


Abstract
Phenobarbital (PB) alters expression of numerous hepatic genes, including genes involved in xenobiotic metabolism. Phenobarbital-dependent induction of cytochrome P-450 2B1 (CYP2B1) is subject to regulation by cytokines [e.g., by epidermal growth factor (EGF)], hormones [e.g., by growth hormone (GH)], or the cellular redox status. To investigate mechanisms involved in regulation of CYP2B1 transcription, we performed promoter activation studies using primary rat hepatocyte cultures transiently transfected with individual CYP2B1 promoter-luciferase reporter gene constructs. The 2679-bp native 5′-flanking region of the CYP2B1 gene conferred reporter gene activation by PB and the potent PB-like inducer permethrin (PM). Furthermore, this region mediated EGF- and GH-dependent repression of gene activation by PB-like inducers. A wide promoter mapping strategy with constructs bearing internal CYP2B1 promoter deletions led to identification of a distal responsive CYP2B1 enhancer region at -2230 to -2170, encompassing the section equivalent to the 51-bp PB-responsive enhancer module situated in the distal mouse Cyp2b10-5′-flanking region. The distal CYP2B1 enhancer region conferred gene activation by PM, repression of PM-dependent activation by EGF, and enhancement of activation by the antioxidant N-acetylcysteine (NAC). Mutational analyses of the region at -2230 to -2170 suggested that the mechanisms of PB-dependent induction of CYP2B1 and the modulating effects by EGF or NAC are closely related.
Hepatic biotransformation plays a key role in elimination and detoxification of foreign compounds (xenobiotics). Many foreign compounds are capable of inducing their own metabolism and/or metabolism of other xenobiotics, which is considered an adaptive response to xenobiotic exposure. Induction of hepatic drug metabolism by barbiturates [e.g., by phenobarbital (PB)] was described more than 40 years ago (Remmer, 1958). The spectrum of hepatic enzymes subject to PB-dependent regulation of expression includes members of the cytochrome P-450 (P-450) subfamilies CYP2A, CYP2B, CYP2C, and CYP3A, and other xenobiotic-metabolizing enzymes, such as UDP-glucuronosyltransferase isoforms (Sueyoshi and Negishi, 2001). In addition, genes not involved in xenobiotic metabolism are regulated by PB (Ueda et al., 2002). An array of lipophilic compounds is known to lead to a comparable alteration in hepatic gene expression as PB. These compounds [e.g., environmental pollutants such as polychlorinated biphenyls (Waxman and Azaroff, 1992) or pesticides such as dichlorodiphenyltrichloroethane (popularly known as DDT) or the pyrethroid permethrin (Heder et al., 2001)], are referred to as “phenobarbital-like” inducers.
Transcriptional activation plays a major role in PB-dependent induction of enzymes belonging to the CYP2B subfamily (Atchison and Adesnik, 1983). Recently, several laboratories have demonstrated the relevance of a phenobarbital-responsive enhancer situated in the distal 5′-region flanking the rat CYP2B1/2 genes (Trottier et al., 1995; Park et al., 1996; Smirlis et al., 2001), the human CYP2B6 gene (Sueyoshi et al., 1999) or the mouse Cyp2b10 gene (Honkakoski and Negishi, 1997). Analyzing the murine Cyp2b10-5′-flanking region, Negishi and coworkers delimited this enhancer to a 51-bp region [the phenobarbital-responsive enhancer module (PBREM)], which is highly homologous to corresponding CYP2B1/2 and CYP2B6 sequences (Sueyoshi et al., 1999; Sueyoshi and Negishi, 2001). The PBREM encompasses a nuclear factor 1-(NF1-) binding site flanked by two nuclear receptor-binding sites (NR1, NR2). A heterodimer consisting of the constitutive active (androstane) receptor (CAR) and the retinoid X receptor (RXR) has been found to bind to the NR1 site (Honkakoski et al., 1998), and CAR has been shown to be crucial for induction of murine Cyp2b10 by PB (Wei et al., 2000). Nevertheless, any efforts to demonstrate a direct binding of PB to the transcription factor CAR have failed (Tzameli et al., 2000).
In addition to adaptive induction in response to xenobiotics, expression of several xenobiotic-metabolizing P-450 isoforms is regulated by endogenous mediators. During liver regeneration (Klinger and Karge, 1987) or hepatocarcinogenesis (Chen et al., 1993), reduced expression of cytochrome P-450 enzymes has been observed. Likewise, expression levels of various P-450 isoforms have been shown to be decreased during infection or inflammation (Morgan, 2001). Endogenous factors leading to diminished PB-dependent inducibility of CYP2B genes have been identified. In particular, hormones such as growth hormone (GH; Schuetz et al., 1990) or the thyroid hormones T3 and T4 (Murayama et al., 1991), but also cytokines such as the epidermal growth factor (EGF; Aubrecht et al., 1995; Kietzmann et al., 1999) or tumor necrosis factor-α (Milosevic et al., 1999) have been found to repress phenobarbital-dependent CYP2B induction. Former studies using primary hepatocyte cultures suggest that EGF, by repressing CYP2B1 induction primarily under periportal conditions, acts as a major determinant of zonated (predominantly perivenous) CYP2B1 induction in the rat liver (Kietzmann et al., 1999). Recently, PB-dependent activation of the rat CYP2B1 promoter has been shown to be repressed by reactive oxygen species (ROS) and enhanced by antioxidants [e.g., by the glutathione precursor N-acetylcysteine (NAC), indicating the relevance of the cellular redox status in regulating CYP2B1 induction (Hirsch-Ernst et al., 2001)]. Proinflammatory cytokines as well as EGF may lead to ROS production via physiological signal transduction (Thannickal and Fanburg, 2000). Nevertheless, it is unclear to what extent different signal transduction pathways separately mediate PB-dependent induction and modulation by cytokines, hormones, or the cellular redox status.
To study molecular mechanisms of modulation of CYP2B1 induction, we employed a system of primary rat hepatocyte cultures in which inducibility of CYP2B1 expression by PB, a hepatocyte-specific function lost in mammalian hepatoma cell lines, was retained. Promoter activation analyses were performed with hepatocyte cultures transiently transfected with CYP2B1 promoter-reporter gene constructs. Applying a wide promoter-mapping strategy covering approximately 2.7 kb of the CYP2B1-5′-flanking region, reporter gene activation by PB-like inducers as well as inhibition by EGF and enhancement by NAC of activation were found to be associated with a distal enhancer region upstream of -2155. Specification of the regulatory region and mutational analysis in heterologous promoter-reporter gene constructs demonstrated that EGF- and NAC-dependent modulation of CYP2B1 induction converged with respect to a region comparable with the mouse Cyp2b10 PBREM.
