


Abstract
To investigate possible species-specificity of aryl hydrocarbon receptor (AhR)-mediated signal transduction pathways, activities of 2,3,7,8-tetrochlorodibenzo-p-dioxin (TCDD) and six synthetic flavonoids were evaluated in mouse hepatoma and guinea pig adenocarcinoma cells transfected with an AhR-responsive luciferase reporter. Rank order potency in these two cell lines was similar for the ability of these flavonoids to antagonize TCDD-induced reporter gene expression. However, in the presence of flavone alone, a species-specific difference in agonist activity was observed. In guinea pig cells, several flavonoids demonstrated agonist activity up to 50% of the maximum TCDD response. In mouse cells, however, no significant agonist activity was observed at the same concentrations based on luciferase enzyme activity, protein expression, and mRNA analysis. Moreover, competitive ligand-binding assays, using [3H]TCDD in cytosolic fractions, demonstrated that 3′-methoxy-4′-nitroflavone had a similar IC50 in both recombinant cell lines, suggesting that the flavone has similar binding affinity to receptors from both species. However, electrophoretic mobility shift assay using the cytosolic fractions demonstrated that this flavone elicited binding to the DRE by guinea pig but not mouse AhR complex. The dependence of the AhR in this differential interaction was further demonstrated using in vitro synthesized guinea pig and mouse Ah receptors and mouse Arnt. Together, these data suggest that the differential agonist/antagonist activity of these flavone derivatives is caused by the efficacy of these flavonoids in eliciting an AhR conformation that recognizes regulatory response elements in a species-specific manner.
The aryl hydrocarbon receptor (AhR), a member of the basic helix-loop-helix protein superfamily, is a ligand-activated transcription factor (Swanson and Bradfield, 1993; Schmidt and Bradfield, 1996). Ligand-free AhR resides in the cytosol as a complex associated with the 90-kDa heat shock protein and several other proteins (Kazlauskas et al., 1999;Whitlock, 1999). Ligand-elicited activation [e.g., 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)] promotes the receptor to undergo a series of processes involving dissociation from cytosolic chaperones, translocation to the nucleus, association with the AhR nuclear translocation partner (Arnt), recognition of the dioxin responsive element (DRE), and modification of gene transcription (Denison et al., 1989; Dong et al., 1996).
Among a variety of species, the AhR possesses similar physiochemical properties, and the interaction between TCDD-bound AhR and DRE has been demonstrated to be highly conserved for AhR-mediated signal transduction (Bank et al., 1992). Nevertheless, significant species-, strain-, tissue-, and developmental stage-specific differences are common features in TCDD responsiveness (Ema et al., 1994; Dohr et al., 1995; Sanderson and Bellward, 1995; Garrison et al., 1996; Kikuchi et al., 1996). For example, in response to acute TCDD toxicity, the difference in LD50 between guinea pig, the most sensitive species, and mouse is 100-fold; for hamster, the most resistant species, it is as high as 1000-fold (Birnbaum, 1994). However, the molecular mechanisms accounting for these differences are still not well understood. These could be because of the differences in ligand-binding affinity (Ema et al., 1994; Poland et al., 1994), or in processes such as nuclear translocation, DRE binding, and/or gene- and tissue-specific transactivation (Abnet et al., 1999; Korkalainen et al., 2000). It is also possible that species-specific differences in amino acid sequence of the receptor could differentially modulate, qualitatively or quantitatively, the molecular signal initiated by ligand interaction with the AhR ligand-binding domain (Ema et al., 1994).
To study the molecular mechanism accounting for these differences, one approach is to determine how changes in ligand structure affect binding affinity and the activity of the receptor in gene regulation. Previous work on TCDD and its congeners has almost exclusively emphasized the agonist activity of these ligands. However, it is becoming increasingly recognized that many AhR ligands, especially certain flavonoids, elicit mainly antagonist activity (Harris et al., 1989; Gasiewicz and Rucci, 1991; Mahon and Gasiewicz, 1992; Kurl et al., 1993; Liu et al., 1993;Lu et al., 1995, 1996; Gasiewicz et al., 1996). For example, the synthetic molecule 3′-methoxy-4′-nitroflavone can compete with TCDD to bind to the AhR and inhibit its nuclear uptake and transformation (Lu et al., 1995; Henry et al., 1999). Notably, several compounds (e.g., bioflavonoid galangin and resveratrol) widely present in plants used for human consumption have been shown to block TCDD-induced gene expression (Casper et al., 1999; Ciolino and Yeh, 1999; Ashida et al., 2000; Quadri et al., 2000). However, the precise structural features of ligand and ligand-binding domains that regulate agonist versus antagonist activities still need to be understood and defined. Limited mechanistic studies of AhR ligands across species have been performed, and it is unknown whether the antagonist activity of flavonoids is a common feature that reflects similar structural and regulatory properties of the AhR.
One of the interests of researchers in this laboratory has been to identify potent AhR ligands and use these compounds to investigate the molecular events that regulate AhR activity. In the present report, we describe studies that examined the relative agonist/antagonist activity of a group of synthetic flavonoids in guinea pig and mouse cells. Results presented herein demonstrate that although these chemicals have the same rank-order potency for AhR binding and ability to antagonize TCDD-induced reporter gene induction at lower concentrations, several flavonoids exhibited species-specific and concentration-dependent agonist activity in guinea pig cells, but not mouse cells. Further mechanistic studies with 3′M4′NF indicate that a differential activation of the AhR by this flavonoid, to interact with the DRE accounts for this species-specific effect.
Materials and Methods
Chemicals.
Six flavonoid compounds, 3′-acetamideflavone (3′AAF), 3′,5′-methoxyflavone (3′5′MF), 4′-nitro-7,8-benzoflavone (4′N7,8BF), 7,8-benzoflavone (ANF), 3′-methoxy-4′-nitroflavone, and 3′-dimethylaminoflavone (3′DMAF) were synthesized in the laboratory of Dr Andrew Kende (University of Rochester, Rochester, NY) by the procedure described previously (Cunningham et al., 1992). The purity of these compounds was determined by 1H-NMR spectroscopy, thin-layer chromatography, and determination of melting points. TCDD was purchased from Cambridge Isotopes (Cambridge, MA), and [3H]TCDD was from ChemSyn (Lenexa, KS). Actinomycin D and cycloheximide were from Sigma (St. Louis, MO). All chemicals were dissolved in DMSO before use.
