


Abstract
Arylamine N-acetyltransferases (NATs) catalyze the biotransformation of a variety of arylamine drugs and carcinogens and may play diametrically opposing roles in enhancing either the detoxification of these chemicals or their metabolic activation into DNA-binding electrophiles. To facilitate the study of these processes, we have generated a Nat1/Nat2 double-knockout mouse model by gene targeting in embryonic stem cells. Nat1/2(-/-) mice were born at the expected frequency and seemed normal and viable with no overt phenotype, indicating that these genes are not critical for development or physiological homeostasis. In wild-type mice, NAT1 and NAT2 transcripts were detectable by RT-PCR in all tissues assayed including liver, kidney, colon, brain, bladder, and spleen. NAT1 and NAT2 transcripts were completely undetectable in the Nat1/2(-/-) mice. The in vitro N-acetylation of p-aminosalicylate was detected at significant levels in liver and kidney cytosols from either wild-type inbred `rapid acetylator' C57BL/6 mice or from outbred CD-1 mice possessing homozygous rapid, heterozygous, or homozygous `slow acetylator' Nat2 genotypes. Activity was undetectable in cytosol preparations from Nat1/2(-/-) mice. Nat1/2(-/-) mice also displayed severely compromised in vivo pharmacokinetics of p-aminosalicylate (PAS) and sulfamethazine (SMZ), with a drastically increased plasma area under the curve for PAS and a complete absence of their acetylated metabolites (AcPAS or AcSMZ) from plasma, confirming the functional absence of these enzymes and impaired drug metabolism capacity. This knockout mouse model should be helpful in delineating the role that variation in acetylating enzymes plays in mediating interindividual differences in susceptibility to arylamine-induced chemical toxicity and/or carcinogenesis.
Exposure to environmental chemicals of the arylamine class is a known causal factor in a multistep process that ultimately leads to the production of tumors both in animal models and in human exposure situations (Windmill et al., 1997; Hein, 2002). Found in hair dyes, food pyrolysis products, and tobacco smoke, arylamines are procarcinogens that require metabolic activation by drug or xenobiotic-metabolizing enzymes to DNA-binding electrophiles. One class of enzymes that has been implicated in this process is the arylamine N-acetyltransferases (NATs) (Grant et al., 2000; Hein, 2002). Two functional isoenzymes, NAT1 and NAT2, are present in humans, and each is capable of detoxifying a range of potential arylamine carcinogens to innocuous metabolites via N-acetylation. Competing with N-acetylation, however, is an N-oxidation reaction to an arylhydroxylamine, which is probably mediated at least in part by one or more isoforms of cytochrome P450. This represents a critical first step in a putative metabolic activation pathway (Eaton et al., 1995), whereby further O-acetylation of these P450-generated metabolites by NATs leads to the production of unstable acetoxy esters that can decompose to highly reactive nitrenium ions (Hein et al., 1993).
Although the human NAT2 polymorphism has historically been associated with impaired N-acetylation of a number of therapeutically useful drugs, it is now known that both NAT1 and NAT2 are highly polymorphic (Grant et al., 2000). Several studies have investigated the relationship between the function of NAT1 and NAT2 and cancers of the colon, bladder, lung, breast, and prostate (reviewed in Grant et al., 2000; Hein et al., 2000), but there seems to be no clear correlation between acetylator status and cancer risk. Such epidemiological studies are confounded by differences in phenotyping and genotyping protocols used, sample sizes, and the amount, type and extent of exposures. Thus, it is still unclear what role the NATs may have in preventing or initiating cancer.
The mouse has been extensively studied as a model for the human acetylation polymorphism and for studying the role of variable acetylation in altering risk for arylamine-induced cancers (Hein et al., 1997). Three NAT isoenzymes are present in the mouse, NAT1, NAT2, and NAT3. The slow acetylator phenotype found in inbred strains such as A/J and A/HeJ is associated with the presence of a N99I amino acid substitution in NAT2 compared with the rapid acetylator strains C57BL/6J, C3H/HeJ, and BALB/c (Martell et al., 1991; Kelly and Sim, 1994; Fretland et al., 1997). The outbred CD-1 strain is truly polymorphic, carrying both rapid (Nat2*8; N99) and slow (Nat2*9, I99) alleles (Estrada et al., 2000). Recently, polymorphisms have also been identified in the coding region of the Nat1 gene from the wild-derived inbred Mus spretus strain. One polymorphism consists of a H232R amino acid substitution, and is associated with a decrease in recombinant NAT1 activity (Boukouvala et al., 2002). Studies with NAT3 have failed to reveal activity toward many common NAT1/2 substrates (Fretland et al., 1997; Estrada-Rodgers et al., 1998; Boukouvala et al., 2002).
In contrast to human NAT1, mouse NAT1 is found primarily in liver and exhibits a substrate profile similar to the human NAT2 isoenzyme, namely a higher catalytic activity for sulfamethazine (SMZ) and isoniazid (Estrada-Rodgers et al., 1998). Similarly, mouse NAT2 displays a more widespread tissue distribution (Stanley et al., 1997) and metabolizes p-aminosalicylic acid (PAS), p-aminobenzoic acid (PABA), and the folate catabolite p-aminobenzoyl-l-glutamate (Estrada-Rodgers et al., 1998; Payton et al., 1999), and thus seems to be the functional equivalent to human NAT1 in terms of substrate selectivity. Interestingly, mouse NAT2 is present early in development, suggesting a possible link to folate metabolism and neural tube development (Stanley et al., 1998; Mitchell et al., 1999). It is likely then that the mouse NAT2 polymorphism cannot be considered the functional equivalent of the human NAT2 polymorphism.
Nonetheless, acetylator congenic inbred mice have been used to study the relationship between acetylator phenotype and arylamine-induced DNA adduct formation. Studies have revealed higher levels of 2-aminofluorene-1 DNA adduct levels in the livers of rapid acetylator mice (Levy and Weber, 1989). Slow acetylator female mice, in contrast, displayed higher levels of 2-aminofluorene-1 DNA adducts in urinary bladder (Levy and Weber, 1992). However, male mice have been shown to be more susceptible to arylamine-induced bladder tumors (Schieferstein et al., 1985). Clearly, the challenges in extrapolating such results to a human risk situation include the known species differences in the arylamine substrate selectivities of the genetically variable forms of NAT and the fact that overlapping substrate selectivities of these enzymes may lead to compensatory acetylation of particular substrates in existing rapid or slow acetylator animals, making it difficult to interpret observed toxicity from short- or long-term exposure studies.
To develop a mouse model that may be useful in dissecting out the roles that acetylation reactions may play in chemical carcinogenesis, we have developed a knockout mouse that is devoid of both NAT1 and NAT2 function. Several knockout animal models have already been made for other drug metabolizing enzymes and have provided some insight into specific drug-induced toxicities (for review, see Gonzalez and Kimura, 2003). The role that NATs may play in chemical carcinogenesis and during development, however, remains to be fully elucidated. The creation of a mouse model lacking functional NAT activity should help to determine whether for particular substrates these enzymes serve a protective role by detoxifying carcinogenic arylamines, or facilitate chemical carcinogenesis in an organ-specific fashion by generating DNA-binding electrophiles.