Materials and Methods
Materials. All chemicals were of reagent grade and purchased from commercial suppliers. Collagenase was obtained from Biochrom KG Seromed (Berlin, Germany) and fetal calf serum from PAA (Coelbe, Germany). Cell culture dishes were purchased from Nunc (Wiesbaden, Germany). DNA-modifying enzymes were supplied by MBI-Fermentas (Vilnius, Lithuania). Expand Long Template DNA-polymerase, T4-polynucleotide kinase, and epidermal growth factor isolated from mouse submaxillary glands were from Roche Applied Science (Mannheim, Germany). Human recombinant GH was purchased from Bachem (Heidelberg, Germany). [γ-32P]ATP was from Amersham Biosciences (Freiburg, Germany). The transfection reagent Effectene was provided by QIAGEN (Hilden, Germany). The pCR-XL-TOPO vector was purchased from Invitrogen (Groningen, The Netherlands). Firefly luciferase expression plasmids (pGL3-Basic and pGL3-SV40-Promoter), the Renilla reniformis luciferase expression plasmid (pRL-CMV) as well as the dual luciferase reporter assay kit were obtained from Promega (Mannheim, Germany).
Generation of CYP2B1 Promoter Reporter Gene Plasmids. A fragment representing approximately 2.7 kb of the native 5′ sequence flanking the rat CYP2B1 gene was amplified by PCR from genomic DNA isolated from Wistar rat hepatocytes using primers corresponding to sites of the promoter sequence published by Shaw et al. (1996) (GenBank accession number U30327.1; bases -2648 to -2623 and +29 to +4, relative to the transcription start site, respectively). To facilitate later cloning, the primers contained an additional 5′-NheI recognition site and a three-base overhang (Table 1). The obtained PCR fragment was ligated into the pCR-XL-TOPO vector (Invitrogen), and Escherichia coli were transformed with the resulting plasmid. After sequence analysis of three individual clone inserts, slight sequence differences in our sequence obtained from the Wistar rat strain were consistently detected compared with the original CYP2B1 promoter sequence obtained from Sprague-Dawley rat (GenBank accession number U30327.1). The Wistar CYP2B1 promoter sequence (submitted to EMBL under accession no. AJ320166.1) was chosen as the basis for further experiments. After digestion of the verified pCR-XL-clone with NheI, the reobtained CYP2B1 promoter fragment was ligated into the NheI site of the pGL3-Basic luciferase expression vector (Promega). The sequence of the resulting CYP2B1 promoter firefly luciferase construct, designated pGL3C2B1, was validated by sequence analysis employing standard sequencing primers of the pGL3-Basic plasmid and internal primers of the insert.
Primer sequences Restriction sites (NheI or EcoRI) are underlined. The numbering of the CYP2B1 promoter primers is in accordance with the promoter sequence deposited under EMBL accession number AJ320166.1.
Generation of CYP2B1 Promoter Deletion Constructs. Deletion constructs bearing successive internal deletions (from 180 to 472 bp in length) of the CYP2B1 promoter were generated by PCR. Using pGL3C2B1 as a template, primer pairs flanking the sequence to be deleted and containing an additional EcoRI recognition site were employed. The orientation of the primers was adjusted to allow complete plasmid amplification with exception of the sequence section to be deleted (Table 1). After digestion of the PCR product with EcoRI, the linear construct was closed to a circular plasmid using T4 DNA ligase (MBI-Fermentas). The sequences of the resulting deletion constructs designated as pGL3CD2 to pGL3CD8 were validated by sequence analyses. The mutated construct pGL3CM2 was generated similarly using the heterologous construct pGL3CS2 as a template.
Generation of Heterologous CYP2B1-SV40 Promoter Constructs. To obtain a heterologous construct in which a proximal SV40 promoter was regulated by a distal fragment of the CYP2B1 promoter containing the rat PBREM region, the pGL3CD7 plasmid was digested with BamHI and EcoRI. The resulting 263-bp fragment (representing the Wistar CYP2B1 promoter sequence from bp -2415 to -2157 and additional 4 bases of the introduced EcoRI site) was treated with T4 DNA polymerase (MBI-Fermentas) to yield blunt ends and further cloned into the SmaI site of the pGL3-SV40-promoter vector (Promega), yielding the construct pGL3CS1. The constructs pGL3CS2, pGL3CS5, pGL3CM3, pGL3CM4 and pGL3CM5 were obtained by ligation of appropriate oligonucleotides (Table 2) into the Acc65I/BglII sites of the pGL3-SV40-Promoter vector.
Comparison of the different CYP2B1 promoter sequences used in heterologous SV40-promoter-luciferase constructs The NR1 element is bold; mutations are lower case. For sticky-end ligation into the pGL3-SV40-promoter vector (Acc65I/BglII sites), double-stranded oligonucleotides with additional four-base, sticky-end overhangs were used.
Hepatocyte Isolation and Culture. Primary hepatocytes were isolated from adult male Wistar rats (180-220 g body weight) by collagenase perfusion (Seglen, 1976). Hepatocyte suspensions showed viabilities >90% as determined by trypan blue exclusion. Cells were plated onto culture dishes at a density of 9 × 104 cells/cm2 in MX-82 medium (Hoffmann et al., 1989) supplemented with 10% fetal calf serum. After an initial attachment period of 3 h at 37°C in a humidified atmosphere of 10% CO2 and 90% air, the medium was replaced with serum-free MX-83 medium that lacked arginine, but contained 1 μM insulin and 20 μM hydrocortisone hemisuccinate. The cells were further cultured (37°C, 10% CO2/90% air) for up to 3 days. Transient transfection and treatment with CYP2B1-inducers (1.5 mM PB or 50 μM permethrin, PM) and/or modulators of induction such as EGF (16 nM), GH (9.6 nM), the catalase inhibitor 3-amino-1,2,4-triazole (AT; 2 mM) or the antioxidant NAC (10 mM) were performed as indicated below. The medium was exchanged daily.
