


Abstract
We have reported that the protein-protein interaction between UDP-glucuronosyltransferase (UGT) 2B7 and cytochrome P450 3A4 (CYP3A4) alters UGT2B7 function. However, the domain(s) involved in the interaction are largely unknown. To address this issue, we examined in more detail the CYP3A4-UGT2B7 association by means of immunoprecipitation, overlay assay, and cross-linking involving 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide. Purified CYP3A4 or glutathione transferase (GST)-tagged CYP3A4 was cross-linked to UGT2B7 in solubilized baculosomes. The formation of the cross-linked complex was detected by immunoblotting using both antibodies against CYP3A4 and UGTs. Although the GST-tagged CYP3A4 containing the region ranging from Tyr25 to Ala503 was cross-linked to UGT2B7, the same did not occur when another construct containing Met145 to His267 was used. This observation was consistent with the result of the overlay assay indicating that CYP3A4 lacking the N-terminal hydrophobic segment retains the ability to associate with UGT2B7, whereas the Met145-to-His267 region loses this capacity. Although the Met145-to-His267 peptide was recognized by one anti-CYP3A4 antibody that has the ability to coimmunoprecipitate UGT2B7, it was not recognized by another antibody incapable of coimmunoprecipitating UGT2B7. The epitope of the latter antibody was mapped to the Leu331-to-Lys342 region, which is located on the J-helix of CYP3A4. Taken together, the results obtained suggest that 1) CYP3A4 and UGT2B7 are a pair of enzymes in proximity to each other and 2) either the Leu331-to-Lys342 domain or the surrounding region plays a role in the interaction with UGT2B7, whereas the hydrophobic Met145-to-His267 region does not contribute to this interaction.
Cytochrome P450 (P450) and UDP-glucuronosyltransferase (UGT) are two major enzyme groups responsible for phase I and II reactions, respectively (Guengerich, 1989; Oguri et al., 1994; Gonzalez and Lee, 1996; Ritter, 2000). These enzymes are localized on the cytosolic (P450) and luminal sides (UGT) of the endoplasmic reticulum (ER) membrane. UGT plays an important role in detoxifying drugs, including potent carcinogenic metabolites formed by P450 (Tukey and Strassburg, 2000). To minimize toxicity, it would be reasonable to expect that the reactive metabolite produced by P450 is directly transferred to the other enzymes participating in its subsequent metabolism (e.g., the UGTs) via protein-protein interactions. Our recent studies have suggested that CYP3A4 and other P450 isoforms interact with UGT2B7 to modulate the activity of the UGT (Ishii et al., 2005; Takeda et al., 2005a,b). The interactions between P450 and UGT occur also in rats, and the UGT in a P450-UGT complex is catalytically active (Ishii et al., 2007). These series of studies suggest that the interaction of P450 and UGT takes place ubiquitously in several P450-UGT couples and animal species. Other workers have also confirmed the CYP3A4-UGT2B7 interaction (Fremont et al., 2005). However, it is still uncertain whether CYP3A4 and UGT2B7 are associated in the ER membrane. The identity of the domains of both enzymes participating in protein-protein interactions is another question of interest.
Microsomal P450s are anchored to the ER via their N-terminal region (Edwards et al., 1991; Williams et al., 2000). Based on a model of the three-dimensional structure and the characteristics of the engineered forms of CYP3A4, another region, the F-G loop (e.g., residues 218-236), located between the F and G helices has been suggested to associate with the ER membrane (Cosme and Johnson, 2000; Williams et al., 2000, 2004; Yano et al., 2004). It has also been reported that CYP3A4 is pulled into the lipid bilayer membrane to change secondary structure and increase catalytic activity (Kim et al., 2003). As in P450, analysis of the UGT structure has suggested that this enzyme is partially embedded in the ER membrane by a short internal helix (Ciotti et al., 1998). Another group of workers has also made the same claim (Ouzzine et al., 1999). These lines of evidence support a hypothesis that P450 and UGT can meet in the ER membrane, although their membrane topologies are quite different. One possible approach for clarification of the in situ association between UGT and P450 is an analysis of the effect of mutations introduced into the interaction domains of both enzymes. However, the possible interaction region(s) of CYP3A4 and UGT2B7 has not yet been elucidated. To better understand the mechanism of the CYP3A4-UGT2B7 interaction and to assist in the design of future experiments, this study tried to identify the CYP3A4 domain(s) that contributes to the interaction with UGT2B7. For this purpose, we have analyzed the formation of a CYP3A4-UGT2B7 complex by means of cross-linking as well as immunoprecipitation and overlay assay, using truncated forms of CYP3A4 and CYP3A4 antibodies with different specificities as far as epitope recognition is concerned.
Materials and Methods
Materials.n-Octylglucoside, cholic acid, and 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide (EDC) hydrochloride were purchased from Sigma-Aldrich (St. Louis, MO). Emulgen 911 and 913 were generous gifts from Kao Atlas Co. (Tokyo, Japan). The purified form of recombinant CYP3A4 was purchased from Panvera (Madison, WI). Human liver microsomal samples were obtained together with an agreement for their use from donors from BD Gentest (Woburn, MA) and stored at -80°C until use. Expression vector pGEX-6P-1 carrying glutathione transferase (GST, Schistosoma japonicum) cDNA and anti-GST polyclonal antibody (lot no. 310702) were purchased from GE Healthcare (Chalfont St. Giles, UK). Protein N-glycosidase (PNGaseF) was purchased from Takara (Tokyo, Japan). Formic acid for amino acid sequence analysis grade was obtained from Wako Pure Chemicals (Tokyo, Japan). Reagents for protein assay were provided by Bio-Rad Laboratories (Hercules, CA). All other reagents were of analytical grade commercially available and used without further purification.
Anti-CYP3A Antibody. Purified form of recombinant CYP3A4 (10 μg/ml saline) was vigorously mixed with an equal volume of complete Freund's adjuvant (Wako Pure Chemicals) to produce a homogenous emulsion. Emulsified antigen (1 μg/100 μl) was injected intraperitoneally into male BALB/cAnNCrj mice (17-22 g) obtained from CLEA Japan (Tokyo, Japan). Two weeks later, mice received a booster intraperitoneal injection of the same amount of the antigen emulsified in Freund's incomplete adjuvant (Wako Pure Chemicals). Mice were bled 1 week after the booster injection, and serum samples were collected. The titer and specificity of the antiserum obtained (anti-3A4#1) was verified by immunoblotting, using three P450s (CYP3A4, CYP1A2, and CYP2C9; Supersomes) obtained from BD Gentest. In this study, we also used two other anti-CYP3A antibodies: one antibody (anti-3A4#2) was a mouse anti-CYP3A4 monoclonal antibody obtained from a commercial source (BD Gentest), and the other antibody (anti-3A4#3) was a rabbit anti-CYP3A4 polyclonal antibody (Ogino et al., 2002).