Materials and Methods
Materials. The pPNT plasmid vector was generously provided by Dr. Janet Rossant (Mount Sinai Hospital, Toronto, ON). R1 embryonic stem (ES) cells were from Dr. Andras Nagy (Mount Sinai Hospital, Toronto, ON). Most ES cell culture reagents (nonessential amino acids, l-glutamine, sodium pyruvate, Dulbecco's modified Eagle's medium, G418, ganciclovir) were purchased from Invitrogen (Burlington, ON, Canada) with the exception of β-mercaptoethanol (Sigma-Aldrich Canada Ltd., Oakville, ON, Canada), fetal bovine serum (Hyclone, Logan, UT) and the leukemia inhibitory factor ESGRO (Chemicon International Inc., Temecula, CA). Acetyl-dl-carnitine, carnitine acetyltransferase, acetyl-CoA sodium salt, PAS, and SMZ used for NAT activity assays were acquired from Sigma-Aldrich Canada Ltd. AcPAS to use as a standard was produced in definable quantities from PAS using wild-type recombinant human NAT1 as a catalyst, whereas AcSMZ was chemically synthesized (Dupret et al., 1994). Restriction enzymes for cloning and restriction digests were purchased from Invitrogen. Oligonucleotide primers were synthesized by Invitrogen. CD-1 and C57BL/6 mice were obtained from Charles River Laboratories (Wilmington, MA). All procedures involving animals were in accordance with the Canadian Council for Animal Care guidelines for use and care of animals.
Construction of Targeting Constructs. A Nat1/Nat2 double-knockout targeting construct was generated using the vector pPNT (Tybulewicz et al., 1991), which contains a bacterial phosphoribosyltransferase II gene conferring neomycin resistance (neo) and a herpes simplex virus thymidine kinase (tk) gene, each under the transcriptional control of the mouse phosphoglycerate kinase promoter. To generate the congenic DNA sequences needed to create the targeting vector, a BAC 129S6/SvEvTac RPCI-22 library (BACPAC Resources, Children's Hospital Oakland Research Institute, Oakland, CA) was screened by the MRC Genome Resource Facility at the Hospital for Sick Children (Toronto, ON) with specific NAT1 and NAT2 probes corresponding to the open reading frames. Several BAC clones were isolated containing all three Nat genes. The Nat1 and Nat2 genes are separated by a distance of 9 kb, with Nat3 located approximately 46 kb 3′ of Nat2. From BAC clone 438N17, a 2.0-kb BglII/HincII fragment corresponding to the 5′ untranslated region of the Nat1 gene was blunt-end ligated into the XhoI site of pPNT, located upstream of the neo gene (Fig. 1A). A 3.9-kb PstI/SacI fragment consisting of 0.3 kb of Nat2 coding sequence and 3.6 kb of Nat2 3′ UTR was blunt-end ligated into the KpnI site of pPNT, 5′ to the tk gene. Restriction digests and DNA sequencing confirmed insertion and correct orientation of the sequences. The resulting construct was purified using a QIAGEN MaxiPrep kit (Qiagen Inc., Mississauga, ON, Canada).

Targeted simultaneous knockout of mouse Nat1 and Nat2 genes by homologous recombination. A, schematic representation of the wild-type Nat1/Nat2 gene region, the targeting vector for the simultaneous disruption of both Nat1 and Nat2 genes, and the predicted homologous recombinant locus or disrupted allele. Location of the probe used for genotyping the ES cell colonies and mice is shown by the stars in the 3′ flanking region. The wild-type allele yields a band of 23 kb when digested with BamHI, whereas the targeted allele is represented by a 7.3-kb band. neo, neomycin resistance gene. hsv-tk, herpes simplex virus thymidine kinase gene. Restriction endonuclease sites in parentheses were lost during cloning/ligation. B, Southern blot analysis of genomic DNA samples from representative ES cell clones after homologous recombination. Wild-type ES cell clones (+/+) display a single band of 23 kb. Targeted clones (+/-) show two bands of 23 and 7.3 kb. C, Southern blot analysis of genomic DNA from tail clips from representative wild-type, heterozygous, and null mice. Wild-type mice (+/+) display a single band of 23 kb. Heterozygotes (+/-) are demarcated by the presence of two bands of 23 and 7.3 kb, and null mice (-/-) display only one band at 7.3 kb.
Production of Chimeric Mice. R1 embryonic stem (ES) cells were grown in Dulbecco's modified Eagle's medium supplemented with 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 100 μM β-mercaptoethanol, 2 mM l-glutamine, 20% fetal bovine serum, and 1000 U/ml leukemia inhibitory factor. The plasmid DNA used for targeting was linearized with NotI and introduced into early passage R1 ES cells by electroporation (500 μF, 0.24 kV) using a Bio-Rad Gene Pulser II (Bio-Rad Laboratories Ltd., Mississauga, ON, Canada). After selection in medium containing G418 (200 μg/ml) and ganciclovir (2 μM), individual drug-resistant colonies were isolated, expanded, and genotyped for the appropriate targeting event (see below). Targeted ES cells were provided to the Transgenic Mouse Facility at the Hospital for Sick Children (Toronto, ON) for morula aggregations using host CD-1 females. Chimeric offspring identified by coat color to be greater than 95% 129/SvJ contribution were bred with CD-1 mice to confirm germline transmission, determined by the presence of an agouti coat color in offspring. Chimeric male mice were subsequently bred with C57BL/6 female mice, and F1 heterozygotes were intercrossed to produce the homozygous null and wildtype F2 mice used in our experiments. F1 heterozygotes with a CD-1/129 background, procured from the germline transmission confirmation breedings, were also intercrossed to produce F2 offspring for use in preliminary experiments.
Genotyping of ES Cells and Mice. Genomic DNA was isolated from either ES cell clones or from mouse tail biopsies. ES cells were lysed overnight at 37°C in lysis buffer containing 0.2 M NaCl, 5 mM EDTA, 50 mM Tris-HCl, pH 7.5, 0.2% SDS, and 400 μg/ml proteinase K. Genomic DNA was precipitated with ethanol, washed with 70% ethanol, air-dried, and resuspended in H20. For mouse tail biopsies, approximately 1 cm of tissue was digested overnight at 55°C in lysis buffer containing 200 μg/ml proteinase K, extracted with phenol/chloroform, and ethanol-precipitated as described. To identify genetargeted events and homozygous knockout mice, genomic DNA was digested with BamHI and analyzed by Southern blot analysis. A 407-bp screening probe was generated by PCR using the primers listed in Table 1 and 30 cycles of amplification consisting of 94°C for 10 s, 61°C for 10 s, and 72°C for 30 s on a DNA Engine Tetrad PTC-225 thermal cycler (MJ Research, Inc., Waltham, MA). The probe, corresponding to the region indicated in Fig. 1A, was labeled with [32P]dCTP by random priming and hybridized overnight at 55°C in ExpressHyb buffer (BD Biosciences Clontech, Palo Alto, CA). The blots were washed 2 times in 2× SSC and 0.1% SDS, once in 0.2× SSC and 0.1% SDS, and once in 0.1× SSC and 0.1% SDS for 15 min at 60°C. Alternatively, an AlkPhos direct labeling kit (Amersham Biosciences, Baie D'urfe, QC, Canada) was used for the labeling and hybridization of the probe per the manufacturer's instructions. A 23-kb band indicated the presence of the wild-type allele, whereas a targeted allele was evident as a 7.3-kb band. Genotyping of the naturally occurring Nat2*8 and Nat2*9 allelic variants in CD-1 outbred mice was performed as described by Payton et al. (1999).
