


Abstract
7-Ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin [CPT-11 (irinotecan)] is a water-soluble camptothecin-derived prodrug that is activated by esterases to yield the potent topoisomerase I poison SN-38. We identified a rabbit liver carboxylesterase (CE) that was very efficient at CPT-11 metabolism; however, a human homolog that was more than 81% identical to this protein activated the drug poorly. Recently, two other human CEs have been isolated that are efficient in the conversion of CPT-11 to SN-38, yet both demonstrate little homology to the rabbit protein. To understand this phenomenon, we have characterized a series of esterases from human and rabbit, including several chimeric proteins, for their ability to metabolize CPT-11. Computer predictive modeling indicated that the ability of each enzyme to activate CPT-11 was dependent on the size of the entrance to the active site. Kinetic studies with a series of nitrophenyl and naphthyl esters confirmed these predictions, indicating that activation of CPT-11 by a CE is constrained by size-limited access of the drug to the active site catalytic amino acid residues.
7-Ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin [CPT-11 (irinotecan)] is a prodrug that is activated by esterases to generate 7-ethyl-10-hydroxycamptothecin (SN-38), a potent topoisomerase I poison (Tanizawa et al., 1994). Because CPT-11 has demonstrated remarkable antitumor activity in human tumor xenograft models, this drug is currently undergoing Phase II/III trials in both adults and children (Houghton et al., 1993, 1995; Bugat et al., 1994, 1995;Fujiwara et al., 1994; Escudier et al., 1997; Thompson et al., 1997a,b;Furman et al., 1999). Additionally, the FDA recently approved CPT-11 in combination with 5-fluorouracil for front line treatment of colon cancer.
The enzymes involved in the metabolism of CPT-11 in humans have not been fully characterized; hence the ability to tailor drug doses for individual patients is difficult. This issue is complicated by the fact that in rat and mouse model systems, more than 50% of CPT-11 is converted to SN-38 (Morton et al., 2000), whereas in humans, depending on the dose and scheduling, as little as 1% of the drug is activated (Rivory et al., 1997). It is clear that either the levels of the enzymes responsible for CPT-11 metabolism in humans are low or that these proteins have significantly diverged in structure from rodent counterparts.
Recently, we isolated a rabbit liver carboxylesterase (CE) that could efficiently convert CPT-11 to SN-38 (Potter et al., 1998a). Searches of the GenBank database indicated that a human homolog of this CE (hCE1) with more than 81% amino acid identity had been identified previously. However, expression of hCE1 in human tumor cells did not alter their sensitivity to CPT-11, and in in vitro experiments, extracts derived from these cells metabolized the drug poorly (Danks et al., 1999). Very recently two other human CEs, hCE2 and hiCE, have been demonstrated to efficiently convert CPT-11 to SN-38 (Humerickhouse et al., 2000; Khanna et al., 2000). These proteins are highly homologous to each other and differ only in the 10 N-terminal residues. In contrast, the amino acid sequences of human hiCE and hCE2 differ dramatically from that of the rabbit CE (Khanna et al., 2000).
Unfortunately, no three-dimensional structure of any mammalian CE is known. Hence, a structure-based understanding of the differences in the sequences of the various CEs and their effect on CPT-11 metabolism remains elusive. In an attempt to address why each CE exhibited different abilities to activate CPT-11 that were not predicted from the amino acid sequences, we used computer modeling studies to generate likely structures for rCE, hCE1, and hiCE. These modeling experiments suggested that the entrances to the active site in the CEs that cannot effectively metabolize CPT-11 are smaller than those in enzymes that can activate the drug. Kinetic data for activation of a series of substrates by the rCE and hCE1 were consistent with the computer models. Our combined modeling and experimental data indicate that steric constraints in the protein structures that limit the access of CPT-11 to the catalytic amino acids are probably responsible for the observed differences in prodrug activation.
Materials and Methods
CPT-11.