Western Blotting. Proteins separated by SDS-polyacrylamide gel electrophoresis (PAGE) (7.5-12.5% gels) were electroblotted onto a polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA). Anti-3A4#1, -#2, and -#3 described above were used as primary antibodies to detect CYP3A4. Rabbit anti-UGT2B7 antibody (BD Gentest) and rabbit anti-calnexin (CNX) antibody (Nventa Biopharmaceuticals, San Diego, CA) were used as primary antibodies to detect UGT2B7 and CNX, respectively. In all the blotting experiments, staining of primary antibody associated with target proteins was performed using alkaline phosphatase-labeled secondary antibody.
Immunoprecipitation. Solubilization of human liver microsomes with sodium cholate was performed according to the method described previously (Taura et al., 2000). UGT and/or P450 in solubilized human liver microsomes were immunoprecipitated according to the method described previously (Takeda et al., 2005a). The immunoprecipitates were subjected to SDS-PAGE followed by Western blotting to detect CYP3A4 and/or UGT2B7.
Expression of GST-CYP3A4 Fusion Proteins. GST-tagged CYP3A4 spanning the region from Tyr25 to Ala503 (abbreviated as probe A) was prepared by a method described previously (Takeda et al., 2005a). As indicated above, this fusion protein was designed so that it lacked the N-terminal membrane-anchoring region. The other fusion protein (probe B) in which GST was connected to an internal region (Met145-His267) of CYP3A4 was prepared as follows. The cDNA corresponding to the peptide sequence ranging from Met145 to His267 was amplified by polymerase chain reaction using a full-length CYP3A4 cDNA cloned into pCMV4 vector (Takeda et al., 2005a) as a template and Pfu polymerase (Stratagene, La Jolla, CA) as the enzyme. To generate the restriction sites at both ends of the amplified cDNA, we added additional sequences to the primers. The forward primer (5′-TCC CCC GGGATG GTC CCT ATC ATT GCC C-3′) was designed to have a SmaI site (underlined) in the coding region of the CYP3A4 cDNA. The coding sequence is shown by bold characters. The reverse primer (5′-A TAA GAA TGC GGC CGC TCA GTG CTT TTG TGT ATC TTC GAG-3′) was designed to have a NotI site (underlined) downstream of the CYP3A4. The polymerase chain reaction product obtained was purified, digested with SmaI and NotI, purified again, and then ligated in frame to the GST cDNA cloned into the pGEX-6P-1 plasmid at the SmaI and NotI sites, resulting in the expression of chimeric GST-CYP3A4 protein. The absence of extraneous mutations was verified by sequencing. Heterologous expression of GST-CYP3A4 fusion proteins in Escherichia coli was performed by a method described previously (Takeda et al., 2005a).