PCR primer pairs
PCR primers were used to generate the Southern blotting screening probe for identifying the Nat1/2(-/-) mice, and for RT-PCR of NAT1-, NAT2-, and β-actin-specific products from the various tissue sources. Conditions used for PCR amplification are described in the text.
RNA Isolation and RT-PCR. Total RNA was prepared using an RNeasy mini kit (Qiagen Inc., Mississauga, ON) from various tissue sources (liver, kidney, colon, bladder, cortex, cerebellum, and spleen) stabilized in 10 volumes of RNAlater reagent. Approximately 1 μg of total RNA was first treated with DNase for 30 min at 37°C and heat-inactivated in the presence of 2.5 mM EDTA for 10 min at 65°C. This was followed by reverse transcription using a RevertAid cDNA synthesis kit (MBI Fermentas Inc., Burlington, ON, Canada) and oligo(dT). Briefly, the RNA was incubated with 0.5 μg oligo(dT) for 5 min at 70°C followed by addition of reaction buffer (50 mM Tris-HCl, pH 8.3, 50 mM KCl, 4 mM MgCl2, and 10 mM DTT), 20 U of ribonuclease inhibitor, and 200 μM dNTPs. The reaction was incubated for 5 min at 37°C before the addition of 200 U of RevertAid H Minus Moloney murine leukemia virus reverse transcriptase (RT) and incubation for 60 min at 42°C. Of this 20-μl RT reaction, 3 μl was subjected to PCR amplification (94°C for 30 s, 56°C for 30 s, and 72°C for 45 s; 35 cycles) with the primers listed in Table 1. A no-RT control in which a mock reaction was carried out in the absence of RT (to control for genomic DNA contamination) and a no-template control with RT to account for DNA contamination in the reagents were performed in parallel. In addition, β-actin primers spanning two introns were used as an additional genomic DNA control and to serve as a positive control. PCR amplification reactions with the NAT1, NAT2, and β-actin primers were all run in parallel using the same cDNA sample.
Preparation of Tissue Cytosols. Knockout (n = 6; three male mice, three female mice) and wild-type (n = 6; three male mice, three female mice) siblings (11–15 weeks) were sacrificed by cervical dislocation. Individual tissues were immediately removed, trimmed, snap-frozen in liquid nitrogen, and stored at -80°C. Tissues were homogenized on ice in 10 volumes of lysis buffer (10 mM triethanolamine-HCl, pH 7.0, 1 mM EDTA, 1 mM DTT, 50 mM KCl, 100 μM phenylmethylsulfonyl fluoride, and 100 μM leupeptin) with a Polytron PT 1200C homogenizer (Kinematica AG, Littau/Lucerne, Switzerland) on setting 5 for two 20-s bursts. The homogenate was centrifuged at 4°C for 60 min at 100,000g. Aliquots of the supernatant were snap-frozen and stored at -80°C until used for NAT activity assays. Protein concentrations were determined using a Bio-Rad protein assay dye reagent (Bio-Rad Laboratories Ltd., Mississauga, ON, Canada), with bovine serum albumin as a standard.
Determination of in Vitro NAT Enzyme Activity. NAT activity for PAS N-acetylation was determined using various tissue cytosols. Initial rates for the cytosolic fractions were performed in duplicate with 0.1 mM PAS, 0.1 mM acetyl-CoA, and 20 μl of regenerating system. The regenerating system consisted of 5 mM acetyl-dl-carnitine and 1 U of carnitine acetyltransferase per milliliter of assay buffer (250 mM triethanolamine-HCl, 5 mM EDTA, and 5 mM DTT, pH 7.5). Reactions were preincubated for 3 min at 37°C and initiated with the addition of lysate (50 μg). The reactions (100 μl) were carried out for 10 min at 37°C and terminated by the addition of 10 μl of 15% perchloric acid. After precipitation of the denatured protein, the supernatant fractions were assayed for the N-acetylated product (AcPAS) by HPLC using a Shimadzu isocratic system (Shimadzu Scientific Instruments Inc., Columbia, MD) and a reversed-phase Beckman Ultrasphere ODS 5 μM column (Beckman Instruments Inc., Fullerton, CA). A flow rate of 2 ml/min with a mobile phase consisting of 7% acetonitrile/1% acetic acid/0.1% triethylamine and an ultraviolet detector setting of 270 nM were used.
For kinetic determinations, PAS (1–50 μM), 0.1 mM acetyl-CoA, and 20 μl of regenerating system were incubated in duplicate with liver or kidney cytosol (2–5 μg of total protein) as described above. Apparent Km and Vmax values were determined using nonlinear regression of the standard Michaelis-Menten kinetic equation with the computer program KaleidaGraph (Synergy Software, Reading, PA). Statistical analyses were performed using a Student's t test with the computer program Prism (GraphPad Software Inc., San Diego, CA).
Drug Administration and Plasma Elimination Kinetics. PAS (50 mg/kg) or SMZ (50 mg/kg) dissolved in saline was administered by i.p. injection to 7- to 9-week-old age- and sex-matched Nat1/2(-/-) mice (n = 6; three male mice, three female mice) and wild-type sibs (n = 6; three male mice, three female mice). Blood samples were drawn from the saphenous vein using heparinized Microvettes (Sarstedt Inc., Montreal, QC, Canada) at four different time-points (15, 30, 60, and 90 min for PAS; 2, 6, 22, 24 h for SMZ) and centrifuged to separate plasma. Plasma samples were diluted 1:50 in 50 μl of HPLC mobile phase and analyzed for parent and acetylated metabolites by HPLC as described above for the in vitro assays. For SMZ and AcSMZ, a mobile phase consisting of 88% sodium perchlorate buffer/12% acetonitrile and an ultraviolet detector setting of 254 nM were used. Blank plasma samples were spiked with known amounts of PAS and AcPAS or SMZ and AcSMZ to quantify the amount of parent and acetylated products present in the samples. Area under the curve (AUC) values were determined by the trapezoidal rule using GraphPad Prism.
Results
Generation of Nat1/2(-/-) Mice. The simultaneous targeted disruption of both the mouse Nat1 and Nat2 genes was successfully achieved by insertion of the neomycin resistance (neo) gene and resultant deletion of ∼11 kb of sequence, including the entire coding region of the Nat1 gene and most of the coding region of the Nat2 gene (Fig. 1A). The two genes were simultaneously ablated by virtue of their relatively close proximity (∼9 kb) within the BAC clone (Fig. 1B). Homologous recombinant clones with the correct targeting event were identified by Southern blot analysis (Fig. 1B) using a probe derived from the 3′ flanking region. A correctly targeted ES clone was distinguished by the presence of a 7.3-kb band versus 23 kb for a wild-type clone. Similarly, mice possessing the targeted allele were identified by a band of 7.3 kb, whereas a wild-type allele was evident by the presence of a 23-kb band (Fig. 1C). One ES cell line was isolated from 134 colonies and used to generate a mouse line. A total of 24 male chimeric mice, distinguished by agouti coat color carried by the R1 ES cell genome, were generated from two aggregation experiments. Of these, 12 male chimeras were bred, with 8 showing germline transmission.