CPT-11 was kindly provided by Dr. J. P. McGovren (Pharmacia, Peapack, NJ).
Transient Transfection of COS7 cells.
COS7 cells were transfected with mammalian expression vectors as described previously (Potter et al., 1998a,b). After 48 h, cells were harvested by trypsinization and extracts were prepared by sonication in minimal volumes of 50 mM HEPES, pH 7.4, on ice.
Molecular Modeling of Carboxylesterase Proteins.
Homology models for hCE1, hiCE, and rCE were generated using the crystal structure of Torpedo californica acetylcholinesterase (AChE) containing the drug E2020 bound within the active site gorge (Kryger et al., 1999). Models were constructed using Modeller 4 software (Andrej Sali, Rockefeller University, New York, NY) (Sali and Blundell, 1993). Sequences showing 30 to 50% identity/similarity have been shown to give homology models that have 75 to 90% overlap of the α carbon traces (within 3.5 Å) with the corresponding crystal structures (Sanchez and Sali, 1998). Hence, the homology models should be viewed as low-resolution structures that may be used to aid in the interpretation of our kinetic data. These and all subsequent calculations reported here, unless otherwise noted, were performed under the Linux 2.2 for x86 kernel.
Initial models were minimized with respect to energy using TINKER (Jay W. Ponder, Washington University School of Medicine;http://dasher.wustl.edu/tinker) to a root-mean-square gradient of 1.0 kcal/mol/Å. The resulting model structures were analyzed using the online versions of Procheck and What_Check model evaluation software (Rodriguez et al., 1998). Neither software package indicated any significant problems in the structures of the models (Procheck average scores for hCE1, hiCE, and rCE were −0.43, −0.48, and −0.41, respectively). The models were further minimized with a short (1 ps) molecular dynamics run using the Molecular Modeling Toolkit (Hinsen, 2000). Molecular surfaces for the models were constructed using the molecular graphics program spock (Jon A. Christopher, Texas A&M University, College Station, TX).
Molecular Surface Calculations.
Analysis of the active site gorges in the models was performed with the Molecular Modeling Toolkit (Hinsen, 2000) using a procedure modified from the work of McCammon and colleagues (Tara et al., 1999). Molecular surfaces were calculated for each model using a probe radius that was incremented by 0.1 Å until the surface no longer contained contributions from the active site serine and histidine residues. As the active site serine and histidine residues are proposed to make initial contact with the ester bond to be cleaved (Satoh and Hosokawa, 1998), the largest probe radius that could contact both residues was considered to provide the diameter of the active site gorge.
Purified Carboxylesterases.
Pure rCE was prepared from baculovirus-infected cell culture media as described previously (Morton and Potter, 2000). hCE1 was purified in an identical fashion. CE activity for all enzymes was measured as described previously usingo-nitrophenyl acetate (o-NPA) as a substrate (Beaufay et al., 1974; Potter et al., 1998a).
Kinetics of Nitrophenyl Esters Catalysis.
K m, K cat, andV max values were determined by monitoring conversion of nitrophenyl esters to nitrophenol spectrophotometrically at 420 nm. Substrates were dissolved in methanol and reactions performed in 50 mM HEPES, pH7.4. A minimum of eight concentrations of substrate was used, ranging from 10-fold less than the observedK m value to 5-fold greater. Data were collected over 45 s and reaction velocities determined from linear regressions of the initial data points. Kinetic parameters were determined from hyperbolic plots of reaction velocities versus substrate concentrations using the Prism software (GraphPad, San Diego, CA). r2 values were obtained from these curve fits.
Kinetics of Naphthyl Esters Catalysis.
K m, K cat, andV max values were determined as above except that substrates were dissolved in 2-methoxyethanol and the liberation of naphthol was measured at 230 nm. Linear regression of the initial data points was used to provide reaction velocities that were plotted against substrate concentration. Kinetic parameters were then calculated as described previously.
Kinetics of CPT-11 Metabolism.