Cell Culture and Stable Transfection.
Hepa.1c1c7 cells were a gift from Dr. Oliver Hankinson (UCLA). The luciferase reporter plasmid p2DLuc containing two copies of the DREDconsensus sequence and a minimal promoter was described previously (Gasiewicz et al., 1996). Hepa.2DLuc.3A4 (Hepa.2D) cells, stably transfected with this reporter construct, were established from Hepa.1c1c7 cells as described previously (Henry et al., 1999). GPC.2DLuc.1C12 (GPC.2D) cells, containing this same p2Dluc construct, were established from GPC.16 cells (American Type Culture Collection, Manassas, VA) using a similar procedure. All experimental cells were grown at 37°C, 5% CO2 in modified Eagle's medium (Sigma) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA), sodium pyruvate, l-glutamine, sodium bicarbonate, and gentamicin, defined as modified Eagle's medium+.
Luciferase Activity Assay.
Methods described previously (Gasiewicz et al., 1996; Henry et al., 1999) were modified to detect luciferase reporter gene activity in a high throughput format. The use of a long-lasting luminescent substrate (Steady-Glo; Promega, Madison, WI) provides a stable luminescent signal (t1/2 ∼ 5 h) and enables cell lysis and luciferase activation directly in the culture medium. The use of microcarrier beads increases the overall surface area for cell adhesion; this allows for the introduction of more cells per well, increasing the sensitivity and reproducibility of the assay in a 96-well plate format (S. D. Dertinger, C. M. Palermo, and T. A. Gasiewicz, unpublished observations).
Cytodex 1 microcarrier beads (Sigma) were hydrated and autoclaved in phosphate-buffered saline (PBS) at a dry weight of 3 mg/ml. For each experiment, 10 ml bead/PBS suspension was transferred to a 50-ml polypropylene tube, and PBS was diluted out with the prewarmed modified Eagle's medium+. Hepa.2D cells (7.5 × 105) or 8.0 × 105 GPC.2D cells were added to the 10-ml bead/media suspension. The cell/bead/media suspension was then transferred to a 100-mm nontissue culture dish and incubated for 48 h. After that, the suspension was transferred to a 50-ml polypropylene tube, mixed, and divided into several 1-ml aliquots. The flavonoids were added to these aliquots to achieve final concentrations of 0, 0.001, 0.01, 0.1, 1 μM with or without nonsaturating TCDD, to assay for antagonist or agonist activities, respectively. Quadruplicate 100-μl aliquots of cell beads were then transferred immediately from each group into an opaque 96-well culture plate (PerkinElmer Life Science, Boston, MA). Plates were incubated for 4 h for Hepa.2D and 5h for GPC.2D cells. Steady-Glo Luciferase Assay reagent (100 μl; Promega) was added to each well and luminescence was read using a LumiCount (PerkinElmer) plate reader. Background value luminescence (in wells with media only) was subtracted from each experimental value. For antagonist studies, the data were plotted for each flavone concentration as a percentage of the TCDD response in the same group in the absence of the antagonist. For agonist activity, the data were plotted as a percentage of 1 nM TCDD response in the same cell lines.
Western Blotting Analysis of Luciferase.
The luciferase protein used as positive control in blotting was synthesized using in vitro TnT coupled reticulocyte lysate system (Promega) following the recommended procedure.
Equal numbers of cells were grown in six-well plates (VWR, Bridgeport, NJ) to 80% confluence. Cells were treated with either vehicle (DMSO, 0.1%) or increasing concentrations of test compounds for 5 h and lysed with 150 μl of SDS-polyacrylamide gel electrophoresis loading buffer (0.063 M Tris, 2% SDS, 10% glycerol, 5% mercaptoethanol, and 0.05% bromphenol blue, pH 6.8). Lysates were heated in boiling water for 10 min before loading. 30 μl of lysate for each sample were resolved using SDS-polyacrylamide gel electrophoresis (7.5% acrylamide resolving gel) and transferred to polyvinylidene difluoride membrane (Millipore, Bedford, MA). The membrane was blocked in bovine lacto-transfer optimizer buffer (5% nonfat dry milk, 10 mM Tris, 50 mM NaCl, 1 mM EDTA, and 30 mM sodium azide, pH 7.4) for 1 h, and then probed with rabbit anti-luciferase IgG fraction of antiserum (Sigma) in Tris-buffered saline/Tween 20 (10 mM Tris, 150 mM NaCl, and 0.1% Tween 20, pH 7.2) for 5 h. The membrane was washed for 3 × 10 min with PBS containing 0.05% Tween 20, blocked for 0.5 h, and incubated with anti-rabbit IgG coupled to horseradish peroxidase (Jackson ImmunoResearch, West Grove, PA) for 1 h. Proteins were visualized by chemiluminescence (KPL, Gaithersburg, MD).
Total RNA Extraction.
Total RNA was isolated from cells grown in six-well plates and treated with compounds for 4 h, using SV Total RNA Isolation System (Promega) with minor modifications. Cells were washed twice with ice-cold PBS and lysed with 175 μl of SV RNA lysis buffer per well. SV RNA dilution buffer (350 μl) was added to the lysates on each well followed by heating the plate at 70°C for 3 min. Lysates were transferred to microcentrifuge tubes and total RNA isolated following the recommended procedures. The concentration was measured with GeneQuant pro (Amersham Biosciences, Piscataway, NJ).
Real-Time Quantitative Polymerase Chain Reaction.
