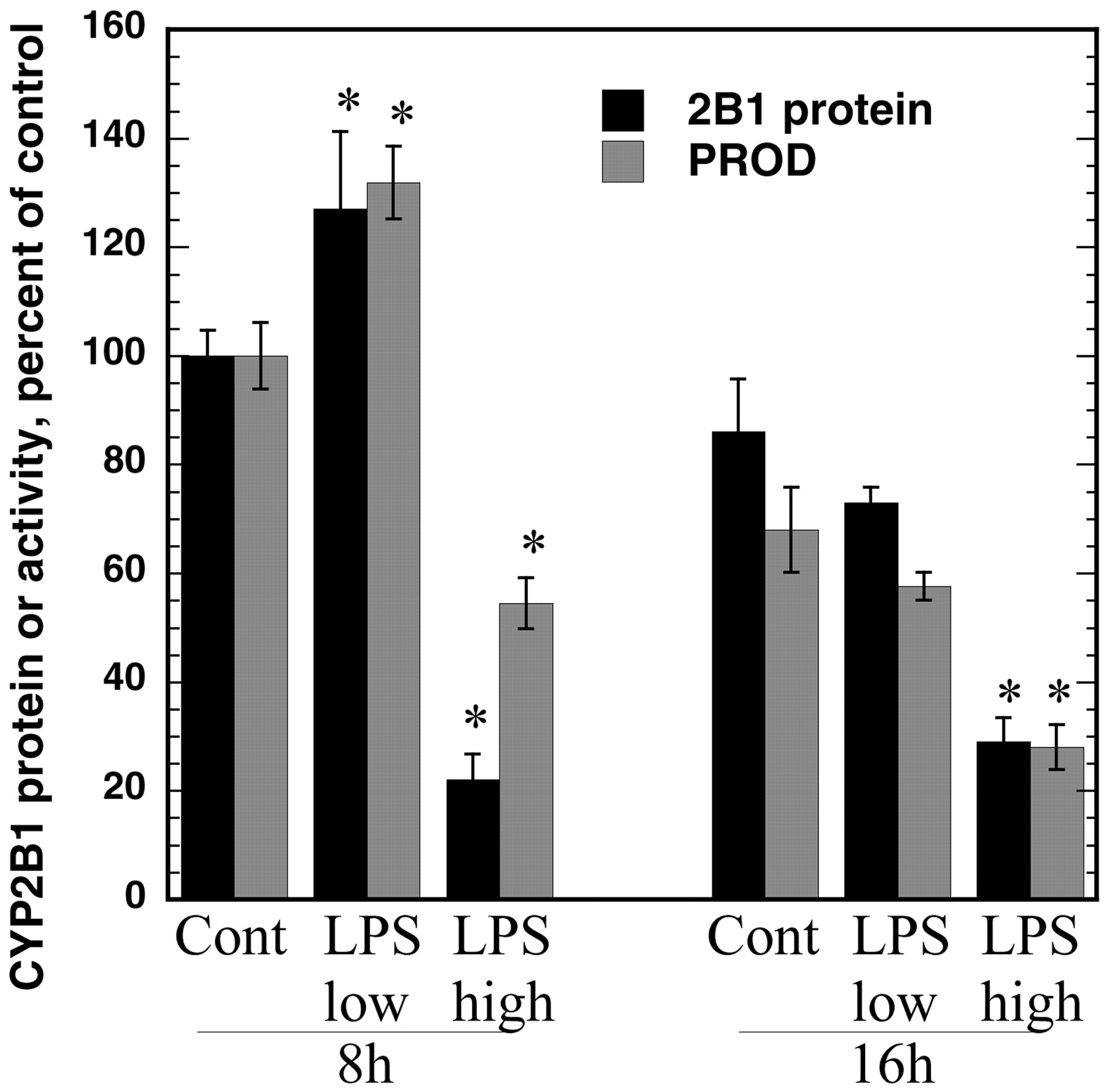
Concentration-dependent effect of LPS on CYP2B1 protein and PROD activity. Hepatocytes were treated with 1 mM PB for 48 h; then, LPS was added at either 10 pg/ml (low) or 10 μg/ml (high) concentrations. Hepatocytes were harvested 8 or 16 h after LPS addition for preparation of microsomes. CYP2B1 protein levels and PROD activities were as described in the text. Values represent the means ± S.E. of four independent samples for each group and are expressed as a percentage of the mean of the 8 h group treated with medium (PB, no LPS). The mean PROD activity of this group was 0.22 nmol/mg/min. *p < 0.05, significantly different from respective control.