Chimeric male mice showing germline transmission were subsequently bred with C57BL/6 mice. Heterozygous F1 mice (+/-) displayed normal viability and fertility and were intercrossed to produce the homozygous mutants. Mice homozygous for the disrupted alleles were born normally and seemed indistinguishable (appearance, mortality rate, behavior, reproductive capacity) from their wild-type littermates. No change in the sex ratio of Nat1/2(-/-) mice was noted, with 12 male and 10 female null mice born out of 79 F2 offspring. In addition, there seemed to be no in utero lethality of the homozygous null mice as determined by their birth frequency of about 28% in litters from heterozygote crosses (+/+, 19 offspring; +/-, 38 offspring; -/-, 22 offspring), and gross pathological examination revealed no apparent abnormalities.
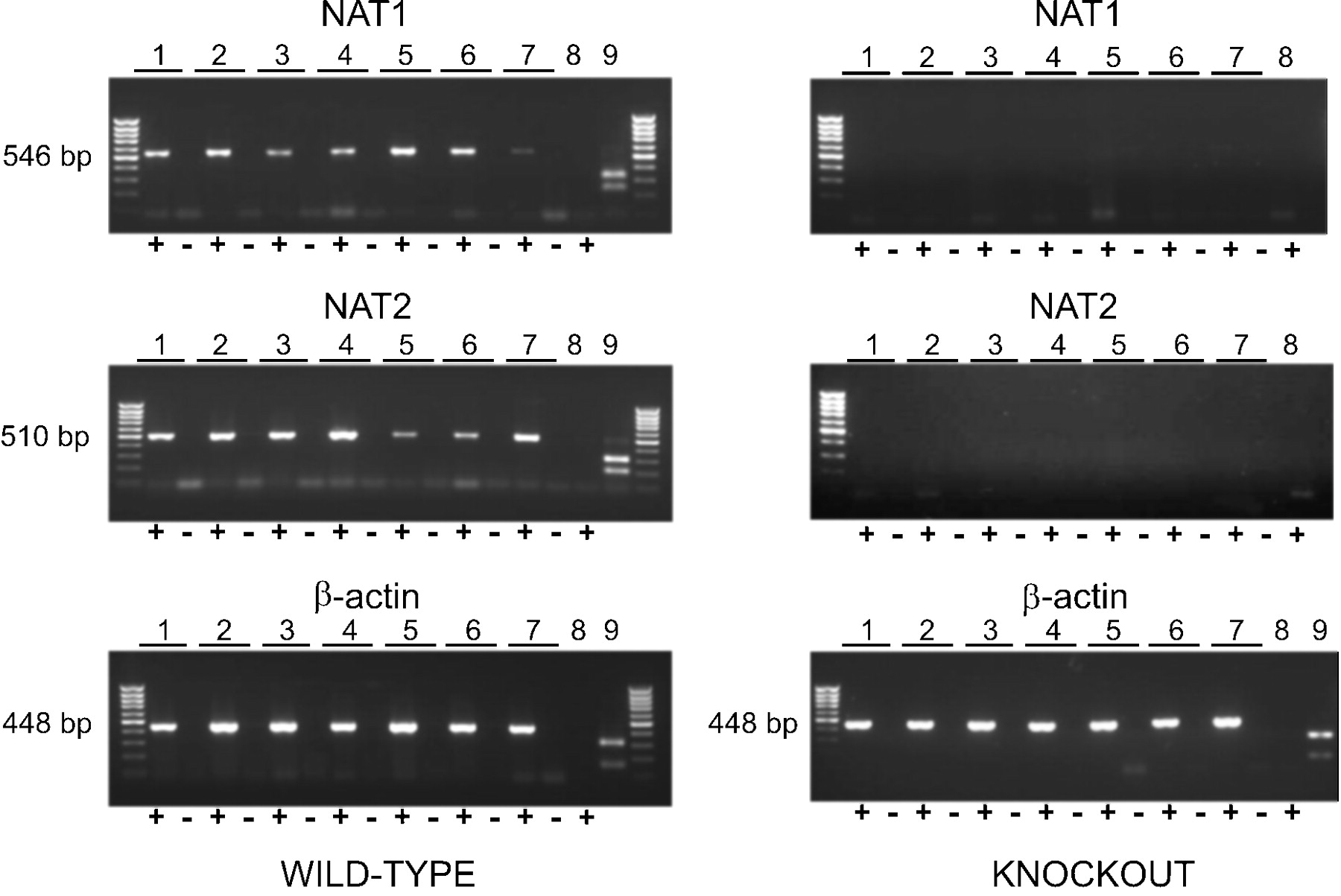
NAT1 and NAT2 Expression in Wild-Type and Nat1/2(-/-) Mice. RNA isolated from several tissues from both wild-type and Nat1/2(-/-) mice was subjected to a highly sensitive RT-PCR assay to confirm the absence of gene expression. RT-PCR confirmed the absence of both NAT1 and NAT2 transcripts in the Nat1/2(-/-) animals (Fig. 2). In wild-type animals, NAT2 expression was present in all tissues assayed, confirming the widespread distribution pattern of NAT2. A somewhat lower expression of NAT2 was present in brain samples (both cortex and cerebellum) compared with peripheral samples. NAT1 expression was also found in all tissues assayed, and lower amounts of NAT1 transcript were consistently found in bladder and spleen samples. Restriction digests of the products verified the specificity of the PCR products. Digestion of the NAT1 product with BglII, the NAT2 product with PstI and the β-actin product with BglII yielded the appropriately sized fragments (Fig. 2).