The mRNAs for luciferase were determined by real time quantitative PCR on iCycler iQ Multi-Color Real Time PCR Detection System (Bio-Rad, Hercules, CA). All probes were designed from the published cDNA database on GenBank (http://www.ncbi.nlm.nih.gov/), synthesized by Synthegen (Houston, TX) or IDT (Coralville, IA), and diluted to 20 μM before use. The sequences for primers and probes of luciferase and GAPDH are listed in Table 1. The EZ real-time PCR reaction mix from Applied Biosystems (Foster City, CA) was used in the reaction. Each tube contained 3 mM manganese acetate, 300 μM dNTPs, 200 nM concentrations of probe and each primer, 0.1 U/μl recombinant Thermus thermophilus DNA polymerase (Applied Biosystems), 0.01 U/μl AmpErase (Applied Biosystems), 100 ng of total RNA (or same volume of nontemplate control) in 1× EZ buffer (Applied Biosystems) in a final volume of 25 μl. The real-time PCR reaction for luciferase and GAPDH were carried out in separate tubes. Triplicate samples from each treatment group were run under the following conditions: 50°C for 2 min, 60°C for 30 min, 95°C for 5 min, and 40 cycles of amplification between 94°C for 20 s and 62°C for 1 min. The relative standard curve method was used and produced by using samples from serial dilution of total RNA extracted from GPC.2D or Hepa.2D cells. Luciferase input, calculated from the experimental cycle threshold value based on its standard curve, was normalized to GAPDH obtained similarly from the same sample. The values were then normalized to DMSO from the same cell line to obtain the fold induction.
Primer and probe sequences used in mRNA analysis
Competitive Ligand Binding Assay and Electrophoretic Mobility Shift Assay.
Cytosolic extracts from Hepa.1c1c7 and GPC.16 cells were prepared as described previously (Henry et al., 1999). Briefly, confluent cells were harvested and homogenized in HEDG buffer containing 1 μM leupeptin, 4 μg/ml aprotinin, and 0.3 mM phenylmethylsulfonyl fluoride. Membranes were removed by a 45-min centrifugation at 100,000g and the supernatant was stored at −70°C until use.
The ability of 3′M4′NF to compete with [3H]TCDD for binding to the receptor was assessed by incubating aliquots of cytosol (2.1–2.5 mg protein/ml) from guinea pig or mouse cells with a range of concentrations (0–1000 nM) of the flavone and 3 nM [3H]TCDD for 2 h at room temperature. TCDD (3 nM) is nonsaturating at these protein concentrations. Specific binding of [3H]TCDD was determined in duplicate aliquots by the hydroxylapatite assay (Gasiewicz and Neal, 1982), with correction for nonspecific binding measured in the presence of 150-fold excess of unlabeled 2,3,7,8-tetrachlorodibenzofuran. Data were plotted for each antagonist concentration as a percentage of the specific binding of [3H]TCDD in the absence of the competitor.
Aliquots of cytosol from the same incubations were mixed with nonspecific DNA (herring sperm), 0.08 M NaCl, and 25,000–45,000 cpm of32P–end-labeled oligonucleotide. The annealed oligonucleotide contained a single consensus DRE that is recognized by the transformed AhR (for complete sequence, see Gasiewicz et al. (1996). Samples were subjected to nondenaturing electrophoresis (4% acrylamide), and 32P associated with the AhR-retarded band was quantified using a PhosphorImager (PSI; Amersham Biosciences). The amount of radioactivity detected at the equivalent position in a lane containing vehicle-treated cytosol was used as background and subtracted from each sample value. Values were expressed as a percentage of that observed in cytosol treated with TCDD alone (Henry et al., 1999).
Construction of a Mammalian Expression Vector of the Guinea Pig AhR.
The full-length construct of guinea pig AhR [pPCR-Script-Amp-SK(+)/AhR] was a kind gift from Dr. Raimo Pohjanvirta (Kuopio, Finland). Two PCR-derived errors within the actual sequence (A to G at 378 and G to A at 687) were corrected sequentially using QuikChange site-directed mutagenesis kits (Stratagene, La Jolla, CA) with the primers listed in Table 2. The correction was confirmed by sequencing. The corrected sequence was cut sequentially with restriction enzymes NotI andApaI. The fragment containing the guinea pig AhR was gel-purified and inserted into the vector pcDNA3.1(+) (Invitrogen) predigested with the same enzymes. The incorporation of guinea pig AhR into pcDNA3.1(+) was confirmed by restriction digestion and sequencing analysis. The resultant product was called pcDNA3.1/AhR.
Primers used for mutagenesis (with the corrected nucleotide underlined)
Reconstitution of Mouse AhR and Guinea Pig AhR with Mouse Arnt.
The mouse AhR, guinea pig AhR and mouse Arnt proteins were synthesized in vitro by the TnT coupled reticulocyte lysate system (Promega) following the recommended procedure. Briefly, 0.5 μg of pcDNA3/AhR (mouse), 0.5 μg of pcDNA3.1/AhR (guinea pig), and 1 μg of pcDNA3/Arnt (mouse) expression vectors were incubated separately with the reticulocyte lysates and other components in a final volume of 25, 25, and 50 μl, respectively. The reactions were incubated at 30°C for 90 min, then stopped by incubating the reaction tubes on ice. The expressed mouse Arnt was divided into two 25-μl aliquots; one was mixed with the mouse AhR and the other with guinea pig AhR. The resultant solutions were diluted with HEDG to obtain a final volume of 200 μl, which were further divided into five aliquots of 40 μl. The aliquots were incubated for 90 min at room temperature with DMSO, 3′M4′NF (0.1, 1, 10 μM), or TCDD (10 nM). Samples were analyzed for DRE binding as described above.
Results
TCDD Dose Response for Luciferase Induction.
To begin to approach issues of species-specific differences and/or similarities in response to AhR ligands, we used guinea pig adenocarcinoma (GPC.2D) and mouse hepatoma cells (Hepa.2D) stably-transfected with an AhR-responsive luciferase gene construct. The TCDD dose-response indicated that both cell lines were sensitive bioassay systems (Fig.1). The minimum effective concentration for luciferase induction was approximately 10 pM, and maximum response was reached at 1 nM for both cell lines, consistent with previously published data (Aarts et al., 1995). These data indicate that the AhR in both species is able to recognize and respond to TCDD, the most potent agonist known, in a similar manner. Interestingly, the vehicle [0.1% DMSO (v/v)] alone produced elevated light units (above background) to about 10% of the saturating TCDD response in GPC.2D cells, although it had no detectable effect in Hepa.2D cells. This phenomenon was observed consistently, but the exact mechanism is unknown.