To determineK m, K cat, andV max values, rCE and hCE1 were incubated with CPT-11 for 2 and 10 min, respectively, at 37°C in 200 μl of 50 mM HEPES, pH 7.4. The amounts of SN-38 in the reaction were quantitated using high-performance liquid chromatography. Separation and detection of CPT-11 and SN-38 were performed by high-performance liquid chromatography as described previously (Guichard et al., 1998). Data were fitted to a one-site binding hyperbolic function using GraphPad Prism software and K m andV max values were determined from the equation describing the curve fit. Because of the very highK m value for hCE1, CPT-11 concentrations ranged from 1 to 250 μM for studies with this enzyme.
Results
Metabolism of CPT-11 by Chimeric Rabbit and hCE1 Proteins.
In an attempt to identify domains responsible for CPT-11 recognition and catalysis by CEs, we generated a series of chimeric proteins based upon unique restriction sites present within identical regions of the rabbit and hCE1 cDNAs. This produced three chimeric enzymes as indicated in Fig. 1 and Table 1. To determine the ability of these CEs to metabolize CPT-11, we ligated the cDNAs into pCIneo and transiently transfected COS7 cells. Extracts were prepared by sonication of cells 48 h after transfection and their ability to hydrolyze o-NPA and CPT-11 were determined. As can be seen, all proteins retained their ability to metabolizeo-NPA; however, RH2 activated CPT-11 with approximately one tenth the activity of RH1 and RH3 (Table 1). RH2 contains amino acids residues 121 to 453 of hCE1, indicating that the domains affecting CPT-11 metabolism are located within this region. However, because this represents a significant proportion of the enzyme, we thought it unlikely that we could identify specific residues responsible for CPT-11 recognition and catalysis by mutagenesis procedures. Hence, we adopted an alternative approach using computer predictive modeling of the CEs.

Schematic indicating the amino acids residues present in the chimeric hCE1 (■) and rCE (▪) proteins. The active site residues, serine (S), glutamic acid (E), and histidine (H), are indicated.