Hepatocyte Transfection and Luciferase Reporter Gene Assay. Primary adherent rat hepatocyte cultures, plated onto six-well plates (Nunc), were transiently transfected 24 h after seeding with 0.5 μg/well of one of the firefly luciferase constructs (pGL3-derivatives) and 0.03 μg/well of the R. reniformis luciferase construct pRL-CMV using the Effectene reagent (QIAGEN) according to the manufacturer's instructions. Six hours later, the medium was replaced with fresh MX-83 medium containing inducers of CYP2B1 expression or modulators of induction as indicated. After 48 h of treatment (including one medium exchange), hepatocytes were lysed with 150 μl of passive lysis buffer (Promega), and firefly and R. reniformis luciferase activities were measured in 20 μl of cell lysate using the dual luciferase reporter assay kit (Promega) and a Lumat LB 9501 luminometer (Berthold Technologies, Bad Wildbad, Germany). Firefly luciferase activity was normalized according to R. reniformis luciferase activity in the same sample (“relative luciferase activity”). Student's t test for unpaired values was applied in statistical analyses.
Preparation of Nuclear Extracts and Electrophoretic Mobility Shift Assays. Nuclear extracts from 107 cultured hepatocytes or 100 mg of liver tissue were prepared according to the method of Schreiber et al., 1989. The electrophoretic mobility shift assay binding reaction (30 min, room temperature) was performed with 0.5 to 20 μg of nuclear protein extract, 5 × 105 cpm of 32P-end-labeled, double-stranded NR1-oligonucleotide probe (Honkakoski et al., 1998), and 1 μg of the unspecific competitor poly(dI·dC) in 20 μl of assay buffer, pH 7.6, containing 20 mM Tris, 5 mM HEPES, 100 mM NaCl, 50 mM KCl, 2 mM EDTA, 2 mM MgCl2, 1 mM dithiothreitol, and 15% (v/v) glycerol. In competition experiments, a 200-fold molar excess of unlabeled double-stranded NR1 oligonucleotide was added before the addition of the nuclear extract. To resolve the nuclear proteins, the incubation mixture was subjected to electrophoresis through a nondenaturating polyacrylamide gel (8%). Oligonucleotide-protein-complexes were visualized using the BAS 1500 Bio-Imaging Analyzer (Fujix, Tokyo, Japan).
Results
EGF and GH Repress CYP2B1 Promoter Activation by PB-Like Inducers in Primary Hepatocytes. Former studies have demonstrated that transcriptional activation largely contributes to CYP2B1 induction by PB-type inducers (Atchison and Adesnik, 1983; Heder et al., 2001; Hirsch-Ernst et al., 2001). To investigate the possible effect of the inhibitors of CYP2B1 induction EGF and GH on CYP2B1 promoter activation, we used an established model system of transiently transfected primary rat hepatocyte cultures (Heder et al., 2001; Hirsch-Ernst et al., 2001) in which inducibility of CYP2B1 expression by PB was retained. The plasmid pGL3C2B1 which contained the firefly luciferase reporter gene under the control of approximately 2.7 kb of the native 5′-flanking region of the CYP2B1 gene was employed as the basis for further promoter mapping experiments. Slight promoter sequence differences observed compared with the originally published CYP2B1-5′-flanking sequence (Shaw et al., 1996; GenBank accession number U30327.1) were detected; therefore, a modified entry was submitted (EMBL accession no. AJ320166.1), and numbering of the promoter sequence was re-adjusted. Primary hepatocytes cotransfected with the promoter reporter gene constructs pGL3C2B1 and pRL-CMV (expressing R. reniformis luciferase) were subsequently treated with the potent phenobarbital-like inducer PM (50 μM; Heder et al., 2001), PB (1.5 mM) alone or with a phenobarbital-like inducer in combination with the cytokine EGF (16 nM) or GH (9.6 nM) for 48 h. Firefly luciferase activity was determined in hepatocyte lysates as a measure of promoter activation, and R. reniformis luciferase activity served as an internal standard of transfection efficiency in the same samples.
In cells transfected with the recipient vector for CYP2B1 promoter fragments (pGL3-Basic) alone, basal luciferase expression was not inducible by PB or PM (data not shown). In hepatocytes transfected with the pGL3C2B1 construct (bearing the native 2679 kb CYP2B1-5′-flanking region), significant activation of luciferase expression by PM or PB was observed (Fig. 1). EGF repressed PB-dependent gene activation via the native CYP2B1 promoter (Fig. 1). Accordingly, EGF and GH also significantly decreased PM-dependent CYP2B1 promoter activation, whereas EGF or GH alone had no effect on basal promoter activation in the absence of an inducer (Fig. 1). These results on repression by EGF of CYP2B1 promoter activation complement previous studies concerning EGF-dependent repression of CYP2B1 induction by PB on the mRNA level (Aubrecht et al., 1995; Kietzmann et al., 1999), and indicate that repression of transcriptional activation plays a major role in EGF-dependent modulation of CYP2B1 induction. Furthermore, the repression by GH of CYP2B1 promoter activation is in agreement with a former study according to which GH inhibited PB-induced CYP2B1/2 mRNA expression on the transcriptional level. (Schuetz et al., 1990).

CYP2B1 promoter activation by PB or PM and modulation by EGF or GH. Primary rat hepatocytes cultured for 1 day were transiently cotransfected with the CYP2B1 promoter luciferase-reporter gene construct pGL3C2B1 (containing approximately 2.7 kb of the 5′-flanking CYP2B1 promoter region) and the R. reniformis luciferase construct pRL-CMV. Six hours after transfection, cells were incubated with 1.5 mM PB or 50 μM PM and/or 16 nM EGF or 9.6 nM GH, as indicated, for an additional 48 h. Firefly and R. reniformis luciferase activities were subsequently determined in cell lysates as described under Materials and Methods. Relative luciferase activity (firefly luciferase activity, standardized according to R. reniformis luciferase activity) is shown as a measure of promoter activation. Data represent mean values of five parallel culture wells ± S.D. of experiments representative for three independent hepatocyte preparations. Mean control values in the absence of inducer or modulator of induction (C) were set to 1. Significant induction (p < 0.05) compared with control cells (C). *, significant (p < 0.05) modulation of induced promoter activity; ⋄↓ decreased induction.