Effects of TCDD on luciferase induction in Hepa.2D and GPC.2D cells. Hepa.2D and GPC.2D cells were treated with increasing concentrations of TCDD for 4 or 5 h, respectively. Luciferase enzymatic activities were measured and background luminescence from DMSO-treated cells was subtracted from each sample value. Data are expressed as percentage of 1 nM TCDD response as described underMaterials and Methods. The figure is a representative of three experiments and the error bars are ± S.D. calculated from quadruplicate samples in a representative experiment.
Similar Rank-Order Potency of Antagonist Activity of Substituted Flavonoids.
To study the ability of six flavonoids to block TCDD-induced reporter gene activation, approximate EC50 concentrations of TCDD (150 and 100 pM in Hepa.2D and GPC.2D, respectively) were used. Durations of treatment of 4 h for Hepa.2D and 5 h for GPC.2D cells were chosen based on the observation that the luciferase in Hepa.2D cells responds to TCDD more rapidly than that in GPC.2D cells (data not shown) and that flavonoid compounds may be metabolized after 5 h (Henry et al., 1999). 3′AAF, 3′5′MF, and 4′N7,8BF demonstrated a consistent rank-order potency in both cell lines for the ability to block TCDD-induced reporter gene expression (Fig. 2, A and B). Of these, 4′N7,8BF was the most potent antagonist, followed by 3′AAF and 3′5′MF. Likewise, the use of ANF, 3′M4′NF, and 3′DMAF also indicated evidence of a similar rank-order potency for AhR antagonism in both species, with 3′M4′NF > 3′DMAF > ANF (Fig. 2, C and D). However, at higher concentrations, these compounds demonstrated species-specific differences in their quantitative ability to antagonize the TCDD-induced luciferase response. These flavonoids could antagonize TCDD-induced reporter response completely in mouse cells up to 1 μM, suggesting “pure” antagonism (Fig. 2C). In guinea pig cells (Fig. 2D), however, although there was an apparent antagonism of TCDD-induced luciferase at lower concentrations; at higher concentrations, luciferase activity was increased.
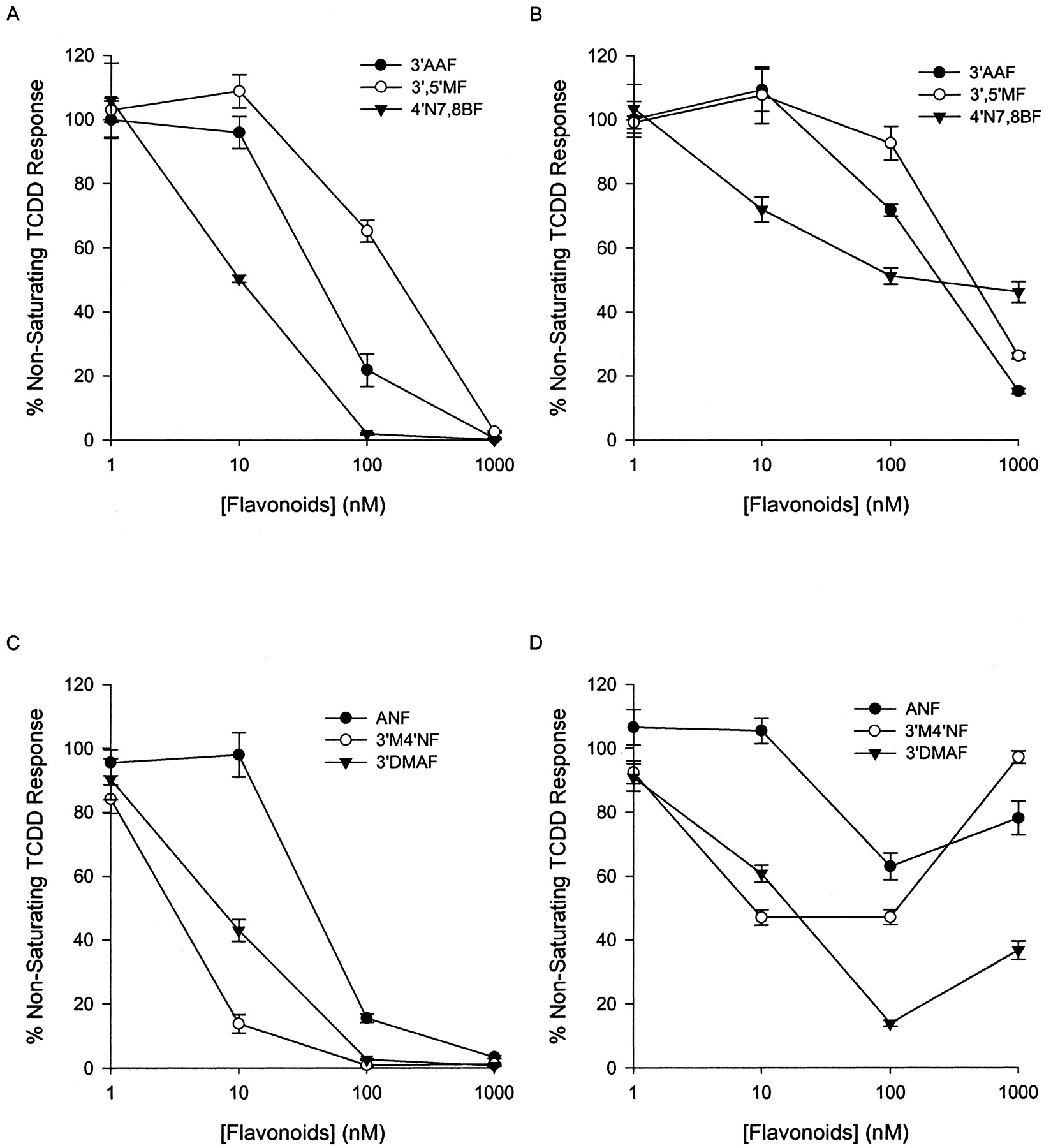
Antagonist activity of substituted flavonoids in Hepa.2D (A and C) and GPC.2D (B and D). Cells were treated with nonsaturating TCDD (100 pM for GPC.2D and 150 pM for Hepa.2D) and increasing concentrations of flavonoids. Luciferase enzymatic activities were measured and the data were plotted for each flavone concentration as a percentage of the TCDD response on the same group in the absence of the antagonist. The data are representative of two separate experiments. The error bars are ± S.D. calculated from quadruplicate samples of a representative experiment.