Overlay Assay with GST-CYP3A4 Fusion Proteins. The microsomes of insect cells transformed with baculovirus carrying UGT2B7 cDNA (Supersomes; BD Gentest) were used as the source of UGT2B7. The UGT2B7 microsomes were thoroughly washed and solubilized with n-octylglucoside as described previously (Takeda et al., 2005a). These solubilized UGT2B7 microsomes were used for the overlay assay with GST-tagged CYP3A4 fusion proteins by the method described previously (Takeda et al., 2005a). In brief, UGT2B7 microsomes were electrophoresed on SDS-PAGE, electrically transferred to PVDF membrane, and treated with GST-tagged CYP3A4. The association of UGT2B7 and CYP3A4 was detected by antibody against GST. Purification of GST protein (Schistosoma japonicum) was performed according to standard protocols using glutathione-Sepharose (Amersham-Pharmacia).
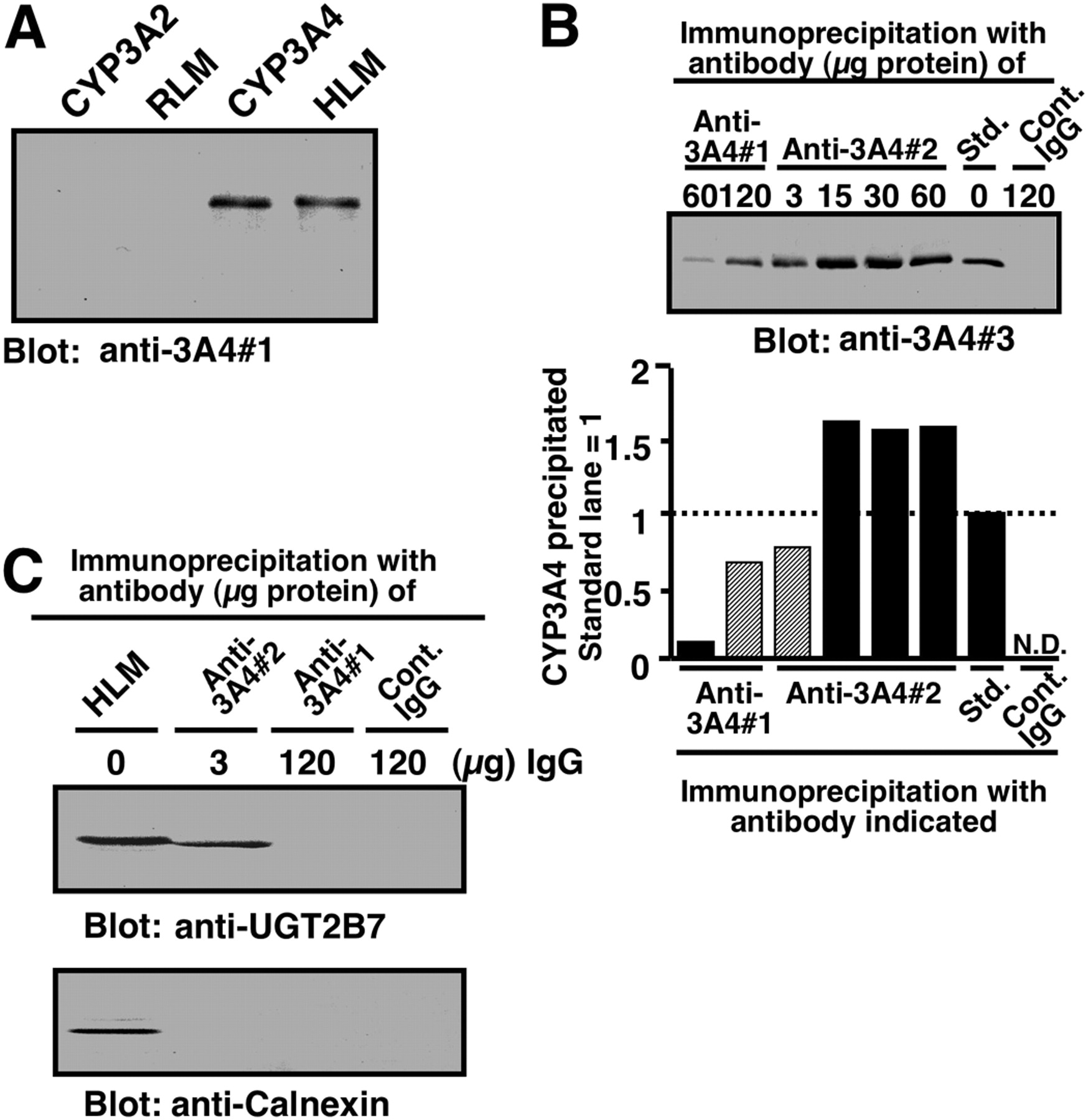
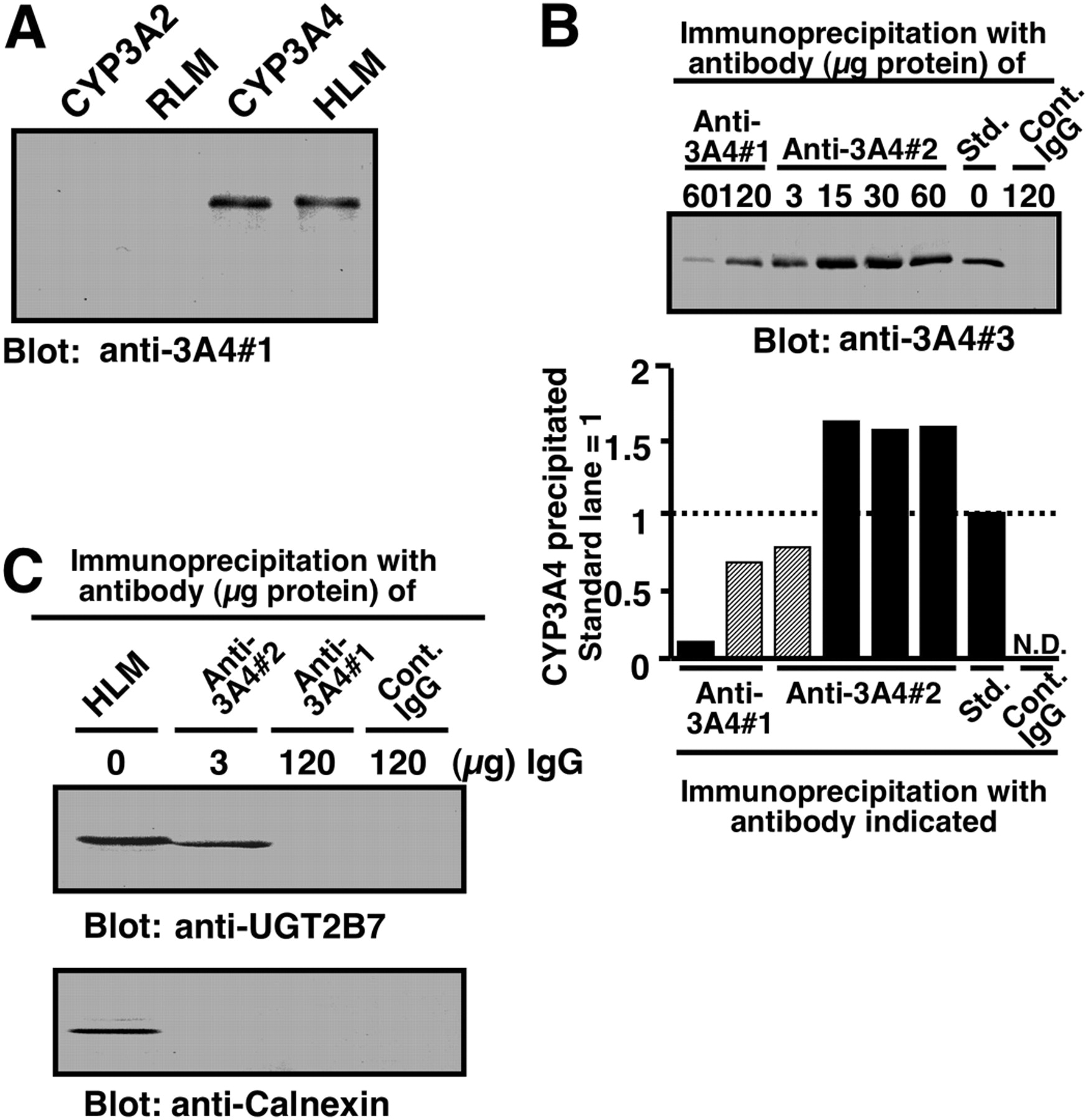
Comparison of two anti-CYP3A antibodies with regard to their specificity and the ability to coimmunoprecipitate UGT2B7. A, Western blotting performed with an anti-3A4#1 as a primary antibody. Samples were purified human CYP3A4 and rat CYP3A2 (each 0.5 pmol) and human liver microsomes (HLM) and rat liver microsomes (RLM) (each 0.25 μg of protein). B, immunoprecipitation of CYP3A with two types of anti-CYP3A antibodies is shown. Sodium cholate-solubilized human liver microsomes (100 μg of protein) were subjected to immunoprecipitation with mouse anti-3A4#1 or mouse anti-3A4#2 antibodies. Immunoprecipitates obtained were subjected to Western blot analysis with rabbit anti-3A4#3 as a primary antibody. The amount of anti-CYP3A4 IgG used for immunoprecipitation is indicated in the figure. A negative control was carried out with nonimmune mouse IgG (120 μg of protein). The band intensity of precipitated CYP3A4, which was quantified by using NIH Image version 1.61 software (http://rsb.info.nih.gov/nih-image/), is shown beneath the blot image. Std. represents purified recombinant CYP3A4 (0.5 pmol) without immunoprecipitation. The experiments shown in this figure were repeated twice and then reproducibility was confirmed. C, coimmunoprecipitation of UGT2B7 with anti-3A4#2 antibody. Solubilized pooled human liver microsomes (100 μg of protein) were subjected to immunoprecipitation with anti-3A4#1 and anti-3A4#2 antibodies. The immunoprecipitates obtained were subjected to Western blot analysis with rabbit anti-UGT2B7 and rabbit anti-CNX as primary antibodies. Pooled HLM represents solubilized pooled human liver microsomes (10 μg of protein) without immunoprecipitation procedures. The amount of anti-CYP3A4 IgG used in the immunoprecipitation experiments is indicated. A negative control for the immunoprecipitation was carried out using nonimmune IgG (control IgG). Details are described under Materials and Methods.