Metabolism of CPT-11 by chimeric rabbit and hCE1 proteins
Molecular Modeling of Carboxylesterase Proteins.
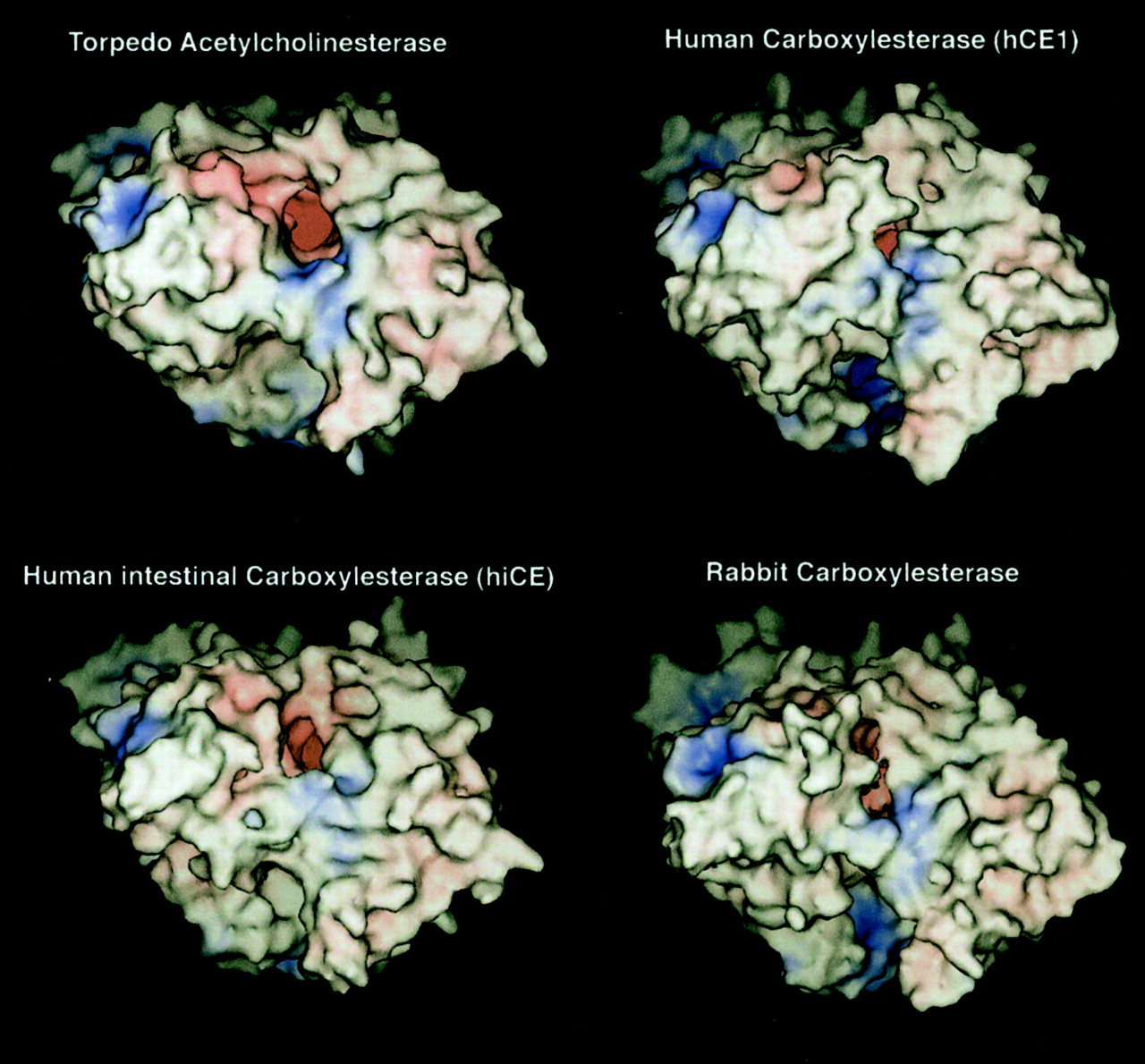
To perform the computer homology studies, a template structure was required to allow modeling of the CEs. To identify such a template, searches of the protein database with the rCE amino acid sequence were performed. The greatest homology was observed with T. californica AChE. Subsequent alignments of the sequences of hCE1, hiCE, and rCE indicated 37.7%, 38.0%, and 32.5% identity, respectively, with AChE (Fig.2) and 47.0%, 46.0%, and 40.5% similarity, respectively, as computed by GAP (Devereux et al., 1984). As shown in Fig. 2, the homology of the sequences extends over the length of the sequences; therefore, the global folding of the proteins is likely to be similar. This is also expected because the α/β-hydrolase tertiary fold is highly conserved among members of this superfamily (Oakshott et al., 1999). To generate models of the CE proteins, the amino acids sequences were modeled upon the T. californica AChE crystal coordinates. The resulting molecular surface representations of the initial homology models of hCE1, hiCE, and rCE, after energy minimization with TINKER, are shown in Fig.3. Overall, the models suggest that the three proteins fit well into the folding pattern of T. californica AChE and consequently have similar overall molecular shape.

Sequence alignment of T. californicaAChE, rCE, hCE1, and hiCE. Sequences were aligned using the GAP program (Devereux et al., 1984). For clarity, the last 31 amino acids ofT. californica AChE are not shown.

Computer predicted homology models of hCE1, hiCE, and rCE. Models were constructed as described in the text using T. californica AChE as the template structure. Molecular surface representations of the final, minimized models are shown. Areas of negative charge are shown in red, positive charge in blue, and neutral in white. Surfaces were generated from the van der Waals radii of the atoms. The view is looking directly into the active site gorge (the dark red area near the center of the model).
Analysis of Carboxylesterase Models.