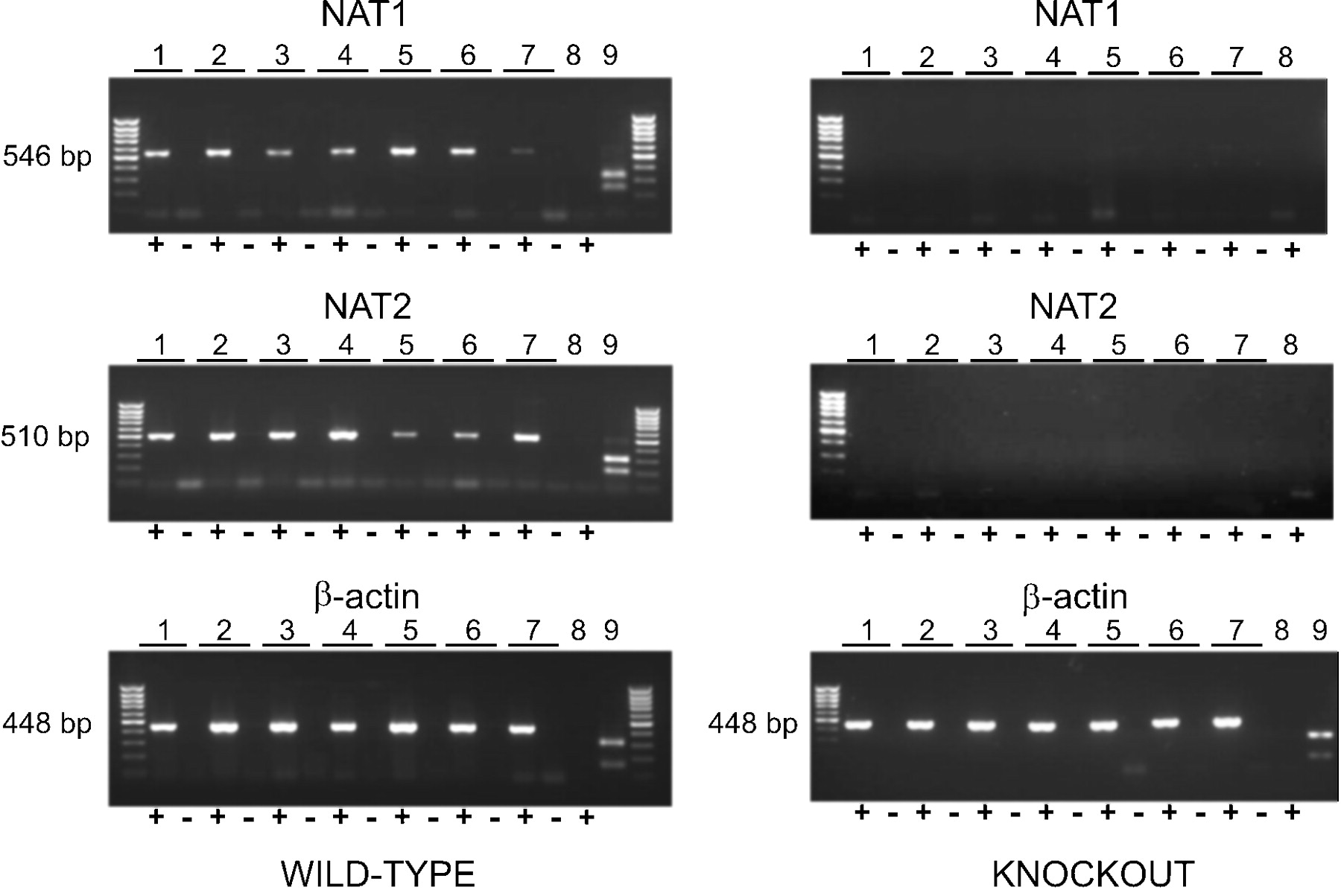
RT-PCR detection of NAT1, NAT2, and β-actin transcripts in tissues from representative wild-type mice (left) and Nat1/2(-/-) mice (right). Lane pairs: 1, liver; 2, kidney; 3, colon; 4, bladder; 5, cortex; 6, cerebellum; 7, spleen. Lane 8 is a diethyl pyrocarbonate-treated water negative control. Lane 9 is a diagnostic restriction digest with BglII for NAT1 and β-actin and PstI for NAT2. In each of the lane pairs, 1 to 8, the `+' lane is a PCR reaction after reverse transcription, whereas the `-' lane is a negative control in which PCR was performed after a mock reverse transcription reaction in which RT was omitted, to control for false positive signals caused by genomic DNA contamination.
In Vitro NAT Enzyme Activity.Nat1/2(-/-) mice displayed no measurable cytosolic PAS-NAT activity in either liver or kidney (<0.03 nmol/min/mg), whereas their wild-type siblings displayed high levels of NAT activity at least 60- to 180-fold above this detection limit in these tissues (Fig. 3). Similarly, no PAS-NAT activity could be detected using Nat1/2(-/-) tissue cytosols prepared from colon, spleen, and cortex, whereas activities in wild-type animals (mean ± S.E.M., n = 2) were 2.3 ± 0.1 nmol/min/mg for colon, 2.6 ± 0.02 nmol/min/mg for spleen, and 0.4 ± 0.01 nmol/min/mg for cortex.
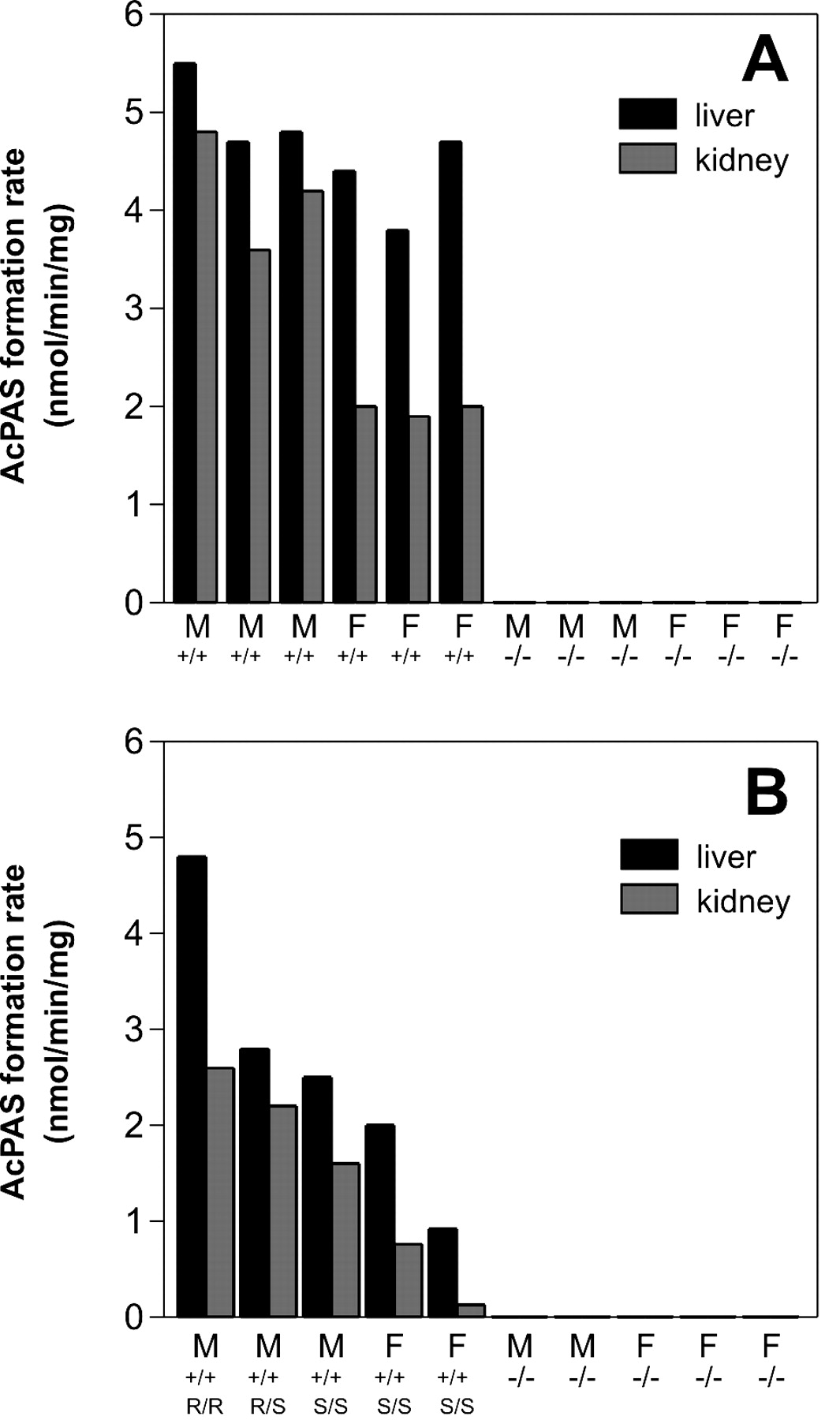
Cytosolic PAS N-acetylation rates from wild-type and Nat1/2(-/-) mice. A, B6/129 genetic background. B, CD-1/129 genetic background. Liver and kidney cytosols from wild-type and Nat1/2(-/-) mice were assayed for NAT activity with 0.1 mM PAS in the presence of 0.1 mM acetyl-CoA as described under Materials and Methods. AcPAS was separated and quantified by HPLC. Product formation rates from individual wild-type and knockout mice are shown, and these represent the mean of duplicate determinations, which varied by less than 5%. Limit of detection for AcPAS formation under these assay conditions was 0.03 nmol/min/mg. M, male; F, female; +/+, wild-type; -/-, knockout; R, rapid Nat2*8 allele; S, slow Nat2*9 allele.