Differential Agonist Activity of Substituted Flavonoids.
To explain the different dose-response curves shown in Fig. 2, we hypothesized that the flavonoids, although they were competitive AhR ligands, might possess species-specific agonist/antagonist activity for the modulation of gene transcription. To test this, the agonist activities were determined in both cell types after treatment with 1 μM flavonoid alone compared with 1 nM TCDD (Fig.3). Upon flavonoid exposure, guinea pig cells demonstrated higher luciferase induction in the range of 0 to 50% of the saturating TCDD-induced effect, whereas in mouse cells, no agonist activity was detectable at the same concentration (Fig. 3A). Further study of 3′M4′NF demonstrated that its agonist activity in guinea pig cells is concentration-dependent; a maximum response at about 2 μM is approximately 50% of that induced by saturating TCDD. In mouse cells, the response even at 10 μM is less than 5% of 1 nM TCDD (Fig. 3B).

Agonist activity of synthetic flavonoids in Hepa.2D and GPC.2D cells. A, Hepa.2D and GPC.2D cells were treated with DMSO, 1 μM flavonoid, or 1 nM TCDD. B, cells were treated with increasing concentrations of 3′M4′NF. The data were plotted as a percentage of 1 nM TCDD response in the same cell lines. The data are means ± S.D. calculated from three separate experiments.
Differential Enhancement of Luciferase Protein and mRNA Expression by 3′M4′NF.
To verify the result of luciferase reporter activity described above, the relative levels of luciferase protein were determined by Western blotting. Increased luciferase protein expression in guinea pig cells after 3′M4′NF treatment was dose-dependent, whereas no luciferase protein was detected in mouse hepatoma cells under the same conditions (Fig. 4A).

Differential expression luciferase protein and mRNA. A, Western blotting analysis of luciferase protein in GPC.2D cells and Hepa.2DLuc cells. Cells were treated with DMSO (D), 3′M4′NF (0.01, 0.1, 1, 10 μM), or 1 nM TCDD (T) for 5 h. In vitro-synthesized luciferase protein was used as a positive control (Cntr). The data are representative of three separate experiments. B, luciferase mRNA was measured in GPC.2D and Hepa.2D cells by real-time quantitative PCR. The data were expressed as fold induction compared with DMSO. The data are means ± S.D. calculated from three separate experiments. (*,p < 0.01; significantly different from vehicle control by t test).
To investigate whether the elevated protein expression by 3′M4′NF in guinea pig cells is a result of transcriptional activation, total mRNA was extracted from cells treated with DMSO, increasing concentrations of 1 μM 3′M4′NF, or 1 nM TCDD, and luciferase mRNA was measured using real-time quantitative PCR. With increasing concentrations of 3′M4′NF, there was a dose-dependent increase in luciferase mRNA levels in guinea pig cells (Fig. 4B). In mouse cells, however, treatment with 3′M4′NF had no detectable effect on the mRNA level of this reporter gene, suggesting that the compound is unable to trans-activate the same reporter gene. These data further support the hypothesis that the compound has species-specific ability to activate the receptor and enhance transcription of this DRE-dependent reporter gene.
To rule out the possibility that the enhanced luciferase activity is caused by increased stabilization of luciferase mRNA or protein by flavonoids, the guinea pig reporter cells were cotreated with 1 μM 3′M4′NF plus increasing concentrations of actinomycin D (transcription inhibitor) or cycloheximide (protein synthesis inhibitor) for 5 h, and the luminescence was measured as described under Materials and Methods. Cotreatment with either inhibitor abolished the 3′M4′NF-induced luciferase activity, consistent with the hypothesis that increased luciferase was caused by increased transcriptional activity (data not shown).
Competitive Ligand Binding of 3′M4′NF and TCDD to the Ah Receptor.
We hypothesized that 3′M4′NF has a similar binding affinity for the receptors from guinea pig and mouse cells. To test this, cytosolic fractions were obtained from each cell line and used to study the ability of 3′M4′NF to competitively inhibit [3H]TCDD binding to the AhR (Fig.5). The data indicate that the ability of the compound to compete with [3H]TCDD for binding to the receptor is similar between species (IC50 ∼ 50 nM), suggesting that at least the ligand binding domain in both receptors has a similar ability to interact with 3′M4′NF.

Competitive ligand binding of 3′M4′NF and [3H]TCDD to AhR from cytosol of Hepa.1c1c7 and GPC.16 cells. The ability of 3′M4′NF to compete with [3H]TCDD for binding to the receptor was assessed by incubating aliquots of cytosol from guinea pig or mouse cells with a range of concentrations of 3′M4′NF as described under Materials and Methods. Data were plotted for each antagonist concentration as a percentage of the specific binding of 3 nM [3H]TCDD (nonsaturating concentration) in the absence of the competitor. The data are means ± S.D. calculated from three separate experiments.
Species-Dependent DRE Binding.
Although similar in ligand binding affinity, it is possible that different ligands may induce variant conformational changes, which in turn may modify the interaction of the active AhR/ARNT complex with the regulatory DRE sequence, leading to differential reporter gene induction. To test this, EMSA was performed to identify whether 3′M4′NF could transform cytosolic receptors to an active form to interact with the DRE enhancer sequence, which is the same as that present in the upstream region of the luciferase reporter gene. Using mouse cytosol, 3′M4′NF antagonized the TCDD-induced AhR/Arnt/DRE complex formation in a concentration-dependent manner and nearly completely at 1 μM (Fig.6A). However, in guinea pig cytosol, low concentrations partially antagonized TCDD-induced DRE binding, but at 1 μM, DRE binding was increased. Further analysis with 3′M4′NF alone demonstrated a species-specific, concentration-dependent agonist activity in promoting the interaction of AhR/Arnt complex with the DRE (Fig. 6B). These patterns are notably similar to those observed for the induction of luciferase in whole cells (Figs. 2D and 3B).