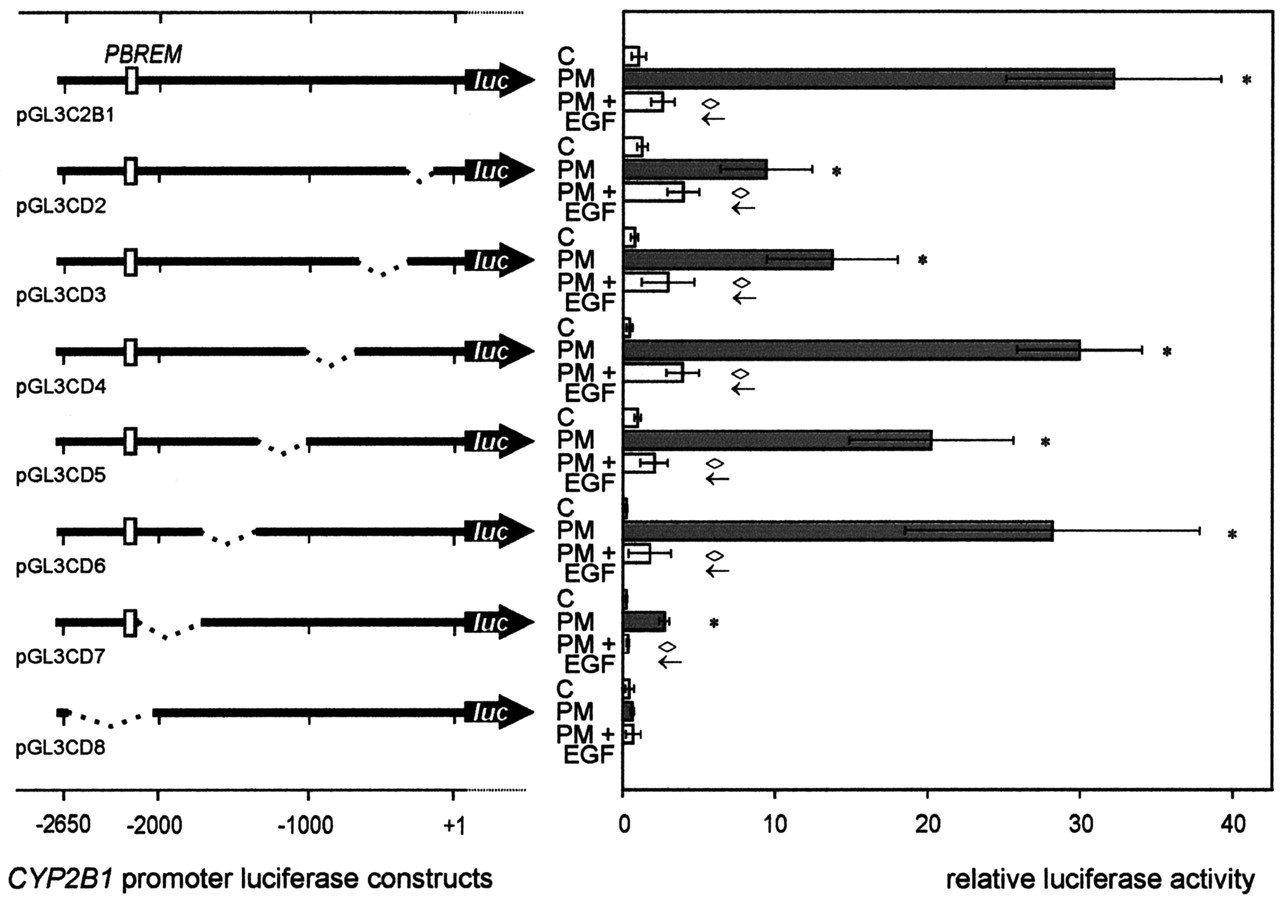
CYP2B1 promoter mapping was performed to define possible regions responsive to CYP2B1 inducers and to modulators of induction. The major phenobarbital-responsive region corresponding to the previously characterized murine PBREM (Honkakoski et al., 1998) was expected to be located upstream of -2000 bp. A promoter deletion strategy was implemented to confirm the role of a distal PB-responsive enhancer and to examine whether further responsive promoter regions might be involved in conferring modulation of induction. A set of seven reporter gene constructs was generated comprising successive internal deletions (Fig. 2) within the CYP2B1 promoter sequence, thus allowing both the detection of potential proximal elements by deletion experiments and simultaneous retention of PB- or PM-dependent induction. Promoter screening was performed using PM as the inducer and EGF as an inhibitor of induction. Although the extent of induction and inhibition was influenced by the sequence deletions, in all but one (pGL3CD8) of the tested CYP2B1 promoter constructs, both activation by PM and modulation by EGF were retained (Fig. 2). Consequently, internal deletions in the promoter range of -2157 to -175 revealed no essential EGF-responsive region, which might be responsible for conferring the inhibitory effect on PM-dependent CYP2B1 promoter activation. Deletion of the distal promoter region from bp -2626 to -2156 (pGL3CD8) led to a complete loss of PM-dependent promoter activation (Fig. 2). The loss of inducibility by PM in the pGL3CD8 construct is in accordance with the loss of the expected distal PB-responsive region encompassing the PBREM.

CYP2B1 promoter mapping employing constructs with successive internal promoter deletions. Primary rat hepatocytes cultured for 1 day were transiently cotransfected with individual CYP2B1 promoter-luciferase-reporter gene constructs (pGL3C2B1, pGL3CD2 to pGL3CD8) and the R. reniformis luciferase construct pRLCMV (for standardization). Six hours after transfection, cells were incubated with 50 μM PM or with both 50 μm PM and 16 nM EGF for additional 48 h. Firefly and R. reniformis luciferase activities were subsequently determined in cell lysates as described under Materials and Methods. Relative luciferase activity (firefly luciferase activity, standardized according to R. reniformis luciferase activity) is shown as a measure of promoter activation. Data represent mean values of five parallel culture wells ± S.D. of experiment representative for three independent hepatocyte preparations. Mean standardized basal luciferase activity (C) in cells transfected with the pGL3C2B1 construct was set to 1. *, significant induction (p < 0.05) compared with control cells cultured in the absence of inducer or modulator of induction (C); significant (p < 0.05) modulation of induced promoter activity: ⋄←, decreased induction.