A gender difference in acetylation was noted in the B6/129 F2 wild-type mice in kidney cytosol, with male kidney cytosol displaying a 2-fold higher acetylation rate (p < 0.001) than female kidney (Fig. 3A). The apparent Km and Vmax values for PAS acetylation using liver and kidney cytosol are listed in Table 2 for the wild-type male and female B6/129 F2 mice. The overall average Km was 4.0 ± 0.14 μM, whereas the average Vmax was 5.7 ± 1.0 nmol/min/mg protein for liver cytosols.
Kinetic constants for liver and kidney cytosolic PAS-NAT activity from wild-type B6/129 mice
Liver and kidney cytosols were incubated at 37° C with an acetyl-CoA regenerating system, 0.1 mM acetyl-CoA, and concentrations of PAS ranging from 1 to 50 μM for 10 min as described under Materials and Methods. The values represent the mean ± S.D. from three male or female wild-type B6/129 F2 mice. Apparent Km and Vmax values were determined using nonlinear regression of the standard Michaelis-Menten kinetic equation with the computer program KaleidaGraph (Synergy Software).
CD-1/129 F2 wild-type mice displayed a wider variation in activity (Fig. 3B). This is because of the outbred background of the CD-1 mice and the presence of both rapid (*8) and slow (*9) Nat2 alleles in CD-1 mice (Estrada et al., 2000). Genotyping of the F2 wild-type CD-1/129 mice for the presence of the Nat2*8 and Nat2*9 allelic variants indicated the presence of homozygous rapid, heterozygous, and homozygous slow mice, even among the small sample size tested. This genetic variation is reflected in their respective activities. The CD-1/129 mice genotyped by MseI RFLP analysis to be homozygous for the naturally occurring slow acetylator (Nat2*9) allele displayed generally lower PAS-NAT activities for both liver and kidney cytosol. However, even these homozygous `slow acetylator' mice displayed much higher PAS acetylation capacities than their corresponding Nat1/2(-/-) siblings, in which no PAS-NAT activity could be measured.
In Vivo NAT Activity and Plasma Pharmacokinetics. In accord with the in vitro results from tissue cytosol preparations, Nat1/2(-/-) mice displayed no ability to metabolize PAS upon its in vivo administration. Wild-type male and female mice (Fig. 4, A and C) showed roughly equivalent amounts of PAS and AcPAS in plasma by 15 min after PAS administration and similar elimination profiles for the parent PAS and its metabolite. By 60 min, virtually all PAS and AcPAS were eliminated from plasma in wild-type mice. In the Nat1/2(-/-) mice, there was a complete absence of AcPAS in the plasma samples (Fig. 4, B and D) and a 3-fold increased AUC for the parent compound (Table 3). Mean PAS AUC values for wild-type (n = 6) and knockout (n = 6) mice were 5.3 ± 2.7 and 15.7 ± 3.6 mmol min/l, respectively (p < 0.001). The mean AcPAS AUC for all wild-type mice (n = 6) was 6.95 ± 3.2 mmol min/l, whereas AUC values could not be determined for the Nat1/2(-/-) mice because AcPAS concentrations were below the limit of assay detection. Urinary excretion of AcPAS accounted for up to 80% of the total administered dose of PAS in wild-type mice but was undetectable in urine collected from Nat1/2(-/-) mice (data not shown).
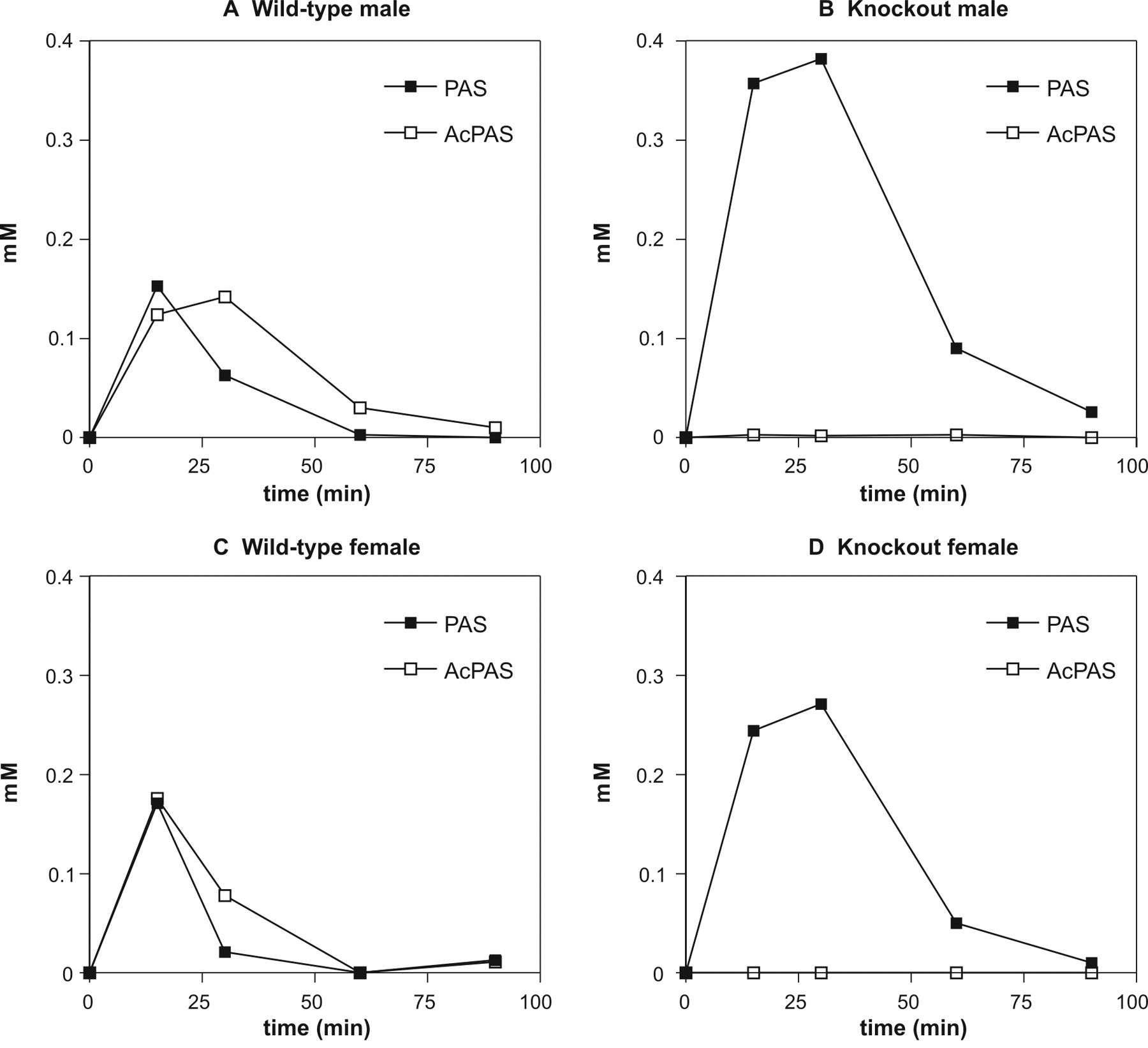
Plasma pharmacokinetics of PAS and AcPAS in B6/129 wild-type and Nat1/2(-/-) mice. A, plasma kinetics of PAS and AcPAS from a representative wild-type male mouse. AUC values for PAS and AcPAS were 3.8 and 6.1 mmol min/l. B, plasma kinetics of PAS and AcPAS from a representative knockout male mouse. AUC for PAS was 17.0 mmol min/l. C, plasma kinetics of PAS and AcPAS from a representative wild-type female mouse. AUC values for PAS and AcPAS were 3.2 and 4.6 mmol min/l, respectively. D, plasma kinetics of PAS and AcPAS from a representative knockout female mouse. AUC for PAS was 11.4 mmol min/l. For all mice, PAS (50 mg/kg) was administered by i.p. injection, and four sequential blood samples were drawn from the saphenous vein of each mouse at the indicated time points. Plasma was isolated from each blood sample, diluted 1:50, and analyzed for PAS and AcPAS by HPLC. The limit of detection for AcPAS was 0.008 nmol or 8 μM. AUC values were determined by the trapezoidal rule using the Prism software.
AUC values for PAS and SMZ elimination
AUC values (mean ± S.D., n = 3) were determined by the trapezoidal rule using Prism software. PAS or SMZ (50 mg/kg) was dosed by i.p. injection to age- and sex-matched wild-type and knockout mice. Four sequential blood samples were drawn and analyzed for parent and metabolite compounds as described under Materials and Methods.