In contrast to its lack of effect on CYP2B1 mRNA suppression by LPS, concentrations of NMA that blocked NO production also blocked the down-regulation of CYP2B1 protein by LPS in hepatocytes at both 6 and 24 h after LPS addition (Fig. 6, A–C). NMA also blocked the suppression of CYP2B1-dependent PROD activity (Fig. 6D). AG also attenuated CYP2B1 protein down-regulation by LPS, but it was not as effective as NMA in this regard, especially at the 24-h time point (Fig. 6, B and C).

NOS inhibitors prevent CYP2B1 protein suppression by LPS treatment. After 48-h treatment with 1 mM PB, hepatocytes were treated with fresh medium containing PB ± 10 μg/ml LPS in the presence or absence of 300 μM AG or NMA. Cells were harvested 6 or 24 h later, and microsomes were prepared. CYP2B1 protein levels in the samples were assayed by Western blotting. NOx in the media was measured as described in the text. A, Western blot showing inhibition of CYP2B1 protein suppression by NMA, 24 h after LPS addition. Each band represents an independent sample consisting of the pooled microsomes from two cell culture plates. B and C, quantitative analyses of the effects of NMA and AG on LPS suppression of CYP2B1 protein at 6 and 24 h, respectively. Data represent the means ± S.E. of five independent samples for each group. D, PROD activities of the samples harvested at 24 h. Data represent the means ± S.E. of four independent samples. The data in B, C, and D are expressed as a percentage of the mean of the control group treated with medium (PB, no LPS). a, significantly different from control; b, significantly different from LPS-treated group, p < 0.05.
To affirm that the inhibition of CYP2B1 protein suppression by NMA and AG was caused by their inhibition of NOS activity, and not by nonspecific effects of the drugs, we tested the effect of a chemically unrelated drug that inhibits NO production by a different mechanism. LY83583 is a quinolone compound that prevents NO production by inhibiting the induction of NOS2, rather than by inhibiting its activity (Geng et al., 1998). The mechanism of inhibition of NOS2 induction seems to be related to the inhibition of particulate guanylate cyclase by LY83583 (Geng et al., 1998). As reported for human chondrocytes, we found that 3 μM LY83583 effectively blocked NOx production stimulated by either LPS or IL-1β in rat hepatocytes (Fig.7). Furthermore, in agreement with our experiments using NOS inhibitors, LY83583 blocked the suppression of CYP2B1 protein caused by 24 h of LPS treatment (Fig. 7). Like LPS, IL-1 suppressed CYP2B1 protein in hepatocytes, and this effect was also blocked by LY83583 (Fig. 7).

Inhibition of CYP2B1 suppression by LY83583. Hepatocytes were treated for 48 h with 1 mM PB to induce CYP2B expression. Then, they were treated with fresh medium containing PB in the presence or absence of LPS (10 μg/ml), IL-1β (5 ng/ml), or LY83583 (3 μM) as indicated. Cells were harvested 24 h later, and microsomal CYP2B1 protein levels were measured by Western blotting. Because LY83583 was delivered in ethanol, an equivalent concentration of ethanol (20mM) was added to all cultures that did not receive LY83583. This concentration of ethanol did not affect either CYP2B1 protein or NOx levels (not shown). Data represent the means ± S.E. of four independent samples. a, significantly different from control; b, significantly different from LPS-treated group,p < 0.05.