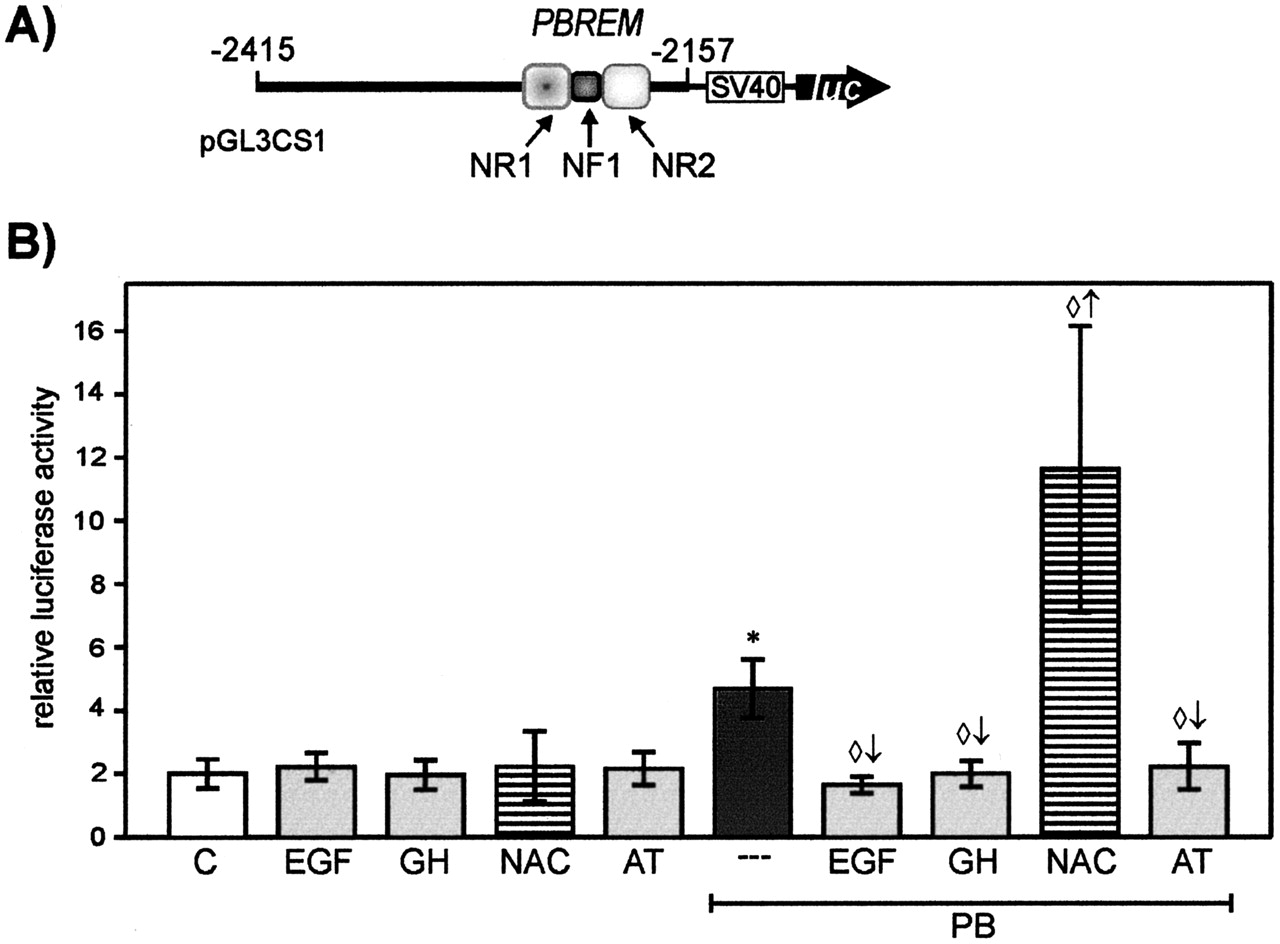
The Distal CYP2B1-PBREM Region Plays a Central Role Not Only for Gene Activation by PB-Like Inducers, but Also for Modulation of Induction. To further determine the relevance of the isolated distal promoter region for modulation of PB-dependent induction, we employed heterologous promoter-reporter gene constructs expressing the luciferase gene under control of a distal part of the CYP2B1-5′-flanking region and the proximal SV40 promoter. Experiments with hepatocytes transfected with the pGL3-SV40 promoter-luciferase vector alone (without the CYP2B1 promoter insert) revealed that the SV40 promoter was insensitive to PB-type inducers or to EGF (data not shown). We initially analyzed the activity of the CYP2B1-5′-distal section between -2415 and -2157 (pGL3CS1; Fig. 3A), which represents a large segment of the deleted region in the inactive pGL3CD8 construct. This distal region in the heterologous pGL3CS1 context was capable of conferring promoter activation by PB and PM as well as the inhibitory effects elicited by EGF and GH, respectively (Figs. 3 and 4), in a manner comparable with the native CYP2B1 promoter (pGL3C2B1; Fig. 1). Furthermore, as we have shown previously in studies demonstrating regulation of CYP2B1 gene activation by the cellular redox status (Hirsch-Ernst et al., 2001), the pGL3CS1 construct conferred enhancement of PB- or PM-dependent reporter gene activation by NAC and repression by the catalase inhibitor AT (Figs. 3 and 4). Thus, promoter activation experiments with the heterologous promoter construct pGL3CS1 substantiated the results of the CYP2B1 promoter deletion analyses and underlined the particular relevance of this distal promoter region (-2415 to -2157) in mediating both CYP2B1 induction and modulation of induction by EGF, GH, AT, and NAC.

Activation of the distal CYP2B1 promoter region (-2415/-2157) by PB and modulation by EGF, GH, or modulators of the cellular redox status (AT or NAC). Primary rat hepatocytes cultured for 1 day were transiently cotransfected with the heterologous CYP2B1 promoter-luciferase-reporter gene construct pGL3CS1 [containing 259 bp of the CYP2B1 promoter followed by the proximal SV40-promoter (A)], and the R. reniformis luciferase construct pRL-CMV. Six hours after transfection, cells were incubated with or without 1.5 mM PB in the presence or absence of 16 nM EGF, 9.6 nM GH, 10 mM NAC, or 2 mM AT for an additional 48 h. Firefly and R. reniformis luciferase activities were subsequently determined in cell lysates as described under Materials and Methods. Relative luciferase activity (firefly luciferase activity, standardized according to R. reniformis luciferase activity) is shown as a measure of promoter activation. Data represent mean values of five parallel culture wells ± S.D. of a representative experiment. Mean standardized basal luciferase activity in cells transfected with the pGL3C2B1 construct was set to 1. *, significant induction (p < 0.05) compared with control cells transfected with pGL3CS1 (C); significant (p < 0.05) modulation of induced promoter activity: ⋄↑, enhanced induction; ⋄↓, decreased induction.