Cross-Linking Experiments. Triton X-100-solubilized lysate of E. coli expressing GST-tagged CYP3A4 and n-octylglucoside-solubilized UGT2B7 (Supersomes) were prepared as described previously (Takeda et al., 2005a). Solubilized GST-tagged CYP3A4 fusion proteins (or the purified form of recombinant CYP3A4) and UGT2B7 proteins were suspended at a protein concentration of 0.5 mg/ml in buffer I (100 mM NaCl, 0.5 mM N-hydroxysuccinimide, and 20 mM MES, pH 6.0) and buffer II (400 mM NaCl and 230 mM HEPES-NaOH, pH 7.5), respectively. Buffers I/II and cross-linker stock solution (50 mM EDC in buffer I) were prepared just before use. GST-tagged CYP3A4 and purified recombinant CYP3A4 (each 10 μg of protein) in buffer I were activated with several concentrations of EDC (see Results) for 20 min at 25°C. Preliminary experiments indicated that a reaction time of 20 min gives a maximum cross-linking (data not shown). A control experiment performed in the absence of EDC was run simultaneously. The reaction was terminated by the addition of 100 mM 2-mercaptoethanol and then UGT2B7 (20 μg of protein) in buffer II was added to the mixture, followed by incubation for 2 h at 25°C. The cross-linking reaction was quenched by adding an equal volume of ice-cold 50% trichloroacetic acid. Proteins were allowed to precipitate on ice for 30 min and then centrifuged for 8 min at 9000 rpm. The resulting precipitates were washed with ice-cold acetone, centrifuged again, and analyzed by Western blotting.
Cleavage of CYP3A4 with Formic Acid. Cleavage at the aspartyl-prolyl bonds of CYP3A4 was performed according to the method of Landon (1977).
Mapping of the CYP3A4 Epitope. The epitope of CYP3A4 toward anti-CYP3A4#1 antibody was mapped by SPOTs method according to the Custom SPOTs Technical Manual (version 1.1; Sigma-Aldrich). Twenty-eight synthetic peptides that cover the candidate region of CYP3A4 epitope ranging from Arg268 to Asp404 were synthesized by coupling with β-alanyl-β-alanines, spacers, which were immobilized on a nitrocellulose membrane (derivatized membrane; Sigma-Aldrich). This membrane conjugated with peptides was supplied from Sigma-Aldrich. Each peptide consisting of 12 amino acid residues was designed to overlap at the both ends with neighboring peptides by seven residues. In addition, the N-terminal of all peptides was acetylated. A derivatized nitrocellulose membrane conjugated with synthetic peptides was washed with methanol and blocked overnight at 4°C with blocking buffer (Sigma-Aldrich). The membrane was washed with Tris-buffered saline containing 0.05% Triton X-100 for 10 min at room temperature and then incubated overnight at 4°C with 0.1% anti-serum (mouse anti-CYP3A4#1). The IgG associated with peptides was detected with sheep anti-mouse IgG coupled with horseradish peroxidase, using enhanced chemiluminescence Western blotting detection reagents (GE Healthcare). The membrane was regenerated according to the manufacturer's protocol. To assess the nonspecific binding, the regenerated membrane was processed and treated with 0.1% preimmune serum in a similar way as described above.
Results
Coimmunoprecipitation of UGT2B7 with Anti-CYP3A Antibodies. To coimmunoprecipitate UGT2B7 together with CYP3A4, we used two anti-CYP3A4 antibodies, including one (3A4#1) that was prepared in this study. Although the 3A4#1 antibody detected a single band of human liver microsomal P450, the mobility of which was the same as CYP3A4, none of the P450 isoforms, including CYP3A2 in rat liver microsomes, could be detected by this antibody (Fig. 1A). The specificity of anti-3A4#1 antibody was further tested by Western blotting using two human CYP1A2, and CYP2C9 forms. The results indicated that neither CYP1A2 nor CYP2C9 can be detected by anti-3A4#1 antibody (data not shown). As shown in Fig. 1B, anti-3A4#1 and -#2 antibodies precipitated CYP3A in solubilized human liver microsomes. However, anti-3A4#2 obtained from a commercial source gave a more effective precipitation than anti-3A4#1. The amount of CYP3A4 immunoprecipitated with 120 μg of protein of anti-3A4#1 was comparable with that obtained with 3 μg of anti-3A4#2.
We then compared the abilities of two CYP3A4 antibodies to coimmunoprecipitate UGT2B7. As shown in Fig. 1C, anti-3A4#2 antibody efficiently precipitated UGT2B7 as well as CYP3A in solubilized human liver microsomes. However, anti-3A4#1 failed to precipitate UGT2B7 (Fig. 1C), although CYP3A4 was precipitated by this antibody (Fig. 1B). The failure to detect an interaction between CYP3A4 and UGT2B7 by anti-3A4#1 seems to be because of the overlap of an epitope recognized by this antibody and a region binding to UGT2B7. This nature of the 3A4#1 antibody is expected to be useful for assessing the domain of CYP3A4, which contributes to the association with UGT2B7 (see below). To exclude the possibility of nonspecific interactions between CYP3A4 and non-UGT proteins, we investigated whether CNX, an ER membrane protein that has the same topology as UGT (Oda et al., 2003), could interact with CYP3A4. Western blotting showed that no CNX was present in both anti-3A4#1- and 3A4#2-derived immunoprecipitates
CYP3A4 Does Not Require Its Met145-to-His267 Region for Interaction with UGT2B7. To investigate the region(s) of CYP3A4 required for interaction with UGT2B7, we prepared two deletion mutants of CYP3A4 that are connected to GST at the N terminus (Fig. 2A). One of them was a fusion construct designated as probe A, consisting of GST and CYP3A4 (Tyr25-Ala503) lacking the N-terminal membrane-anchoring region. The other fusion protein designated as probe B was designed to contain the region ranging from Met145 to His267, which involves the possible domain for association with the ER membrane (Johnson, 2003). We expected that this hydrophobic region would contribute to the protein-protein interaction with UGT2B7. The molecular masses of probes A and B were confirmed to be of the expected sizes (Fig. 2B). The abilities of these probes to interact with UGT2B7 were examined by overlay assay and cross-linking. In these experiments, anti-CYP3A4 antibodies (anti-3A4#1 and -#2) that exhibit different potential in the coimmunoprecipitation of UGT2B7 (Fig. 1C) were used as primary antibodies to detect the CYP3A4-UGT2B7 complex by Western blotting (Fig. 2C). Before the experiments for the CYP3A4-UGT2B7 complex formation, we first examined whether probes A and B could be recognized with anti-3A4#1 and -#2 antibodies. The results of immunoblotting showed that both anti-CYP3A4 antibodies recognize probe A (Fig. 2C). In contrast, although anti-3A4#2 antibody detected probe B, anti-3A4#1 antibody did not recognize this probe (Fig. 2C). Even when increasing amounts of probe B were electrophoresed and immunoblotted, anti-3A4#1 antibody failed to detect this probe (Fig. 2D).