To verify the absence of NAT1 activity, SMZ was used as a selective substrate. In contrast to PAS, clearance of SMZ was much lower, as reflected by the larger SMZ AUC values (Table 3). In addition, SMZ clearance was less dependent on the formation of its acetylated metabolite as determined by the smaller AUC values for AcSMZ compared with SMZ in the wild-type animals (Fig. 5, A and C; Table 3). Mean SMZ AUC values for all wild-type (n = 6) and knockout (n = 6) mice were 388 ± 93 and 547 ± 166 mmol min/l. Most notably, however, the knockout animals displayed a complete absence of AcSMZ in plasma (Fig. 5, B and D), thereby confirming the abolition of functional NAT1 activity. The mean AcSMZ AUC for all wild-type mice (n = 6) was 59 ± 21 mmol min/l. Urinary excretion of AcSMZ accounted for approximately 40 to 55% of the administered dose within the first 6 h in wild-type mice, but could not be detected in the urine from the knockout animals (data not shown).

Plasma pharmacokinetics of SMZ and AcSMZ in B6/129 wild-type and Nat1/2(-/-) mice. A, plasma kinetics of SMZ and AcSMZ from a representative wild-type male mouse. AUC values for SMZ and AcSMZ were 416 and 89 mmol/min/l. B, plasma kinetics of SMZ and AcSMZ from a representative knockout male mouse. AUC for SMZ was 744 mmol/min/l. C, plasma kinetics of SMZ and AcSMZ from a representative wild-type female mouse. AUC values for SMZ and AcSMZ were 546 and 57 mmol/min/l. D, plasma kinetics of SMZ and AcSMA from a representative knockout female mouse. SMZ AUC was 767 mmol/min/l. For all mice, SMZ (50 mg/kg) was administered by i.p. injection and four sequential blood samples were drawn from the saphenous vein at 2, 6, 22, and 24 h. Plasma was isolated from each blood sample, diluted 1:50, and analyzed for SMZ and AcSMZ by HPLC. The limit of detection for AcSMZ was 0.003 nmol or 3 μM. AUC values were determined by the trapezoidal rule using Prism software.
Discussion
We have generated Nat1/Nat2 double knockout mice using gene targeting. Despite the presence of both Nat1 and Nat2 genes early in mouse development (Mitchell et al., 1999) and the presence of NAT2 transcript in preimplantation ES cells (Payton et al., 1999), our Nat1/2(-/-) mice seemed normal, viable, and fertile. It has been suggested that the murine NAT2 may be important during development, given the finding that this enzyme catalyzes the acetylation of the folate catabolite p-aminobenzoyl-l-glutamate and that NAT2 is expressed early in neuronal development (Stanley et al., 1998; Payton et al., 1999). Our results indicate that NATs are not essential for normal development or physiological homeostasis, but this is not an unexpected finding. Naturally occurring polymorphisms in humans exist that render NAT1, the functional equivalent to mouse NAT2, severely impaired (Hughes et al., 1998; Lin et al., 1998), and both canids and felids have been shown to lack one or more NATs (Trepanier et al., 1997, 1998). Furthermore, it has been suggested that the existence of polymorphisms would indicate that a drug metabolizing enzyme is not essential for development or physiology (Gonzalez and Kimura, 2003). In contrast, a well conserved drug metabolizing enzyme with no known functional polymorphisms and/or one that is capable of metabolizing endogenous hormones may be important for development or physiological homeostasis.
It has been proposed that most if not all cytochrome P450 enzymes seem to act on many endogenous substrates (Nebert and Russell, 2002). Nevertheless, deleterious phenotypes have not been associated with any of the cytochrome P450 knockouts (Cyp1a1, Cyp1a2, Cyp1b1, Cyp2e1) or the microsomal epoxide hydrolase knockout (Gonzalez and Kimura, 2003), although knockout of the microsomal NADPH-cytochrome P450 oxidoreductase gene did result in embryonic lethality (Shen et al., 2002). The nondeleterious knockout animals, however, have served as useful models to study the mechanisms of drug metabolism, chemical toxicity and carcinogenesis, and putative endogenous roles not previously realized. Potential endogenous functions have been alluded to, particularly for the Cyp1a2 gene, whereby its ablation leads to an up- and down-regulation of several mouse genes involved in cell-cycle regulation and lipid metabolism (Smith et al., 2003).