EMSA from Hepa.1c1c7 and GPC.16 cells. A, gel shift assay of antagonist activity of 3′M4′NF. 32P–End-labeled oligonucleotides were added to cytosol incubated with 3 nM [3H]TCDD (nonsaturating concentration) plus increasing concentrations of 3′M4′NF. D, DMSO; T1, 3 nM [3H]TCDD; T2, 3 nM [3H]TCDD + 300 nM 2,3,7,8-tetrachlorodibenzofuran; F1, 1 nM 3′M4′NF; F2, 10 nM 3′M4′NF; F3, 100 nM 3′M4′NF; F4, 1000 nM 3′M4′NF. The AhR- and ligand-dependent bands are labeled by arrows shown on the figure. B, quantification of Fig. 6A using PhosphorImager. Data were plotted for each flavone concentration as a percentage of that obtained with 3 nM [3H]TCDD alone. The data are means ± S.D. calculated from three separate experiments. C, gel-shift assay of agonist activity of 3′M4′NF. 32P–End-labeled oligonucleotides were added to cytosol incubated with D (DMSO), T (10 nM TCDD, saturating concentration) or increasing concentrations of 3′M4′NF (F1, 10 nM; F2, 100 nM; F3, 1,000 nM; F4, 10,000 nM). D, quantification of Fig. 6C. Data were plotted for each flavone concentration as a percentage of that obtained with 10 nM TCDD. The data are an average of two separate experiments.
AhR Dependence in the Species-Specific Activity of 3′M4′NF.
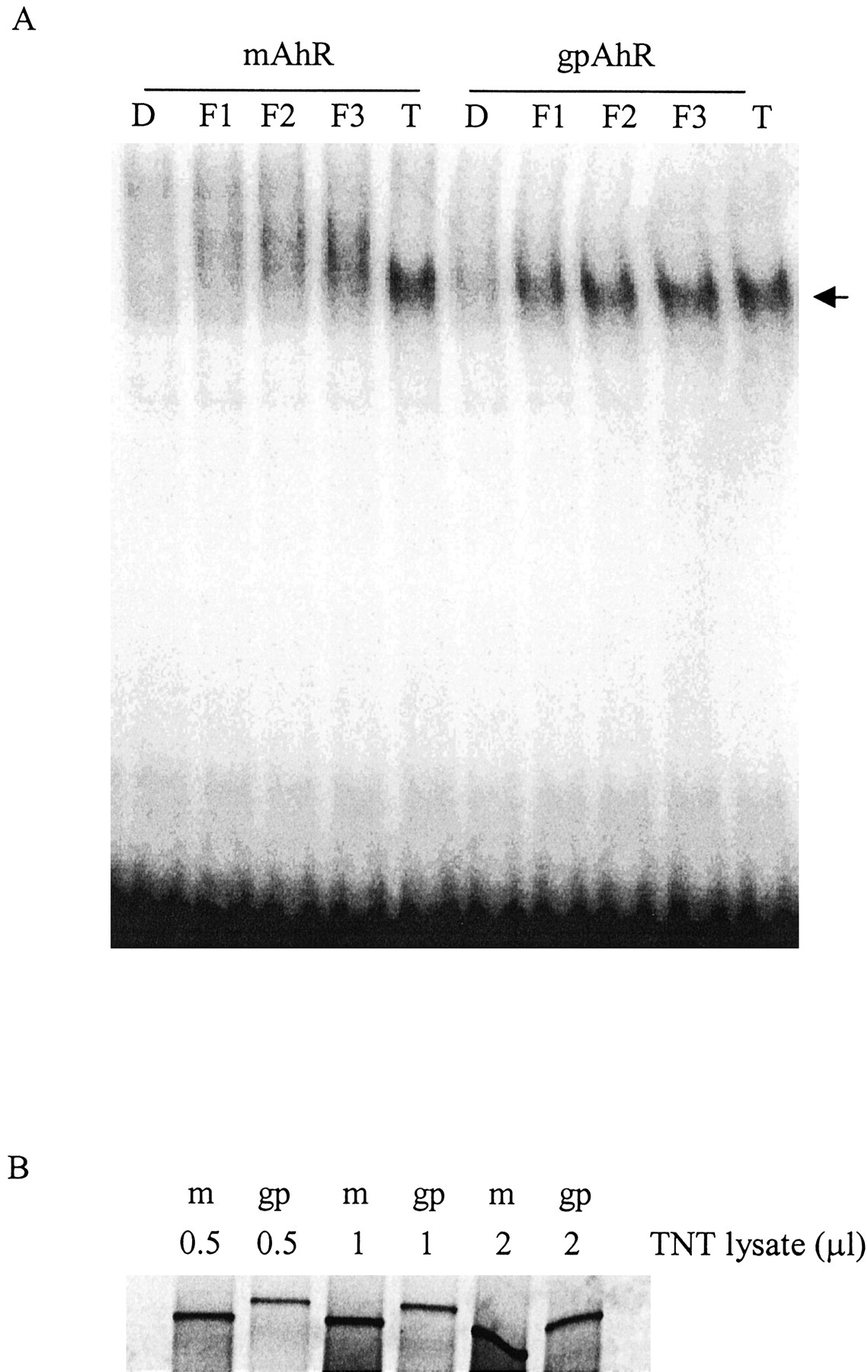
Together, the above data suggest that 3′M4′NF is a pure antagonist in mouse cells, but a partial agonist in guinea pig cells, and that this difference is caused by some ability of 3′M4′NF to transform the guinea pig AhR to a conformation that is able to form a heterodimer with Arnt and recognize the cognate DRE sequences. However, whether this differential interaction induced by 3′M4′NF is exclusively mediated through the AhR or some other species-specific cofactors (e.g., Arnt) is unknown. We hypothesized that this is caused by the differences in the amino acid sequence of the AhR between these two species and the ability of 3′M4′NF to elicit an active conformation of the AhR. To test this, we used a system in which only the AhR is altered. A reconstitution experiment was performed by incubating in vitro synthesized mouse or guinea pig receptors with mouse Arnt, followed by ligand activation and EMSA analysis. With incubation of the guinea pig AhR and mouse Arnt, there was a clear concentration-dependent formation of the guinea pig AhR/mouse Arnt/DRE complex after treatment with 3′M4′NF (Fig. 7). With the mouse AhR, TCDD produced a distinct band, but 3′M4′NF failed to transform the protein to a conformation that was capable of interacting with the DREs in a manner similar to that of TCDD. Instead, there was a concentration-dependent appearance of a smeared band, as seen previously (Gasiewicz and Rucci, 1991; Santostefano et al., 1993), suggesting a very weak interaction of the 3′M4′NF-bound mouse AhR complex with the DRE. The possibility that the increased protein-DNA complex seen in 3′M4′NF-treated guinea pig AhR might be caused by the presence of a higher amount of guinea pig AhR protein was ruled out by comparing [35S]methione incorporation of the in vitro-synthesized AhRs (Fig. 7B). The guinea pig AhR was translated less efficiently than mouse AhR in this system.