Schematic representation (A) of the construction of GST-CYP3A4 chimera proteins used in this study and their immunoblotting (B-D). As shown in A, probe A is a chimeric protein in which the N-terminal membrane anchor region of CYP3A4 was deleted. Probe B is a chimeric protein in which the internal region of CYP3A4 was conjugated with GST. Gray and filled boxes represent GST and CYP3A4 moieties, respectively. The scheme of recombinant CYP3A4 used in this study is also shown. The small hatched box is a region that has been reported to be recognized by anti-CYP3A4 peptide (Val253-Gln273) antibody (Fremont et al., 2005). See Discussion for the role of this region in the coimmunoprecipitation of UGT2B7. Probe B includes a CYP3A4 region that is the most hydrophobic (hydropathy plot not shown). In the experiment in B, E. coli JM109 cells expressing GST or GST-CYP3A4 were lysed and subjected to Western blotting with anti-GST antibody. E. coli lysate (5 μg of protein) was loaded onto each well. Details are described under Materials and Methods. C, comparison of the reactivity of anti-CYP3A4 antibodies with probe A (Met145-His267) and probe B (Tyr25-Ala503). In experiment C, E. coli JM109 cells expressing probe A and probe B were lysed and subjected to Western blotting with anti-3A4#1 and 3A4#2 antibodies. E. coli lysate (5 μg of protein) was applied to each well. In experiment D, different amounts of cell lysate from E. coli JM109 expressing probes A and B were subjected to Western blotting with anti-3A4#1 antibody. Details are described under Materials and Methods.

Detection of the interaction between CYP3A4 and UGT2B7 by overlay assay. UGT2B7 in baculosomes was separated by SDS-PAGE and transferred onto PVDF membrane. The membrane was overlaid with either GST alone (lanes 3 and 4), probe B (lanes 5 and 6), or probe A (lanes 7 and 8) and then treated with anti-GST antibody. In the “none” experiment for overlay, the membrane was treated with anti-GST antibody without overlay pretreatment. The “con.” lanes represent baculosomes lacking UGT2B7.
In an overlay assay, proteins in baculosomes containing UGT2B7 were separated by SDS-PAGE, transferred to a PVDF membrane, and overlaid with GST-tagged CYP3A4 (probes A and B). Binding of the fusion protein to UGT2B7 was then analyzed with an antibody against GST. As the results show, a single band was detected only when UGT2B7 was overlaid with probe A (Fig. 3, lane 7). The apparent molecular size of this band corresponded to that of UGT2B7 (data not shown). Because the appearance of the band in the overlay assay was inhibited by the addition of nontagged CYP3A4, but not by GST, in a concentration-dependent manner (data not shown), it is highly likely that the band seen in Fig. 3 (lane 7) is because of the complex of GST-tagged CYP3A4 and UGT2B7. GST alone and probe B did not exhibit any binding to UGT2B7 (Fig. 3, lanes 3 and 5). Overlay with GST-CYP3A4 fusion proteins on the membrane blotted with baculosomes not containing recombinant UGT2B7 did not exhibit any bands (Fig. 3, lanes 6 and 8). In addition, the absence of a signal in the blots not being overlaid was also observed (Fig. 3, lanes 1 and 2). These results suggest that the CYP3A4-UGT2B7 interaction is specific and that the Met145-to-His267 region of CYP3A4 is not involved in the interaction.

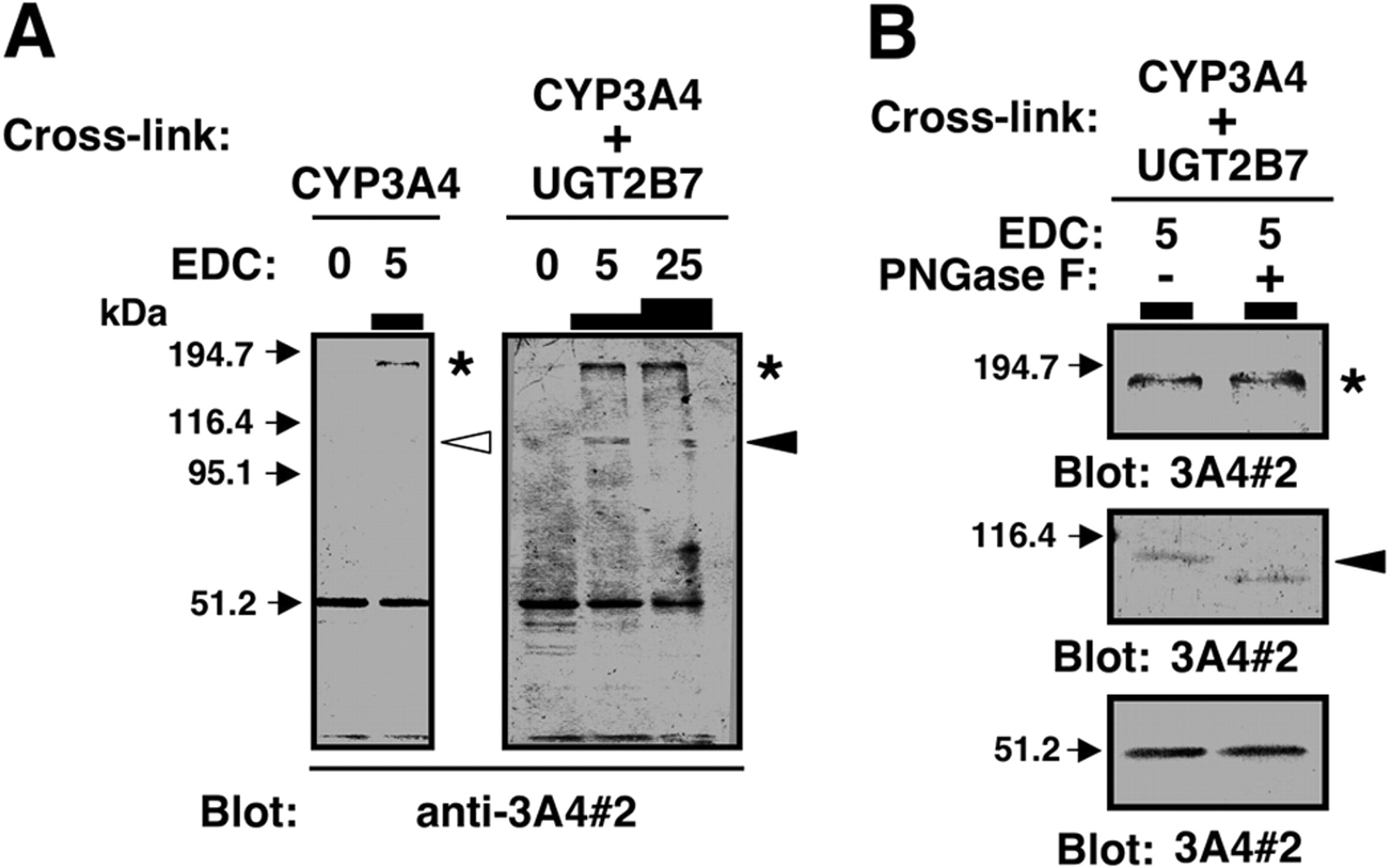
Western blots of UGT2B7 and GST-tagged CYP3A4 after treatment with a cross-linking reagent, EDC. E. coli JM109 cells expressing probes A and B were lysed, and the soluble fraction obtained (10 μg of protein) was subjected to cross-linking with UGT2B7 in solubilized baculosomes (20 μg of protein). Cross-linking was carried out with 5 mM EDC. In the experiments shown by asterisks (lanes 7 and 8), 100 mM 2-mercaptoethanol, an inactivator of cross-linker, was added to the GST-tagged CYP3A4 preparation before treatment with EDC. Western blotting was performed using anti-GST (lanes 1-10), anti-3A4#2 (lanes 11 and 12), and anti-3A4#1(lanes 13 and 14) antibodies. E. coli JM109 cells lysate and solubilized baculosomes, both of which did not contain any recombinant enzymes, were used as controls for GST-CYP3A4 chimeras (lanes 3, 4, 17, and 18) and UGT2B7 (lanes 3-6), respectively. When the mixtures of UGT2B7 and probe A or B were reacted in the absence of EDC followed by immunoblotting with anti-GST, anti-UGT, and anti-CYP3A4 antibodies, only free probe was detected at approximately 40 or 80 kDa (lanes 1, 9, 11, 13, and 15).