RT-PCR analysis established that neither NAT1 nor NAT2 is present in our Nat1/2(-/-) mice, whereas both transcripts displayed a widespread expression pattern in wild-type animals. The NAT2 distribution pattern in mice has previously been characterized by immunohistochemistry (Stanley et al., 1997) and PABA acetylation (Chung et al., 1993), but less is known about the NAT1 tissue-specific expression profile. Northern blot analysis and isoniazid acetylation have confirmed the presence of NAT1 in liver and colon (Levy et al., 1993); however, one recent RT-PCR study found NAT1 only in liver (Boukouvala et al., 2002). Because our RT-PCR method is only semiquantitative at best, further investigation using quantitative RT-PCR or other methods is necessary to determine more accurately the relative amounts of NAT1 and NAT2 present in various mouse tissues.
To determine whether Nat1/2(-/-) mice can metabolize PAS, both in vitro activity assays using tissue cytosol preparations and in vivo elimination studies were performed. PAS-NAT activity assays with various tissue cytosols verified a complete lack of activity for this substrate, and AcPAS could not be detected from the plasma of Nat1/2(-/-) mice after administration of PAS. Although the relative substrate selectivity of mouse NAT2 for PAS most strongly indicates an absence of functional NAT2 in our null mice, recombinant mouse NAT1 does exhibit low but measurable catalytic activity for PAS (data not shown).
To confirm the absence of functional NAT1 activity, in vivo elimination studies were undertaken with the NAT1-selective substrate SMZ. Consistent with our results with PAS, the acetylated metabolite of SMZ was absent in the plasma of our knockout animals. Thus, a complete lack of measurable activity in the Nat1/2(-/-) mice is consistent with our DNA and RNA evidence, suggesting a complete ablation of both of these mouse genes and their expression. Furthermore, the complete absence of activity in our Nat1/2(-/-) mice—in which the Nat3 gene is still intact—suggests that PAS and SMZ are not substrates for mouse NAT3. Preliminary experiments in our laboratory with Nat3 knockout mice suggest that there is no difference between wild-type and Nat3(-/-) mice in the plasma elimination of PAS or SMZ (data not shown). Thus, the Nat1/2(-/-) mice may indeed prove useful in testing arylamine compounds for their ability to be metabolized by NAT3, because studies to date have relied upon recombinant in vitro expression of mouse NAT3 and have observed only marginal levels of function with the substrates tested so far. As such, only low levels of 2-aminofluorene, anisidine, and 5-aminosalicylate acetylation have been observed with recombinant NAT3 (Kelly and Sim, 1994; Estrada-Rodgers et al., 1998).
A gender difference was observed in the wild-type mice with respect to kidney PAS-NAT activity, with male mice displaying 2-fold higher activity. This is in agreement with previous observations using PABA as a substrate (Smolen et al., 1993; Estrada et al., 2000). The higher activity in male kidney has been shown to be modulated by testosterone (Smolen et al., 1993). Whether this higher activity in kidney results in gender-related differences in arylamine adduct levels and a differential susceptibility to arylamine-induced carcinogenesis remains to be investigated. Gender differences have been observed with respect to formation of 2-aminofluorene-DNA adducts in the bladder (Levy and Weber, 1992) and incidence of 4-aminobiphenyl-induced bladder cancers (Schieferstein et al., 1985).
The use of knockout animals lacking enzymes of xenobiotic biotransformation has confirmed their involvement in specific drug-induced toxicities. In the absence of metabolic activation, Cyp2e1 null mice were found to be more resistant to acetaminophen- (Lee et al., 1996), benzene- (Valentine et al., 1996), and carbon tetrachloride- (Wong et al., 1998) induced toxicities, whereas Cyp1b1 and microsomal epoxide hydrolase null mice were more resistant to 7,12-dimethylben-z[a]anthracene-induced toxicity (Buters et al., 1999; Miyata et al., 1999). Interestingly, studies with Cyp1a2 null mice have found no difference in 4-aminobiphenyl-induced carcinomas between wild-type and null mice (Kimura et al., 1999), an augmentation in 4-aminobiphenyl–induced methemoglobinemia in null mice (Shertzer et al., 2002), and little or no change in 2-amino-3-methylimidazo[4,5-f]quinoline adduct levels in the colon and mammary gland, depending on the dose (Snyderwine et al., 2002). These studies all indicate that other enzymes, particularly extrahepatic enzymes, may be involved in the activation process in a tissue-specific fashion, despite the fact that CYP1A2 has been implicated as a primary candidate in the hepatic metabolic activation of environmental carcinogens, including arylamines and heterocyclic amines (reviewed in Eaton et al., 1995; Windmill et al., 1997). The identity of the enzyme(s) involved in the activation process not mediated by CYP1A2 remains to be determined. These results have revealed that studies using in vitro systems as predictive factors must be interpreted with caution, and that some chemicals can be activated by multiple enzymes, perhaps in a tissue-specific manner.
Genetically engineered mouse models are useful for the detection of mutagenic and carcinogenic events and as bioassays for predicting carcinogenicity (Sills et al., 2001). These models, encompassing some of the major participants in the carcinogenesis process, including oncogene activation, tumor suppressor gene inactivation and defects in repair enzymes, are useful in facilitating the extrapolation of animal carcinogenicity studies to human risk assessment. A positive concordance for human carcinogens has been shown, and mechanistic or mode of action information has been revealed using controlled exposure studies with Big Blue transgenic mice, a LacI reporter model (Sills et al., 2001). Our model system with a deficient NAT drug metabolism should provide further insight for investigations into environmental toxicity and carcinogenesis. More importantly, the generation of such a model should help clarify the role that the NATs may play either in the metabolic activation or detoxification of arylamine procarcinogens. Although a human NAT2 prostate-specific transgenic mouse (Leff et al., 1999) displayed no difference in 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine-DNA adducts in the prostate, it was postulated that endogenous mouse NATs may have masked any effect. Our Nat1/2(-/-) model can serve as a starting point for developing more appropriate models to predict human risk and toxicity, and can serve as a background strain for the creation of specific human NAT knock-in models. In addition, crosses with other knockout models should help reveal the interplay between multiple enzyme systems and toxic end-points.
Acknowledgments
We thank Xiaoli Lu and Jean Huang for valuable and excellent technical assistance.
Footnotes
-
This work was supported by a research grant (to D.G. and R.R.) from the National Cancer Institute of Canada, and by a postdoctoral fellowship (to K.S.) from the Canadian Institutes for Health Research.
-
ABBREVIATIONS: NAT, N-acetyltransferase; SMZ, sulfamethazine; PAS, p-aminosalicylic acid; PABA, p-aminobenzoic acid; ES, embryonic stem; AcPAS, N-acetyl-PAS; AcSMZ, N-acetyl-SMZ; kb, kilobase(s); PCR, polymerase chain reaction; SSC, standard saline citrate; DTT, dithiothreitol; HPLC, high-performance liquid chromatography; AUC, area under the curve; RT, reverse transcriptase/transcription; BAC, bacterial artificial chromosome.
-
↵1 Present address: Division of Pediatric Clinical Pharmacology and Medical Toxicology, Children's Mercy Hospital and Clinics, Kansas City, MO 64108.
-
↵2 Present address: Amgen Inc., Thousand Oaks CA 91320-1799.
-
↵3 Present address: Department of Cellular and Molecular Medicine, University of Ottawa, Ottawa, ON K1H 8M5 Canada.
- Received January 31, 2003.
- Accepted April 9, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}