As with AChE, the active site Ser and His residues for rCE, hCE1, and hiCE lie at the bottom of a negatively charged, ∼22-Å-deep gorge in the interior of the CEs. The gorge appears as the red “hole” in the molecular surfaces shown in Fig. 3. The diameter of the distal end of the gorge, where the active site residues are located, was calculated from the molecular surfaces and indicated that all three enzymes can accommodate similarly sized substrates (gorge diameters of 2.9 Å, 3.8 Å, and 3.2 Å for hCE1, hiCE, and rCE, respectively). These data are in good agreement with previous molecular dynamics simulation studies of AChE, in which the active site gorge fluctuates in size, ranging from 2.4 to 5.0 Å (Wlodek et al., 1997; Tara et al., 1999).
In contrast, the external opening to the gorge at the protein surface through which a substrate must pass to access the active site was different for the three CEs. For hCE1 and rCE, we identified three residues around the active site gorge opening near the surface of the proteins. These residues are Asn87, Leu286, and Met341 in hCE1 and Asn87, Leu285, and Met340 in rCE. Using the molecular surface of these residues as a “ring” around the active site gorge, we calculated an approximate diameter for the opening of the gorge at the protein surface. Calculations of active site gorge opening diameter were made for hCE1 and rCE using molecular surfaces at different probe radii, as described above. For rCE, the external opening to the active site gorge was approximately 5.2 Å in diameter, whereas for hCE1, the opening was approximately 4.2 Å. The sequence of hiCE was divergent from hCE1 and rCE; hence, these calculations could not be performed with this protein. However, using the residues with corresponding α-carbon positions in rCE (Phe87, Asn277, and Ile323), a crude estimate of the active site opening for hiCE indicated it was similar to rCE (5–6 Å in diameter). Although this small difference in the gorge openings (4.2 Å vs. 5–6 Å) could be within the error of the homology model, the estimated difference suggested that the sequence divergence between the rCE and hCE1 might result in a smaller entrance to the active site gorge in hCE1 compared with rCE. Even a small change in gorge opening or interior diameter could result in profound differences in the esterase activity of the enzymes. For example, McCammon and colleagues noted that for AcChE, the diameter of the active site gorge substantially altered the rate of hydrolysis of substrates that differ by as little as 0.4 Å in diameter (Zhou et al., 1998). Differences in active site gorge sizes for AcChE vs. butyrylcholinesterase have also been suggested to differentiate inhibitors of these two enzymes for similar steric reasons (Saxena et al., 1997; Saxena et al., 1999). Figure 4 illustrates the way in which CPT-11 must fit into the active site gorge to be activated by a CE. Accordingly, the models suggested that, since CPT-11 is a bulky substrate, the differences in activation of the drug by the different enzymes might be caused by steric constraints on the ability of CPT-11 to access the catalytic amino acids at the base of the active site gorge.

A, schematic of the entry of CPT-11 into the active site gorge of CEs. The surface of the protein is to the top of the figure. The catalytic serine and histidine residues lie near the bottom of the gorge at the interior of the protein, ∼20 Å from the opening, and cooperate in the cleavage of the CPT-11 carbamate ester bond. CPT-11 must fit almost entirely into the gorge to be hydrolyzed. The diameter of the piperidinopiperidine ring system of CPT-11 is 3.1 to 4.3 Å. B, stereogram of the location of CPT-11 within the active site gorge of rCE. CPT-11 was inserted into the gorge such that the active site serine and histidine (shown in green) make contact with the carbamate ester group. The close fit suggests that gorge dimensions only slightly smaller than these would probably prevent entry of the drug into the CE.
Kinetic Analysis of rCE and hCE1 Ester Metabolism.