Finally, the blockade by NMA of LPS-evoked CYP2B1 protein suppression was reversed by inclusion of an excess of the NOS substrate arginine (Fig. 8A), in accord with a mechanism of competitive inhibition of NOS2 by NMA.

Effects of NO donors and arginine on CYP2B1 expression. A, reversal of the effect of NMA by arginine. After 48-h treatment with 1 mM PB, hepatocytes were treated with fresh medium containing PB ± 10 μg/ml LPS in the presence or absence of 60 μM NMA and the indicated concentrations of arginine. Cells were harvested 24 h later, and microsomes were prepared. CYP2B1 protein levels in the samples were assayed by Western blotting, and concentrations of NOx in the media were measured by the Griess reaction. Data represent the means ± S.E. of four independent samples for each group. Western blot data are expressed as a percentage of the mean of the control group treated with medium (PB, no LPS). a, significantly different from control; b, significantly different from NMA-treated group, p < 0.05. B, hepatocytes were treated with the indicated concentrations of NO donors or glutathione for 24 h. Cells were harvested and microsomal CYP2B protein levels were determined by Western blotting. Data represent the means ± S.E. of four independent samples for each group and are expressed as a percentage of the mean of the control group. *, significantly different from control, p < 0.05.
Effect of NO Donors.
SNAP and GSNO each release reactive nitrogen species in aqueous solution. GSNO forms mainly nitrosonium ion, a nitrosylating species, whereas NO and peroxynitrite (a nitrating species) are both formed from SNAP (Patel et al., 1999). Either SNAP or GSNO added to hepatocyte cultures were able to mimic the suppression of CYP2B1 protein caused by LPS or IL-1 (Fig. 8B). The effect was not correlated with the amount of NOx that could be measured in the medium: GSNO produced suppression at a concentration that did not significantly increase detectable NOx, whereas SNAP produced CYP2B1 protein suppression only at a concentration that also resulted in high levels of NOx in the media (Fig. 8).
NO-Independent Suppression of CYP2B1 Protein at Later Time Points.
Because suppression of CYP2B1 mRNA by LPS must ultimately lead to suppression of CYP2B1 protein, and because suppression of CYP2B1 mRNA is not prevented by NMA or AG, we hypothesized that NOS inhibitors would be ineffective in blocking the down-regulation of CYP2B1 protein at later time points. As shown in Fig.9, this proved to be correct. LPS treatment caused a suppression of CYP2B1 protein to 20% of control levels, and this was unaffected by NMA, which effectively blocked NOx production.

NOS inhibitors do not prevent CYP2B1 protein suppression by 48 h of LPS treatment. After 48-h treatment with 1 mM PB, hepatocytes were treated with fresh medium containing PB ± 10 μg/ml LPS in the presence or absence of 300 μM NMA. Cells were harvested 48 h later, and microsomes were prepared. CYP2B1 protein levels in the samples were assayed by Western blotting. Data represent the means ± S.E. of four independent samples for each group and are expressed as a percentage of the mean of the control group treated with medium (PB, no LPS). a, significantly different from control; b, significantly different from LPS-treated group, p< 0.05.