Dissection of the distal CYP2B1 promoter region (-2415/-2157). Primary rat hepatocytes cultured for 1 day were transiently cotransfected with individual heterologous CYP2B1 promoter luciferase-reporter gene constructs (pGL3CS1, pGL3CS2, pGL3CS5, and pGL3CM2 to pGL3CM5) and the R. reniformis luciferase construct pRLCMV. Six hours after transfection, cells were incubated with 50 μM PM in the presence or absence of 16 nM EGF, 10 mM NAC, or 2 mM AT for an additional 48 h. Firefly and R. reniformis luciferase activities were subsequently determined in cell lysates as described under Materials and Methods. Relative luciferase activity (firefly luciferase activity, standardized according to R. reniformis luciferase activity) is shown as a measure of promoter activation. Data represent mean values of five parallel culture wells ± S.D. of experiment representative for three independent hepatocyte preparations. Mean standardized basal luciferase activity in cells transfected with the pGL3C2B1 construct was set to 1. *, significant induction (p < 0.05) compared with cells cultured in the absence of inducer or inhibitor of induction (C); significant (p < 0.05) modulation of induced promoter activity: ⋄→, enhanced induction; ⋄←, decreased induction.
As this distal region contained a sequence very much related to the PBREM of the murine Cyp2b10 promoter, we reduced the distal CYP2B1 promoter region to a 61-bp section (from -2230 to -2170) encompassing the 51-bp PBREM-like sequence (pGL3CS2). To elucidate whether one single regulatory element within the PBREM might be responsible for both induction and modulation of induction or different elements within the PBREM might be essential, several promoter constructs were employed (Fig. 4) that contained different mutations (pGL3CM2-5) or variations (pGL3CS5) of the transcription factor binding sites described previously (NR1, NF1, NR2) within the PBREM. Transfection experiments were performed using PM as the inducer and EGF, AT, and NAC as modulators. The 61-bp sequence encompassing the PBREM (pGL3CS2) still conferred significant activation by PM, which was repressed by EGF and enhanced by NAC (Fig. 4). In contrast, AT did not markedly inhibit gene activation via the 61-bp sequence. Further analyses of this distal promoter region using mutated PBREM sequences (bearing SpeI or EcoRI consensus sequences as mutations) demonstrated that mutation and thus inactivation of one of the three elements within the PBREM resulted in a diminished inducibility by PM compared with pGL3CS1-transfected cultures. PM-dependent reporter gene activation was observed with all constructs bearing mutated PBREM variants, although activation by PM did not reach statistical significance from the construct pGL3CM2 (containing a mutated NR1 element bearing an EcoRI consensus sequence). However, from a construct encompassing the same mutated NR1 element (bearing an EcoRI sequence) in the pGL3CS1 context, significant reporter gene activation by PM was observed (data not shown), indicating that this NR1 mutation per se did not fully abolish responsiveness to PM. Similarly, the inhibitory EGF effect was observed in all mutated PBREM variants, reaching statistical significance with two tested PBREM variants (containing a mutated NR2 site or an NR1 site disrupted by an SpeI site). In contrast, a significant inhibitory effect of AT on PM-dependent induction was not observed for any of the tested constructs containing PBREM analogs. The enhancement by NAC was retained in all cases in which significant induction by PM was observed, indicating that the mechanism of induction by PM is closely related to the mechanism of enhancement by NAC.
The NR1 region is the most conserved element within PBREM-like regions of CYP2B promoters of different mammalian species (Sueyoshi and Negishi, 2001), indicating a pivotal role of the NR1 region in PBREM-dependent gene regulation. We thus examined whether the NR1 element alone might mediate gene activation and modulation by EGF or NAC. Because only one NR1 element inserted upstream of the SV40 promoter (within the pGL3-SV40-promoter vector) was not markedly responsive to PM (data not shown), we employed a triple NR1 construct (pGL3CS5) that contained three concatenated NR1 elements upstream of the SV40 promoter. This artificial enhancer was able to confer distinct inducibility by PM and, furthermore, a marked enhancement of induction by NAC. EGF and AT seemed to repress PM-dependent reporter gene activation from the pGL3CS5 construct to an extent similar to that from the pGL3CS1 construct. Because of variability in the data, however, these effects did not reach statistical significance (Fig. 4). Thus, the triple NR1 element in the heterologous promoter context seems to be sufficient for gene activation by PM and for the intensifying effect of NAC.
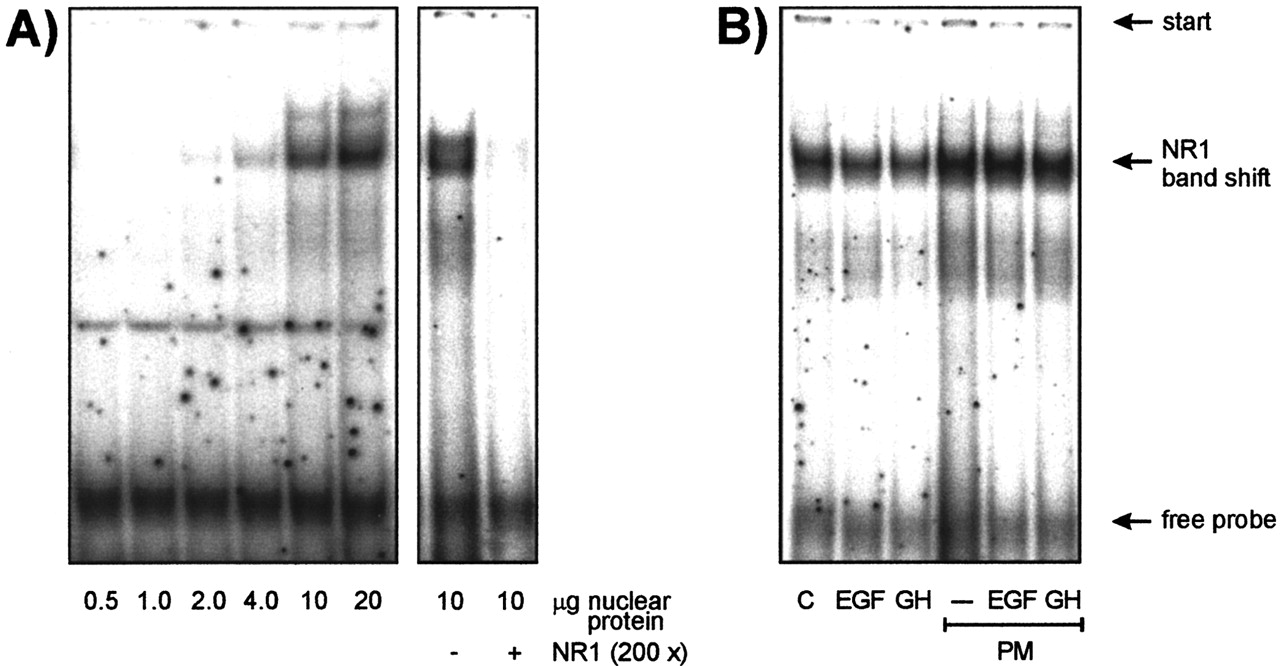
Analysis of Protein Binding to the NR1 Element in Lysates of Hepatocytes Treated with PM as Opposed to PM and EGF/GH. To further examine the role of the NR1 element within the PBREM-like region, electrophoretic mobility shift assays (EMSAs) were performed using nuclear protein extracts prepared from rat liver or primary hepatocyte cultures. Hepatocyte culture conditions were comparable with those used for transfection experiments. A specific DNA-protein interaction was observed that was prevented in the presence of an excess amount of unlabeled NR1 oligonucleotide (Fig. 5A). Interestingly, a substantial specific DNA-protein interaction was found already with extracts of uninduced control hepatocyte cultures (Fig. 5B, lane C). When employing nuclear extracts originating from cells cultured in the presence of PM together with EGF or GH, no marked differences in the band shift pattern compared with nuclear extracts from cells cultured with PM alone were observed (Fig. 5B).