EMSA of reconstituted guinea pig AhR or mouse AhR with mouse Arnt. A, in vitro synthesized mouse AhR (or guinea pig AhR) were mixed with mouse Arnt, then incubated with DMSO (D), 0.1 μM 3′M4′NF (F1), 1 μM 3′M4′NF (F2), 10 μM 3′M4′NF (F3), and 10 nM TCDD (T) for 90 min, followed by the incubation with 32P-labeled DREs. The mixtures were subjected to nondenaturing electrophoresis (4% acrylamide), and 32P associated with the AhR-retarded band was visualized using a PhosphorImager. B, relative levels of synthesized guinea pig AhR and mouse AhR from TNT reactions. Separate reactions supplemented with [35S]methionine were performed simultaneously with the unlabeled receptor synthesis reactions for A. After dilution with the same volume of 2× HEDG, the indicated volumes of lysates were resolved on a 7.5% acrylamide gel, and the radioactivity of the proteins was visualized using a PhosphorImager. m, mouse AhR reaction lysate; gp, guinea pig AhR reaction lysate. The data are representative of two independent experiments.
These data are consistent with those obtained using cytosol and intact cells from the two species (Figs. 3B, 4, and 6B); together, they suggest that the species-specific agonist activity of 3′M4′NF is dependent on the difference in amino acid sequences of the AhR, and not other cofactors, between guinea pig and mouse cells.
Discussion
Using a synthetic reporter gene, we have determined that species-specific differences exist for the ability of certain flavonoids to elicit AhR agonist or antagonist activity. The reporter gene used carries two copies of the synthetic DRED from the mouse CYP1A1 regulatory region. Each DRED consists of a 32-base pair fragment identical to that contained in the upstream regions of the murine CYP1A1 gene. Thus, this is a physiological AhR/Arnt binding site containing both “core” recognition and near flanking sequences. Furthermore, the temporal and dose response of this reporter to TCDD is similar to that containing a 480-base pair intactCYP1A1 regulatory sequence used in other labs (Garrison et al., 1996), suggesting that they function similarly. The advantage of using this synthetic construct in studying AhR transcriptional activity is that there are no other known transcription factor binding sites that may influence reporter gene expression in a species-specific manner (Willey et al., 1998). Using cells from different species stably transfected with the same construct allows us to attribute differential agonist/antagonist activity of compounds to the differences between receptors, the heterodimeric partner Arnt, or ability of other cofactors to interact with these proteins and regulate their transcriptional activity, but not the regulatory sequence of the particular gene.
Using recombinant cell lines, we demonstrated the same rank-order potency of flavonoids to act as AhR antagonists in mouse and guinea pig cells (Fig. 2). This rank order probably reflects the same rank order in ligand binding affinity between these chemicals and the receptors from both species (Safe, 1990). Furthermore, the similar ability of 3′M4′NF to inhibit [3H]TCDD binding to cytosolic receptors from both mouse and guinea pig cells (Fig. 6) suggests that at least some characteristics necessary for ligand recognition by the ligand binding domain of these receptors, such as the critical amino acids or functional groups, are similar in the two species. Of the chemicals tested in this study, 3′M4′NF and 4′N7,8BF are the most potent compounds, and ANF and 3′5′MF are the least potent, in terms of their ability to antagonize the TCDD-induced reporter gene induction in both mouse and guinea pig cells. Limited structure-activity analyses indicate that the electron-rich 3′-methoxy and 4′-nitro groups on the benzene ring seem to play a critical role in ligand-receptor interaction and AhR antagonist activity, consistent with previous data obtained using AhR contained in rat liver cytosol (Henry et al., 1999). 3′5′MF is one of the least potent compounds, which suggests that a bulky group at the 5′-position has a role in decreasing ligand binding affinity, possibly because of steric disturbance or altered electrostatic interaction.
On the other hand, we demonstrated that these flavonoids have species-specific, concentration-dependent agonist activity (Fig. 3). In guinea pig cells, higher concentrations of certain flavonoids promote the AhR through a series of transformation events leading to enhanced luciferase gene induction. In comparison, they were unable to transform mouse receptor and enhance transcription of the same reporter gene at the same concentrations, suggesting that, at least in this species, they act mainly as antagonists in blocking TCDD-induced luciferase induction.
To determine the possible mechanism for the differential agonist/antagonist activity of these flavonoids, we examined several individual steps in the AhR transformation process, including competitive ligand binding, DRE interaction, mRNA transcription, protein expression, and luciferase enzymatic activity. In guinea pig cells, the maximum agonist response produced by 3′M4′NF for both DRE binding and induced enzyme activity was approximately 50% of that produced by saturating TCDD. These data are consistent with 3′M4′NF being a lower efficacy agonist compared with TCDD. However, in mouse cells, these endpoints were less than 5% of those elicited by TCDD (Figs. 3B and 6B). This quantitative consistency between these two endpoints in both species suggests that the differential interaction of 3′M4′NF-activated receptor complex with DREs and the subsequent species-specific differences in induced luciferase enzyme activity might result from differences in the inherent amino acid sequence in the ligand binding domain of the guinea pig and mouse AhR that may differentially transduce an active conformation of the AhR. This mechanism is clearly dissimilar from the strain differences in responsiveness observed between TCDD-susceptible C57BL/6 and TCDD-resistant DBA/2J mice, in which the differential ligand binding affinity between wild-type and defective AhR seems to play the major role in differential enzyme inducibility (Ema et al., 1994).