Because the computer-predicted models suggested that the entrance to the active site gorge was narrower in hCE1 than rCE, we hypothesized that the metabolism of bulky esters by the former protein may be less efficient than by the rabbit enzyme. To determine whether this was the case, we compared the ability of hCE1 and rCE to hydrolyze a series ofortho- and para- isomers of nitrophenyl esters and esters of 1- and 2-naphthol. In nearly all cases, rCE was more efficient at substrate metabolism than hCE1, as determined byK cat/K m values (Table 2). A detailed example is included with NPA and nitrophenyl butyrate (NPB). For both ortho- substrates, the approximate molecular diameter of the compounds is 6.1 Å, whereas for the para- substrates, it is 4.4 Å. The results of enzymatic cleavage of these substrates by hCE1 and rCE are given in Table 2.
Kinetic parameters for the metabolism of nitrophenyl and naphthyl esters and CPT-11 by hCE1 and rCE
For hCE1, the rate of cleavage is similar with both theortho- and para- substrates at ∼ 1 × 105 M−1s−1, indicating a diminished ability to accommodate substrate sizes of 4.4 Å or greater. rCE hydrolyzeso-NPA and o-NPB at rates equivalent to those of the hCE1. However, the smaller p-NPA and p-NPB are hydrolyzed by rCE at a rate at least 10-fold greater than those for the corresponding ortho- substrates. These data are in agreement with the molecular modeling data, which suggest that substrates with a diameter in the 3- to 4-Å range should be more efficiently cleaved by rCE. Because the size of CPT-11 is within this range, the NPA and NPB hydrolysis data suggest that substrate access to the catalytic active sites is likely to be the primary reason for differences in the ability of the CEs to hydrolyze this drug.
Influence of Ester Substituent on Enzyme Catalysis.
We also examined the difference between rCE and hCE1 with a series of CE substrates having various group sizes on either side of the ester bond (Table 2). The majority of nitrophenyl esters interact with both rCE and hCE1 in a similar manner with regard to theV max. Acetate, propionate, and butyrate all have similar V max values for hCE1 (81–120 μmol/min/mg), indicating that esters containing 1-, 2-, and 3-carbon chains have approximately equivalent rates of catalysis. However, as described above, the para- esters were cleaved more efficiently by rCE. Excluding p-NPA, theV max values for rCE for all compounds ranged from 88 to 161 μmol/min/mg, indicating similar rates of cleavage for hCE1 and rCE (Table 2).
Both rCE and hCE1 demonstrated markedly reducedV max values for p-nitrophenyl valerate and p-nitrophenyl trimethylacetate. Hence, as the distance from the carboxyl group and bulkiness of the substrate increases, the rate of cleavage by CEs drops significantly. In agreement with the prediction that the active site gorge is larger in rCE, the decrease in V max with the valerate and methylacetate esters was only about 20% of that observed with the smaller substrates, whereas with hCE1, theV max values for these substrates were 2 orders of magnitude lower.
Hydrophobic Constraints in Esterase-Mediated Catalysis.
TheK m values for the nitrophenyl esters probably reflect hydrophobic “steering” within the active site gorge as well as the actual size of the substrate. With the exception of the butyrate ester, the K m value for hCE1 mirrors the relative hydrophobicity of thep-nitrophenyl esters (trimethylacetate > valerate > propionate > acetate; Table 2). This observation suggests that, similar to AChE, these substrates interact with hydrophobic residues that line the active site gorges of both rCE and hCE1.
Interestingly, with rCE, the trimethylacetate ester has the lowestK m for the p- substituted compounds. However, the K m values for valerate, propionate, and acetate esters are approximately the same for rCE (Table 2). In both enzymes, p-nitrophenyl butyrate is out of order with regard to K m value and hydrophobicity, indicating that it binds poorly to hCE1 but very well to rCE. Clearly, there are physical interactions beyond simple hydrophobic binding for p-nitrophenyl butyrate, but the source of these interactions is not obvious from the models.
Hydrophobic interactions by the butyrate moiety in 1-napthyl butyrate also follow the trend noted above for both hCE1 and rCE, with a lowerK m value for 1-napthyl butyrate than 1-naphthyl acetate. hCE1 shows a decrease inV max value with 1-naphthyl butyrate versus 1-naphthyl acetate, whereas rCE demonstrates very small differences. In contrast, 2-napthyl acetate has a lower K mvalue for both hCE1 and rCE than 1-naphthylacetate, but theV max values drop considerably for this substrate with both enzymes (Table 2).