Cross-Linking of CYP3A4 and UGT2B7. We then investigated whether CYP3A4 and UGT2B7 could be cross-linked using EDC. Figure 4 shows Western blotting analyses of the EDC-treated mixture of CYP3A4 and UGT2B7 using anti-GST, anti-UGT, and anti-CYP3A4 (anti-3A4#1 and -#2) antibodies. Although no cross-linked product was observed between probe B and UGT2B7 even after treating with EDC (Fig. 4, lane 2), treatment with the cross-linker gave rise to the production of a complex of probe A and UGT2B7 (Fig. 4, lanes 10, 12, and 16, indicated by the filled arrowhead). The apparent molecular mass of the cross-linked product was approximately 130 kDa, which is equivalent to the expected size of a complex of probe A and UGT2B7. It should be noted that anti-3A4#1 antibody did not detect probe A-UGT2B7 complex (Fig. 4, lane 14, indicated by empty arrowhead). When 2-mercaptoethanol, an EDC inactivator, was added before the addition of EDC, no complex of probe A and UGT2B7 was observed (Fig. 4, lane 8). To exclude the possibility that CYP3A4 or UGT2B7 associates with endogenous protein(s) in the insect or E. coli lysates, control cross-linking experiments were performed in the following combinations: E. coli lysate (control; no expression of GST-tagged CYA3A4) and solubilized baculosomes (control; no expression of UGT2B7); and probe A and solubilized baculosomes (control). Blotting with an anti-GST antibody showed that there was no observable band at 130 kDa (Fig. 4, lanes 4 and 6). Cross-linking of control E. coli lysate and UGT2B7 in solubilized baculosomes failed to produce any complexes (data not shown). If GST itself can associate with UGT2B7, this would result in cross-linking between GST-tagged CYP3A4 and UGT2B7. However, the observation that probe B was not able to form a complex with UGT2B7 does not support this possibility (Fig. 4, lane 2). The band corresponding to UGT2B7 homodimer was not detected by anti-UGT2B7 antibody under the conditions used (Fig. 4, lane 18).
Figure 5A shows Western blotting analyses of an EDC-treated mixture of untagged CYP3A4 and UGT2B7, using anti-3A4#2 antibody. The cross-linked complex of untagged CYP3A4 and UGT2B7 was detected by anti-3A4#2 antibody. However, the yield of cross-linked product did not depend on the concentrations of EDC (Fig. 5A, indicated by the empty arrowhead). The reason is not clear, but we address this point under Discussion. As can be seen in Fig. 5A, right, when 5 mM EDC was used, three bands were detected by anti3A4#2 at the molecular masses of 51, 115, and 173 kDa, respectively. The latter two proteins are shown by a filled arrowhead and an asterisk, respectively. Although the 51-kDa band can be easily assigned to CYP3A4 monomer, it is uncertain whether UGT2B7 is a component of 115- and 173-kDa proteins. Regarding this issue, UGT2B7 possesses N-linked carbohydrate chains, whereas CYP3A4 does not. If UGT2B7 is present, the anti-3A4#2-detectable band should be shifted to a lower mass by PNGaseF digestion. This was the case for 115-kDa protein (Fig. 5B), and this observation agrees with the view that the 115-kDa protein contains UGT2B7 as well as CYP3A4. Because the 173-kDa protein remained unchanged even after treatment with PNGaseF, it is highly likely that this protein is a CYP3A4 homo-oligomer (Davydov et al., 2005). The 173-kDa protein was also detected after cross-linking CYP3A4 alone (see left blot of Fig. 5A, asterisk), and this observation supports the above-mentioned assumption.
CYP3A4 Domain Contributing to the Interaction with UGT2B7. Because anti-3A4#1 antibody does not have any ability to coimmunoprecipitate UGT2B7 and to detect a CYP3A4-UGT2B7 complex, it is suggested that the epitope of anti-3A4#1 overlaps with the domain involved in the CYP3A4-UGT2B7 interaction. Thus, purified recombinant CYP3A4 was subjected to formic acid-dependent fragmentation, and the epitope of anti-3A4#1 was mapped. In theory, six fragments, a, b, c, d, e, and f, are expected to be formed from CYP3A4 by formic acid treatment (Fig. 6A). Indeed, as shown in Fig. 6B, formation of fragments a, b, d, and f seemed likely, although short peptides c and e could not be detected probably because of the limit of resolution. Among these fragments, only fragment d was detected by anti-3A4#1 but not by anti-3A4#2 (Fig. 6C). Anti-3A4#2 antibody failed to detect not only fragment d but also any other fragments (Fig. 6C). Because anti-3A4#2 recognizes probe B (Fig. 2), the epitope of anti-3A4#2 is thought to be located in the region covered by fragments b, c, and d. If the epitope of anti-3A4#2 is present within the region of fragment c, it might be too small to be detected under the SDS-PAGE conditions used. Otherwise, maintaining a formic acid-degradable site (Asp-Pro) might be necessary for the epitope. No band was observed with control IgG as a primary antibody (Fig. 6C). As expected, the N-terminal sequence of band d was identical to that of fragment d the first residue of which is Pro218 (Table 1). Because anti-3A4#1 can not recognize the region ranging from Met145 to His267 (probe B), the epitope of anti-3A4#1 is assumed to be present in the region between Arg268 and Asp404. To further identify the epitope, 28 peptides covering this region were screened with anti-3A4#1 antibody. Of 28 peptides, peptide 15 (LQEEIDAVLPNK) exhibited marked reactivity toward the antibody (Fig. 7). This sequence occupies the Leu331-to-Lys342 region of CYP3A4, which is located on the J-helix. The evidence obtained suggests that the epitope toward anti-3A4#1 is present within this region.