The combination of differential agonist/antagonist effect of these flavonoids probably accounts for the U-shaped concentration-effect curves shown in Figs. 2D and 6A. At lower concentrations, 3′M4′NF binds to the receptor competitively with TCDD, preventing TCDD from binding the receptor. This decrease in “effective” TCDD concentration on the ligand binding site of the AhR results in decreased TCDD-elicited AhR interaction with DRE and the decreased reporter gene induction correspondingly. Because 3′M4′NF was much less efficacious in eliciting an active AhR conformation, the ‘net’ effect was a block of TCDD-induced luciferase enzyme activity. This effect was qualitatively similar in both guinea pig and mouse cells at lower concentrations. At higher concentrations, 3′M4′NF continues to compete with TCDD to bind to the receptor and antagonize the TCDD-induced gene transcription. However, although 3′M4′NF has a lower efficacy compared with TCDD, an increased occupancy in ligand binding sites of the guinea pig AhR by 3′M4′NF at higher concentrations was apparently able to transform the receptor to a transcriptionally active form, leading to the enhanced luciferase gene transcription (compare the lower and higher concentrations of 3′M4′NF in Figs. 2D and 6A). In comparison, at all concentrations tested, 3′M4′NF demonstrated complete antagonism in mouse cells. This is probably caused by a much lower efficacy of 3′M4′NF to transform the mouse AhR.
The whole-cell and cell-free data presented are consistent with the interpretation that the species differences in the AhR protein sequences are the major factor accounting for the differential activities of the flavonoids (Fig. 7). That is, differences in the efficacy of 3′M4′NF between the two cell types are probably caused by the differential ability of this chemical to elicit ligand-dependent conformational changes of the receptor (Hestermann et al., 2000). It is likely that TCDD, the most potent AhR ligand known, elicits a similar conformational change of the AhR in mouse and guinea pig because of its extremely high efficacy. The change in receptor conformation promotes the dissociation of 90-kDa heat-shock protein, leading to the exposure of the nuclear localization signal on the receptor. Subsequently, the nuclear transport proteins shuttle the receptor actively into the nucleus where it dimerizes with Arnt. This TCDD/AhR/Arnt complex then interacts with the DRE and activates gene transcription. However, 3′M4′NF, a structurally dissimilar ligand, may induce distinct conformational alterations because of the difference in amino acid sequences in the ligand binding domains of the guinea pig and mouse AhR. Although recent cloning of the guinea pig receptor revealed that it is highly homologous to that of human, mouse, and rat AhR in the N-terminal sequence, significant variations in the ligand-binding domain and the C-terminal region were shown to exist (Korkalainen et al., 2001). Additional work is being conducted in our laboratory to define the particular amino acid(s) responsible for the functional differences between mouse and guinea pig AhR in the presence of different ligands.
Although the AhR seems to be the primary factor mediating this flavonoid-induced differential responsiveness, other cofactors may also play a role. Transient transfections of the guinea pig and mouse AhR, along with the p2DLuc reporter gene, into receptor-deficient mouse hepatoma cells (BprC1.Tao cells) and African green monkey kidney cells (COS-7 cells) failed to reveal a consistent and statistically significant difference in their response to 3′M4′NF (data not shown). There are several possible reasons. First, the interaction between the transfected guinea pig AhR and heterologous chaperone proteins from mouse (or monkey) cells may prevent an efficient dissociation of the 3′M4′NF-bound guinea pig AhR from the cofactors. Second, even if 3′M4′NF-bound guinea pig AhR could dissociate from the other proteins, the nuclear localization signal on the guinea pig AhR may not be recognized in the same way as it is in guinea pig cells, resulting in decreased nuclear localization. Third, the trans-activation domain of the guinea pig receptor may interact with mouse (or monkey) transcription machinery differently. All of these possibilities, alone or in combination, may affect the Ah receptor's ability to modulate gene transcription in response to 3′M4′NF. Thus, although the binding of 3′M4′NF to the expressed guinea pig AhR in a cell-free system is clearly able to elicit a conformation that recognizes the DRE, even with mouse Arnt, within the intact cells, this altered conformation may be closely coupled to other events necessary fortrans-activation function. Together, these data suggest that the AhR is a necessary but insufficient condition for transducing species-specific ligand-induced signals, and final consequences are probably dependent on both the AhR and other species-specific cofactors.
In summary, the data presented herein demonstrate that there are clear species-specific, ligand-dependent differences in AhR-mediated gene regulation. For 3′M4′NF, this seems to be caused by different efficacies when bound to receptors from different species, leading to modulated DRE binding. These data imply that the relative degree of agonist/antagonist activity of a particular AhR ligand may be very species-, cell-, gene-, and concentration-specific. This may especially be the case for ligands with lower degrees of efficacy compared with TCDD.
Acknowledgments
We would like to thank Dr. Raimo Pohjanvirta (Kuopio, Finland) for the kind gift of guinea pig AhR construct pPCR-Script-Amp-SK(+)/guinea pig AhR, and Dr. Oliver Hankinson (UCLA) for the kind gift of Hepa.1c1c7 cells. We thank Dr. Andrew Kende (University of Rochester) for the synthesis of flavone compounds. We also thank the members in our lab for the critical review of the manuscript.
Footnotes
-
↵1 Litron Laboratory Ltd., Rochester, New York.
-
The research was funded in part by National Institute of Environmental Health Sciences grants ES09702 and ES01247 and a grant from the American Institute for Cancer Research.
-
The data were presented in part at the 40th Annual Meeting of Society of Toxicology, March 2001, San Francisco, California (Toxicol Sci60:439).
- Abbreviations:
- AhR
- aryl hydrocarbon receptor
- Arnt
- Ah receptor nuclear translocation partner
- TCDD
- 2,3,7,8-tetrochlorodibenzo-p-dioxin
- DRE
- dioxin responsive elements
- 3′AAF
- 3′-acetamideflavone
- 3′5′MF
- 3′,5′-methoxyflavone
- 4′N7,8BF
- 4′-nitro-7,8-benzoflavone
- 3′M4′NF
- 3′-methoxy-4′-nitroflavone
- 3′DMAF
- 3′-dimethylaminoflavone
- ANF
- 7,8-benzoflavone
- DMSO
- dimethyl sulfoxide
- PBS
- phosphate-buffered saline
- PCR
- polymerase chain reaction
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- EMSA
- electrophoretic mobility shift assay
- HEDG
- HEPES/EGTA/dithiothreitol/glycerol
- Received October 31, 2002.
- Accepted January 15, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}