The data suggest that the ability of both hCE1 and rCE to hydrolyze the substrates in Table 2 is controlled both by the steric properties of the substrates and their hydrophobic properties. Overall, rCE can hydrolyze bulkier substrates (including CPT-11) more efficiently than hCE1 because the entrance to the catalytic gorge of rCE is relatively large. However, when substrates are primarily involved with binding to the active site gorge via hydrophobic interactions, both enzymes are less efficient at hydrolysis. The observed properties determined experimentally are consistent with the models of hCE1 and rCE.
Discussion
Computer-assisted searching of both nucleic acid and protein databanks is used extensively to identify homologs of candidate genes. In many cases, such searches yield genes demonstrating identical functions in different species. In our studies with rCE, we identified hCE1 as the closest human homolog of this protein and presumed, therefore, that hCE1 was responsible for CPT-11 metabolism in humans. Unexpectedly, hCE1 was found to be very inefficient at drug activation even though it demonstrated greater than 81% identity and 86% similarity with the rCE amino acid sequence (Danks et al., 1999). The lack of correlation between protein function and sequence initiated the series of studies reported here to try to identify the amino acids critical for CPT-11 recognition and activation by CEs. However, because of the large size of these proteins (∼560 amino acids), the use of random mutagenesis studies was prohibitive. As an alternative, we performed computer predictive modeling of the enzymes based upon theT. californica AChE crystal structure (Kryger et al., 1999) and confirmed results of the modeling experiments with kinetic studies.
Cygler et al. (1993) demonstrated that esterase structures could be determined based upon their amino acid homology to two crystallized proteins, T. californica AChE and Geotrichum candidum lipase. However, these authors indicated that high amino acid sequence homology was required for accurate structure prediction. More recently, the development of computer software, such as Modeler 4 (Andrei Sali, Rockefeller University, New York, NY; Sali and Blundell, 1993), has allowed the prediction of protein structures that demonstrate much lower homology (∼30–50% identity; Xu et al., 1996;Sanchez and Sali, 1998). Detailed analyses of these computer models with actual structures determined from X-ray crystallographic studies indicated the validity of this approach.
CEs can exist as multimers in cells; hence, determining the exact overall structure of the complex will require detailed X-ray crystallographic studies. However, without knowing the stoichiometry and detailed arrangement of the proteins in these aggregates, it is difficult to predict the overall structure of the enzyme complex.
Our computer models of hCE1 and rCE indicated that the entrance to the active site was relatively narrow in the former enzyme and might regulate access of substrates to the catalytic amino acids. Whereas these modeling studies were underway, we and others isolated another human CE that was proficient at CPT-11 activation (Humerickhouse et al., 2000; Khanna et al., 2000). By comparing of molecular models of all three proteins, we assessed possible structural reasons for the inefficiency of hCE1 in converting CPT-11 to SN-38 versus the other two enzymes.
In the homology models, the active site serine and histidine residues of all three CEs lie at the bottom of a ∼22-Å gorge that is open to the exterior of the protein. The piperidinopiperidine group that is hydrolyzed from CPT-11 is, at its narrowest, 3.1 Å in diameter, and is 4.3 Å at its widest (see Fig. 4). Hence, only a molecule with this minimum diameter can pass through the gorge to reach the active site residues of the CE. The active site gorge diameter for all models is in the range of 3 to 4 Å. Hence, substrates in this size range should be the most sensitive to differences in the size of the active site gorge. Smaller substrates should fit equally well into both active sites, whereas larger substrates should have equally difficult access to the active site. In the models, we were unable to clearly identify individual amino acids responsible for the more compact gorge in hCE1 versus rCE. Rather, it is the global folding of the enzyme, with slight displacement of most residues, which leads to the inability of hCE1 to activate CPT-11. Our models of hCE1, hiCE, and rCE suggested that the opening to the active site gorge was slightly smaller in hCE1 than for hiCE and rCE, and that this steric constraint impedes access to the active site residues and affects the ability of the different CEs to hydrolyze CPT-11. Kinetic measurements on substrate metabolism by rCE and hCE1 (Table 2) agree with these models.