Binding of nuclear protein extracts to the NR1 element. A, rats were treated for 48 h with PB (with a daily dose of 80 mg of PB/kg of weight, i.p., for 2 days) and nuclear protein extracts were prepared from liver as described under Materials and Methods. Binding of nuclear proteins to the NR1 element of the PBREM-like CYP2B1 promoter region was examined with the use of electrophoretic mobility shift assays using radioactively labeled, double-stranded NR1 oligonucleotides. Unbound DNA and protein-DNA complexes were separated through a native 8% polyacrylamide gel. Specificity of the observed DNA-protein interaction was demonstrated by using a 200-fold molar excess of the unlabeled NR1 oligonucleotide. B, nuclear protein extracts were prepared from primary rat hepatocytes. After a preculture for 24 h, hepatocytes had been further cultured in the presence or absence of 50 μM PM with or without 16 nM EGF or 9.6 nM GH for 48 h (compare with Fig. 1). Nuclear protein (10 μg/binding reaction/lane) was applied.
Discussion
Induction of xenobiotic-metabolizing enzymes by foreign compounds is considered to be a major cellular defense mechanism. However, mechanisms of repression of induction exist that prevent excessive enzyme overexpression under (patho)-physiological conditions. To investigate by which mechanisms modulators of PB-dependent CYP2B1 induction might take effect, we first performed a systematic screening of approximately 2.7 kb of the 5′-region flanking the CYP2B1 gene to identify potential regulatory enhancer/promoter regions responsive to PB-like inducers. Using a set of reporter gene constructs bearing different internal promoter deletions, we demonstrated that apart from the known PB-responsive region upstream of -2155, no other major regions responsive to PB-like inducers are located within the CYP2B1 promoter section of -2157 to -175. This result is in line with a recent study in which the absence of responsiveness to PB was reported for a reporter plasmid containing 2151 nucleotides of the CYP2B1-5′-flanking region (Kocarek and Mercer-Haines, 2002).
Subsequent mutational analyses of the PBREM-like region at -2222/-2172 involving variation of one of the sites NR2, NF1, or NR1 (within pGL3CM3) are in accordance with a former dissection of the rat CYP2B1/2 PBREM-like region (Liu et al., 2001), in that reporter gene activation by an inducer was not completely abolished by mutation of a single site within the module. Thus, it seems that different elements are involved in the response to PB-like inducers and that within the module, nonmutated elements may compensate for a single mutated element. On the other hand, a triple NR1 site in the heterologous context was responsive to PM, which is in agreement with other studies using a multiple NR1 site as an artificial enhancer (Sueyoshi and Negishi, 1999; Muangmoonchai et al., 2001).
Considering the relevance of the distal PBREM region in gene activation by PB-like inducers, the question was raised whether an additional promoter/enhancer element might be involved in EGF-dependent inhibition of induction. Several putative binding sites of EGF-regulated transcription factors (e.g., signal transducer and activator of transcription or activator protein 1) have been described within the analyzed 2679 kb-CYP2B1 promoter section (Shaw et al., 1996). However, the inhibitory effect of EGF on induced promoter activity was not abolished by successive internal promoter deletions without eliminating PM-dependent inducibility as well. Accordingly, using only a small distal section (-2415/-2157) of the CYP2B1-5′-flanking region, both responsiveness to PB-like inducers and EGF-dependent repression of activation were retained. Finally, detailed mutational analysis of the region from -2230 to -2170 (containing the PBREM region from -2222 to -2172) provided evidence that separate, additional promoter regions, such as more proximal putative activator protein 1 or signal transducer and activator of transcription binding sites of the CYP2B1 promoter, are not required for EGF-dependent inhibition of induction. Apart from EGF, GH has been characterized as an inhibitor of CYP2B1 induction in hepatocytes (Schuetz et al., 1990). Responsiveness of the native CYP2B1 promoter in the pGL3C2B1 context to GH and the sensitivity of the -2415 to -2157 region in the heterologous promoter context support the conclusion that a distal enhancer region is also responsible for conferring GH-dependent repression of CYP2B1 induction. Cross-talk between the GH receptor and the EGF receptor has been described previously [e.g., induction of EGF receptor tyrosine phosphorylation by stimulation of rat adipocytes with GH (Yamauchi et al., 1997)]. Further studies are required to clarify to what extent GH- and EGF-dependent signal transduction pathways converge with respect to repression of CYP2B1 induction.