NH2-terminal amino acid sequence of band d obtained by cleaving CYP3A4 with formic acid
Cleavage of CYP3A4 with formic acid was performed as described in the legend to Fig. 6. The fragments obtained were separated on SDS-PAGE and electroblotted onto PVDF membrane according to the method of Kyhse-Andersen (1984). After staining the proteins on the PVDF membrane with Coomassie Brilliant Blue R-250, band d was removed and washed with MilliQ water (Millipore). Then, the membrane was dried and subjected to N-terminal amino acid sequence analysis by a protein sequencer (Applied Biosystems, Foster City, CA). The amino acid at position 218 is a possible site sensitive to cleavage with formic acid.

Cross-linking of UGT2B7 and purified CYP3A4 with EDC. In experiment A, purified CYP3A4 (5 pmol) and UGT2B7 in solubilized baculosomes (20 μg of protein) were incubated in the presence or absence of EDC at the concentrations (millimolar) indicated in the figure. These samples were applied onto each lane of SDS-PAGE. Western blotting was performed using anti-3A4#2. Details are described under Materials and Methods. In experiment B, purified CYP3A4 (5 pmol) and UGT2B7 in solubilized baculosomes (20 μg of protein) were incubated in the presence or absence of 5 mM EDC. After the samples were precipitated with TCA and washed with acetone, the pellets obtained were treated with PNGaseF. Deglycosylation was performed according to manufacturer's recommendation using 1 mU of PNGaseF. The samples, with or without PNGaseF digestion, were subjected to Western blotting using anti-3A4#2 as primary antibody.

Epitope mapping of anti-CYP3A4#1 antibody. A, schematic representation of the possible fragments, a, b, c, d, e, and f, that are expected to be obtained by cleaving CYP3A4 with formic acid. These fragments were deduced on the basis of the sequence of wild-type CYP3A4. Figures in parentheses represent the corresponding residues of the recombinant CYP3A4 lacking the N-terminal region. The gray area and noted as Met145 to His267 is the region covered by probe B. The possible domain containing the epitope of anti-3A4#1 in fragment d is shown by a solid box. In the experiment in B, proteolysis of CYP3A4 by formic acid was analyzed by SDS-PAGE (15% gel). The bands were stained with Coomassie Brilliant Blue R-250. Purified recombinant CYP3A4 (150 μg) was mixed with 70% formic acid (final protein concentration, 0.2 mg/ml) in a brownish glass test tube. The test tube was capped and then incubated at 37°C for 72 or 96 h. Then, formic acid was evaporated under a stream of nitrogen at 60°C. The resulting residue was dried in vacuo for 3 h and then subjected to SDS-PAGE. From the molecular masses, bands a, b, d, and f were tentatively assigned to the fragments a, b, d, and f shown in A, respectively. In the time 0 lane, purified CYP3A4 (15 pmol) was applied. In experiment C, CYP3A4 fragments obtained by formic acid treatment were subjected to Western blotting using anti-3A4#1 and anti-3A4#2 as primary antibodies. Std represents untreated recombinant CYP3A4. In these cases, 1 and 2 pmol of CYP3A4 was electrophoresed and blotted with anti-3A4#1 and anti-3A4#2, respectively. Formic acid-digested CYP3A4 (96 h) was subjected to SDS-PAGE and immunoblotting. For immunoblotting with anti-3A4#1, CYP3A4-digests (0.3, 0.6, and 1.5 μg of protein) were used (left). For blotting with anti-3A4#2, CYP3A4-digests (0.6, 1.5, 3, and 4.5 μg) were used (middle). For a negative control, CYP3A4 with or without formic acid digestion was subjected to Western blotting using control mouse IgG (right).
Discussion
Protein-protein interactions among P450-P450, P450-NADPH-cytochrome P450 reductase, and P450-cytochrome b5 have been reported previously (Alston et al., 1991; Voznesensky and Schenkman, 1992; Omata et al., 1994; Taura et al., 2000). These interactions are reasonable because of similarities in membrane topology and/or interaction-assisted enhancement in P450 function. From this view point, we investigated the hypothesis that the protein-protein interactions between drug-metabolizing enzymes, including P450 and UGT, would modulate mutual functions (Taura et al., 2000, 2004; Ishii et al., 2005; Takeda et al., 2005a,b; Ishii et al., 2007). We and other workers have already suggested that physical protein-protein interactions occur between CYP3A4 and UGT2B7 (Fremont et al., 2005; Takeda et al., 2005a). In the present study, we confirmed the above-mentioned interaction by providing a series of more reliable pieces of evidence.

Identification of CYP3A4 epitope toward anti-CYP3A4#1 antibody. A, sequences of peptides 1 to 28 are shown. B, position where each peptide was spotted (top) and the membrane probed with 0.1% anti-serum (mouse anti-3A4#1) (middle) and with preimmune serum (bottom).
In the cross-linking with EDC, a CYP3A4-UGT2B7 complex was detected by antibodies against CYP3A4 and UGT (Figs. 4 and 5). However, the Met145-to-His267 region of CYP3A4 gave no complex even in the presence of cross-linker (Fig. 4). The difference in the epitope specificity of two anti-CYP3A4 antibodies (anti-3A4#1 and -#2) helped in the estimation of the CYP3A4 region capable of interacting with UGT2B7. It is reasonable to suppose that the residues of CYP3A4 not involved in the CYP3A4-UGT2B7 interaction are accessible to the antibody, the epitope of which is located at the surface of CYP3A4. On the contrary, when the complex of CYP3A4 and UGT2B7 is once formed, it is likely that the region(s) of CYP3A4 involved in the interaction would not be accessible to CYP3A4 antibody, the epitopes of which are the same as the region for the interaction with UGT2B7. Taking this into consideration, the following evidence helped us assess the interaction region of CYP3A4: 1) the region of Met145 to His267 in CYP3A4 was not recognized by anti-3A4#1 antibody, which has no ability to coprecipitate UGT2B7 (Figs. 1C and 2); 2) overlay experiments using GST-tagged CYP3A4 fusion proteins revealed that the region from Met145 to His267 (probe B) in CYP3A4 does not react with UGT2B7 (Fig. 3); 3) the cross-linked complex of CYP3A4 and UGT2B7 was detected by anti-3A4#2 antibody, the epitope of which is suggested to be present at the region of Met145 to His267; and 4) the cross-linked complex could not be detected by anti-3A4#1 antibody incapable of recognizing the Met145- to-His267 region (Fig. 4). These pieces of evidence suggest that a region(s) distinct from the internal hydrophobic area (Met145-His267) plays a role in the specific binding to UGT2B7. It has been demonstrated that UGT2B7 in solubilized human liver microsomes is coprecipitated with CYP3A4 precipitated with its antibody recognizing peptides from Val253 to Gln273 (Fig. 2, indicated by striped gray box) (Fremont et al., 2005). This fact seems to support our observation that the Met145-to-His267 region of CYP3A4 is not involved in the interaction with UGT2B7.