Kroetz et al. (1993) has demonstrated that inhibition of CE glycosylation by tunicamycin reduces enzymatic activity of the human CEs suggesting that addition of sugar residues is important for protein function. Preliminary evidence demonstrates that the rabbit CE is glycosylated at two Asn residues and that mutation of these residues leads to reduced enzymatic activity (M. A. Espinosa, M. R. Redinbo and P. M. Potter, unpublished observations). However, because hCE1 and rCE were both purified from baculovirus, they would be expected to be glycosylated resulting in active enzyme. This was proved to be the case because both proteins could metabolize simple CE substrates such as o-NPA and o-NPB.
Other factors, such as electrostatic and hydrophobic interactions that affect the cleavage of esters by CEs, may also be involved. However, the CE protein family is highly promiscuous concerning the number of structurally divergent compounds that they hydrolyze, to the extent that classification based on phylogeny rather than substrate specificity has been proposed (Satoh and Hosokawa, 1998). In a study of the ability of esterases in human whole blood to hydrolyze 80 different ester-containing drugs, the overwhelming parameter that affected the rate of hydrolysis was steric hindrance around the ester bond, with lipophilicity and electronic parameters contributing much less to the process (Buchwald and Bodor, 1999). Our results are in agreement with these previous reports; moreover, they suggest that tissue- or species-specific activation of CPT-11 by CEs is controlled by the size of the piperidinopiperidine ring system. Slight modifications to this moiety of CPT-11 should allow optimization of hydrolysis by hCE1 and/or hiCE, such that toxicities attributable to tissues-specific expression of CEs may be minimized.
Ultimately, we would like to perform the detailed kinetic studies described in this manuscript with analogs of CPT-11, where the esterified bipiperidino function present at the 10-position of the molecule is replaced with other substituents. However, because of the unavailability of such reagents, we performed these analyses with commercially available substrates that demonstrated characteristics similar to CPT-11 (e.g., bulkiness, hydrophobicity). With an accurate model of protein structure, the design of novel CPT-11 analogs will be greatly simplified.
In conclusion, our modeling and experimental data indicate that the ability of CEs to metabolize CPT-11 is dependent on the access of the drug to the catalytic amino acids. This access is regulated by the size of the active site entrance. Therefore, hCE1 is less efficient at catalysis of bulky substrates than rCE and hiCE. Further elucidation of precise CPT-11-enzyme contacts in the active site gorge await detailed X-ray crystallographic studies of the CE proteins. However, we conclude that simply adjusting the size of the ester substituent cleaved by CEs could confer specificity of CPT-11 metabolism by esterases from different species or tissue types. This concept may be exploited for tissue-selective production of SN-38 from novel camptothecin analogs that differ only in the size of the esterified side chain.
Acknowledgments
We thank Dr. J. P. McGovren for the gift of CPT-11.
Footnotes
- Received January 29, 2001.
- Accepted April 25, 2001.
-
This work was supported in part by National Institutes of Health Grants CA76202, CA79763, the Cancer Center Core Grant CA21765, and the American Lebanese Syrian Associated Charities.
Abbreviations
- CPT-11
- 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin (irinotecan)
- SN-38
- 7-ethyl-10-hydroxycamptothecin
- CE
- carboxylesterase
- hCE human carboxylesterase
- hiCE, human intestinal carboxylesterase
- AChE
- acetylcholinesterase
- NPA
- nitrophenyl acetate
- rCE
- rabbit liver carboxylesterase
- NPB
- nitrophenyl butyrate
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}