The nuclear receptor CAR seems to be indispensable for PB-dependent activation of the murine Cyp2b10 gene, because CAR knock-out mice exhibited a complete loss of PB-dependent Cyp2b10 inducibility (Wei et al., 2000; Ueda et al., 2002). In mice, nuclear translocation and accumulation of CAR were found to be crucial for PB-dependent Cyp2b10 induction and to be associated with increased protein binding to the NR1 element in EMSA experiments (Kawamoto et al., 1999). Evidence has been provided that the NR1-binding transcription factors represent a CAR/RXR nuclear receptor heterodimer (Honkakoski et al., 1998). Nuclear accumulation of CAR seems not to be the only important step required for PB-dependent induction. In primary mouse hepatocytes, treatment with the Ca2+/calmodulin-dependent protein kinase (CaMK) inhibitors KN-62 and KN-93, respectively, led to suppression of PB-dependent Cyp2b10 induction, although CAR translocation into the nuclei was observed. Thus, a second (phosphorylation-dependent) step seems to be required for Cyp2b10 gene activation by PB in the mouse system (Sueyoshi and Negishi, 2001). Inhibition of PB-dependent CYP2B1 induction by KN-62 has also been demonstrated for primary rat hepatocyte cultures (Joannard et al., 2000), indicating that CaMK-dependent phosphorylation of key target proteins might play a crucial role in CYP2B gene activation in the rat system as well.
EMSA experiments performed in the present study demonstrated a specific NR1-protein interaction for extracts of uninduced cultured rat hepatocytes. Furthermore, treatment of cells with EGF or GH in the presence of PM did not result in a marked alteration in the NR1-mobility shift pattern compared with the pattern observed with extracts of cells treated with PM alone. Provided that the rat transcription factors bound to the NR1 region represent a CAR/RXR heterodimer (for which evidence exists: Muangmoonchai et al., 2001) these results suggest that, in the rat system, CAR/RXR binds to the NR1 site already in the absence of inducers. This is in accordance with a constitutive NR1 band shift observed with extracts of uninduced rat liver (Muangmoonchai et al., 2001). Furthermore, our data suggest that CAR translocation to the nucleus in the presence of PM is not modified by EGF or GH and indicate that under treatment with PM and EGF or PM and GH, CAR is not modified to a form that can no longer bind to the NR1 element. These EMSA experiments, however, do not exclude the idea that EGF or GH might influence the transactivation ability of NR1-bound transcription factors. Coregulator proteins regulate the transactivation ability of nuclear receptor proteins. Coactivators, recruited by nuclear receptor proteins, interact with both the activation function domains of nuclear receptor proteins and the basal transcription machinery, supporting nuclear receptor-dependent transcriptional activation (Robyr et al., 2000). Activation of a reporter gene driven by an enhancer/promoter comprising a distal rat CYP2B1 promoter region (encompassing the PBREM) and the proximal SV40 promoter has been shown to be stimulated by coexpression of CAR and the steroid receptor coactivator SRC-1 (Muangmoonchai et al., 2001). Further research is required to clarify whether CaMK-dependent phosphorylation of nuclear receptor transcription factors themselves or their coregulators might play a pivotal role in PB-dependent CYP2B induction. Interactions between CaMK- and growth factor-dependent pathways have been described previously (Joannard et al., 2000). Thus, PB-like inducers and modulators of CYP2B1 induction might influence gene activation by leading to an alteration in the phosphorylation status of key target proteins, which, however, remain to be revealed in detail.
In addition to the mechanism of inhibition of CYP2B1 induction by EGF, we examined the role of the cellular redox status in CYP2B1 activation. As reported previously (Hirsch-Ernst et al., 2001), the inhibitory effect of the catalase inhibitor AT and the enhancing effect of the antioxidant NAC on PB-dependent gene activation were conferred by the distal CYP2B1 promoter section from -2415 to -2157. The present study demonstrates that on further limitation of the promoter region to a 61-bp segment containing the PBREM/PBREM variants or to a triple NR1 site, enhancement by NAC of PM-dependent promoter activation was retained significantly, whereas the inhibitory effect of AT was variable. Several modes of redox-sensitive regulation may be hypothesized. Transcription factors essential in mediating PB-dependent CYP2B1 induction could be redox-sensitive. Members of the nuclear receptor superfamily have been shown to be regulated by critical thiol residues. For instance, the oxidation of the zinc finger 2 in the estrogen receptor DNA-binding domain prevents dimerization and DNA binding (Whittal et al., 2000). Interestingly, a thiol residue within the NF1 transactivation domain seems to be involved in redoxsensitive regulation of CYP1A1 induction by aryl hydrocarbons (Barouki and Morel, 2001). However, the results of the present study suggest that the NF1 binding site within the PBREM is not essential for conferring redox-dependent regulation of CYP2B1-induction; rather, it seems that the NR1 element is sufficient at least for providing enhancement of PM-dependent gene activation in the presence of NAC.
Besides direct regulation of transcription factors by the cellular redox status, an indirect mechanism remains possible in which the activities of transcription factors, coactivators/corepressors, or upstream regulators are modulated indirectly by their phosphorylation status (Thannickal and Fanburg, 2000). H2O2 has been identified as an effective modulator of the mitogen-activated protein kinase pathway (Guyton et al., 1996), potentially acting via inducing hyperphosphorylation and thus activation of the EGF receptor (Kamata et al., 2000). Because treatment with EGF has been found to lead to an increase in intracellular ROS (e.g., in NIH 3T3 cells; Sundaresan et al., 1996), ROS are expected to play an intrinsic role in EGF-dependent signal transduction.
In summary, the present data support the conclusion that molecular mechanisms leading to CYP2B1 induction by PB-type inducers on the one hand and to modulation by EGF or NAC of CYP2B1 induction on the other hand converge with respect to a distal CYP2B1 enhancer region.
Acknowledgments
We thank C. Schmitz-Salue, S. Blume, and G. Rüdell for excellent technical assistance.
Footnotes
-
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (SFB 402, TP A2) and by a Friedrich-Ebert-Stiftung fellowship (to D.B.).
-
ABBREVIATIONS: PB, phenobarbital; bp, base pair(s); PBREM, phenobarbital responsive enhancer module; PM, permethrin; NF, nuclear factor; NR, nuclear receptor; CAR, constitutive active receptor; RXR, retinoid X receptor; P-450, cytochrome P-450; GH, growth hormone; EGF, epidermal growth factor; ROS, reactive oxygen species; NAC, N-acetyl-l-cysteine; kb, kilobase(s); CMV, cytomegalovirus; SV40; PCR; AT, 3-amino-1,2,4-triazole; EMSA, electrophoretic mobility shift assay; CaMK, Ca2+/calmodulin-dependent protein kinase; SRC-1, steroid receptor cofactor-1.
-
↵1 Current address: Preclinical Safety, Novartis Pharma AG, 4002 Basel, Switzerland.
- Received June 27, 2003.
- Accepted October 6, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}