Discussion
The results of this study clearly indicate that CYP2B1 expression in hepatocytes is down-regulated by LPS via two different mechanisms: one occurs at high concentrations of LPS and is NO-dependent and the other occurs at lower LPS concentrations and is NO-independent. Several lines of evidence argue for an NO-dependent suppression of CYP2B1 protein at high concentrations of LPS and at early (24 h or less) time points in the response: CYP2B1 protein levels and catalytic activities are reduced after only 6 to 8 h of LPS treatment (Figs. 5 and 6), at a time when CYP2B1 mRNA has not yet decreased (Fig. 4); the concentration dependence of CYP2B1 protein suppression is very similar to that of NOS induction (Fig. 2); CYP2B1 suppression is reversed by two classes of compounds that block NO production by different mechanisms (Figs. 6,7); blockade of CYP2B1 suppression by the competitive NOS inhibitor is reversed by the NOS substrate arginine (Fig. 8); and CYP2B1 down-regulation is mimicked by NO donors (Fig. 8). Two major findings argue against a role of NO in the suppression of CYP2B1 mRNA by LPS treatment: LPS suppression of CYP2B1 mRNA is several orders of magnitude more potent than the induction of NO production or the suppression of CYP2B1 protein (Fig. 2) and CYP2B1 mRNA suppression by LPS is not inhibited by concentrations of NOS inhibitors that block NO production in the cultures (Fig. 4). The LPS-evoked decline in CYP2B1 mRNA could be caused either by transcriptional suppression or stimulated mRNA degradation; more work is needed to distinguish between these possibilities.
The rapid NO-dependent suppression of CYP2B1 suppression that occurs at high concentrations of LPS is highly suggestive of an increase in CYP2B1 protein degradation. Estimates of CYP2B1 protein half-life from in vitro (Roberts, 1997) and in vivo experiments (Shiraki and Guengerich, 1984) each give a value of about 30 h. Thus, suppression of CYP2B1 mRNA clearly cannot explain the rapid decline in CYP2B1 protein observed after LPS stimulation. When CYP2B1 protein is incubated with peroxynitrite in vitro, an average of two tyrosines per CYP2B1 molecule are nitrated (Roberts et al., 1998), and this nitration is correlated with a decline in CYP2B1 catalytic activity. Whether nitration of these tyrosines is causative for the observed inhibition, or indeed for the decrease in CYP2B1 protein levels in the present study, remains to be determined. It is known that protein modification by NO-derived species can result in accelerated protein degradation: e.g., iron-regulatory protein-2 is targeted for ubiquitin-dependent proteasomal degradation after S-nitrosylation (Kim and Ponka, 1999). Interestingly, peroxynitrite-dependent nitration of CYP8A1 occurs in rat mesangial cells treated with IL-1 (Zou et al., 1998), concomitant with loss of catalytic activity. However, this is apparently not associated with a loss of the CYP8A1 protein in these cells.
It is interesting that GSNO, a nitrosonium ion donor that mainly results in protein nitrosylation (Patel et al., 1999) achieves its effect on CYP2B1 in the absence of a detectable increase in NOx in the medium. This suggests that the effect of GSNO may be caused by direct reaction of GSNO with thiols on CYP2B1. SNAP is an NO donor that can result in peroxynitrite formation in the presence of superoxide anions. SNAP caused CYP2B1 suppression only at high concentrations when there were detectable levels of NOx in the medium, which is consistent with a requirement of high levels of NO to form peroxynitrite. Peroxynitrite, or species derived from it, in addition to protein nitration, can also cause protein oxidation and thiol nitrosylation (Patel et al., 1999). Whether SNAP causes CYP2B1 suppression via nitration, nitrosylation or oxidation remains to be determined.
Because CYP2B1 protein synthesis is obviously dependent on the level of CYP2B1 mRNA, it is germane to ask why the NO-independent down-regulation of CYP2B1 mRNA does not lead to an NO-independent suppression of CYP2B1 protein at 24 h. In our proposed model, the long half-life of the CYP2B1 protein, coupled with a 6 to 12 h lag before CYP2B1 mRNA suppression occurs (Fig. 4), means that the decrease in mRNA level has little impact on CYP2B1 protein levels up to 24 h after treatment. However, when LPS treatment (and suppression of CYP2B1 mRNA) is prolonged for 48 h, inhibition of CYP2B1 protein degradation by preventing NO production can no longer prevent a decrease in CYP2B1 protein because by this time CYP2B1 synthesis is inhibited because of the decrease in mRNA template. This was confirmed by the experiment in Fig. 9, in which NMA could not prevent CYP2B1 protein suppression after 48 h treatment with LPS.