This study has also suggested that the CYP3A4 region contributing to the association with UGT2B7 is located between Arg268 and Asp404. The crystal structure of CYP3A4 has already been reported (Williams et al., 2004). Although the authors have claimed that the three-dimensional structure of this P450 differs from that of bacterial P450s on the basis of structural overlay, the map of the secondary structures in the primary sequence is thought to be highly conserved in the P450 superfamily (Gotoh 1992). If this is true, a region ranging from Arg268 to Asp404 would contain helices G, H, I, J, and K, and β-sheets 2, 3, and 4. As described in the Introduction, the F-G loop (218-236 residue), a highly hydrophobic region, is suggested to play a role in the interaction of CYP3A4 and the ER membrane (Williams et al., 2004; Yano et al., 2004). Therefore, taking the above-mentioned information into consideration, one of the possibilities is that a region present downstream of the F-G loop catches UGT2B7 with the help of the F-G loop, which facilitates the embedding of CYP3A4 into the membrane. In contrast, CYP3A4 epitope capable of binding to anti-3A4#1 antibody was mapped to the Leu331-to-Lys342 region that is located on the J-helix. The present study provided evidence that 1) binding of anti-3A4#1 IgG to this region of CYP3A4 interferes with the interaction to UGT2B7 and 2) this antibody fails to recognize the epitope when the complex of CYP3A4 and UGT2B7 was formed by either cross-linking or overlay assay. Thus, because the epitopic region of CYP3A4 seems to be masked by complex formation, it is suggested that this epitope and domain of CYP3A4 are needed for the interaction with UGT2B7. However, because IgG is quite a bulky molecule, an alternative possibility is that anti-3A4#1 antibody covers a critical domain different from the epitope to prevent CYP3A4 from its access to UGT2B7. Therefore, further study is needed before drawing conclusions about whether the Leu331-to-Lys342 region is the actual binding site of CYP3A4 to UGT2B7.
As a similar epitope toward anti-3A4#1 is found in CYP3A5 (Aoyama et al., 1989), this antibody would also react with CYP3A5. Although the level of CYP3A5 in the liver is low, its expression is polymorphic (Daly, 2006). Therefore, it cannot be excluded that CYP3A5 plays a role in the modulation of UGT2B7 function in some individuals.
The question whether CYP3A4 and UGT2B7 are in proximity in the ER membrane, allowing contact, should be clarified in future studies. In this study, we used EDC to bridge solubilized CYP3A4 and UGT2B7. Under our experimental conditions (see Materials and Methods), EDC activates the carboxyl groups of CYP3A4 so that it can be condensed with the primary amino groups of UGT2B7. Thus, if CYP3A4 and UGT2B7 form a complex by EDC, this would be because of the amide bonding between their own carboxyl and amino groups. Thus, because EDC gives no bridging arm to linked proteins, CYP3A4 and UGT2B7 are thought to be a pair of enzymes that are in proximity to each other. In support of this, we failed to detect CYP3A4-UGT2B7 association by using dithiobis(succinimidyl propionate), a cross-linker having a 12-Å spacer (data not shown). We have shown that CYP3A4 lacking its N terminus reduces UGT2B7-catalyzed glucuronidation at the 3-OH group of morphine and increases conjugation at the 6-OH group (Takeda et al., 2005a). In agreement with this, overlay and cross-linking experiments in this study showed that CYP3A4 lacking its N terminus binds to UGT2B7 (Figs. 4 and 5). Therefore, even if the N-terminal anchor region of CYP3A4 plays a role in the interaction with UGT2B7, it would not be essential.
With respect to the domain of UGT2B7 participating in the association with CYP3A4, this is largely unknown. As mentioned previously, UGT may interact with the ER membrane via its internal region (Ciotti et al., 1998; Ouzzine et al., 1999). It is therefore possible that CYP3A4 binds to this region of UGT2B7 to modulate the function of the UGT. However, there is an alternative possibility as described below. One of our recent studies has shown that tryptic digestion of rat liver microsomes in the absence of detergent enhances the catalytic activity of UGT (Okamura et al., 2006). Treating detergent-untreated microsomes with trypsin removes the cytosolic tail of UGT, although the main body of this enzyme remains unchanged even after digestion (Okamura et al., 2006). This is because of the membrane topology of UGT. For example, although most of the UGT molecule including the catalytic site is located within the ER, a short peptide protrudes into the cytosol. The above-mentioned observation that removing only the cytosolic tail of UGT enhances UGT function suggests the possibility that the short peptide significantly regulates the catalytic activity of UGT. Meech et al. (1996) have reported that recombinant rat UGT2B1 with a truncated cytosolic tail exhibits lower catalytic activities than the wild-type enzyme. If the same is true for human UGT2B7, this observation seems to suggest a regulatory role of the cytosolic tail itself. However, the cytosolic tail may play a role by interacting with other factors. Because treating detergent-untreated microsomes with trypsin must digest P450 as well as the cytosolic tail of UGT, an alternative possibility is that the cytosolic tail suppresses UGT function by interacting with P450. If this is the case, CYP3A4 would interact with the cytosolic tail of UGT2B7 to modulate its function.
In conclusion, this study has used immunoprecipitation, overlay assay and cross-linking to provide evidence that CYP3A4 and UGT2B7 are a pair of enzymes that can bind to each other. In addition, the CYP3A4 region contributing to the interaction with UGT has been identified by means of the specificity of the antibodies against this P450. The data obtained support the hypothesis that P450 modulates UGT function via a protein-protein interaction. To further confirm the hypothesis, the isoform specificity of the interaction and the binding region/mechanism need to be investigated in future studies.
Footnotes
-
This work was supported in part by the Japan Society for Promotion of Science [Grant-in-Aid for Scientific Research (C) 19590147] and by the National Health and Medical Research Council of Australia.
-
This work was presented, in part, at the Forum 2005: Hygienic Pharmaceutical Science and Environmental Toxicology; Tokushima, Japan; 28 October 2005), J Health Sci51(Suppl):S-100 (abstract).
-
ABBREVIATIONS: P450, cytochrome P450; UGT, UDP-glucuronosyltransferase; ER, endoplasmic reticulum; EDC, 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide; GST, glutathione transferase; NHS, N-hydroxysuccinimide; PNGaseF, protein N-glycosidase; PAGE, polyacrylamide gel electrophoresis; PVDF, polyvinylidene diflouride; CNX, calnexin; MES, 2-(N-morpholino)ethanesulfonic acid.
- Received September 12, 2008.
- Accepted January 21, 2009.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}