Recently, Milosevic et al. reported that LPS in the presence of interferon-γ stimulated the suppression of CYP2B1 mRNA and protein in PB-treated hepatocytes, but only if they were cocultured with Kupffer cells (Milosevic et al., 1999). This down-regulation of both CYP2B1 mRNA and protein occurred at 10 ng/ml LPS, and was completely dependent on TNFα production by the Kupffer cells. Inhibition of NOS activity with the NOS2-specific inhibitorl-N 6-(1-iminoethyl)-lysine did not prevent either LPS- or TNFα-evoked suppression of PROD activity in this system. These findings are in general agreement with our conclusion that at low concentrations of LPS, NO-independent mechanisms act to suppress CYP2B1 expression, primarily at the pretranslational level. The fact that we observed suppression of CYP2B1 mRNA at low concentrations of LPS in the absence of added Kupffer cells might be related to the presence of Kupffer cell contamination in our cultures. However, our hepatocyte isolation procedure yields Kupffer cell contamination of less than 0.5% (Sewer and Morgan, 1997), similar to that reported by Milosevic et al. (1999). The apparent lack of a requirement for Kupffer cells in our system could be because our cells were cultured on Matrigel, which confers a highly differentiated phenotype on rat hepatocytes (Schuetz et al., 1988). Hepatocytes have recently been shown to express CD14, the LPS receptor (Liu et al., 1998); thus, LPS can regulate hepatocyte gene expression in the absence of Kupffer cells (Saad et al., 1995). It may be that CD14-regulated pathways are more active in cells cultured on Matrigel.
Our finding that CYP2B1 protein suppression by high concentrations of LPS (or by IL-1) is blocked by NOS inhibition is in accord with the report by Carlson and Billings (1996), who observed a relatively modest decline in CYP2B1 protein levels (33–60% of control) in short-term cultures of hepatocytes stimulated for 24 h with a cytokine combination or with IL-1 or TNFα. The relatively high concentrations of LPS necessary to induce NO production and suppress CYP2B1 by the NO-dependent mechanism (0.1 μg/ml and above) are relevant to physiological or pathophysiological concentrations: for example, endotoxin levels from 30 to 300 ng/ml can be detected in the blood of children with bacteremia (Scheifele et al., 1981). It should also be borne in mind that CD14-dependent responses to LPS are potentiated by LPS binding to soluble LPS binding protein (Ulevitch and Tobias, 1995). Thus, in the presence of this physiological partner (not available to us), responses of hepatocytes to LPS will be potentiated.
In conclusion, the identification of two different mechanisms (one NO-dependent and one NO-independent) by which LPS can cause suppression of CYP2B1 protein and activity in hepatocytes may provide an explanation for some of the apparently contradictory reports regarding the role of NO in P450 down-regulation by inflammatory mediators. It is possible or likely that other P450 enzymes will be down-regulated by both NO-dependent and -independent mechanisms as well.
Footnotes
-
This work was supported by Grants GM53093 (E.T.M.), ES03619 (J.R.H.) and ES06676 (University of Texas Medical Branch Center Grant) from the National Institutes of Health.
Abbreviations
- P450
- cytochrome P450
- NOS
- nitric-oxide synthase
- LPS
- bacterial lipopolysaccharide
- IL
- interleukin
- PB
- phenobarbital
- TNF
- tumor necrosis factor
- NMA
- Nω-monomethyl-l-arginine
- AG
- aminoguanidine
- GSNO
- S-nitrosoglutathione
- SNAP
- S-nitroso-N-acetylpenicillamine
- SSC
- standard saline citrate
- PROD
- pentoxyresorufinO-dealkylase
- NOx
- nitrate + nitrite
- Received October 12, 2000.
- Accepted April 9, 2001.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}