


Abstract
Breast cancer resistance protein (BCRP/ABCG2) is a membrane-bound efflux transporter important in cellular detoxification and multidrug resistance. Some aryl hydrocarbon receptor (AHR) agonists were reported to induce BCRP expression in human colon carcinoma cells. However, a direct involvement of AHR transcriptional regulation remains unexplored. In this study, we show that BCRP induction by AHR ligands occurs in human intestinal, liver, and mammary carcinoma cells and in primary colonocytes and hepatocytes. Increased BCRP transporter activity consistent with gene induction was also evident in the Caco2 subclone C2bbe1 cells. Using RNA interference and ectopic expression techniques to manipulate cellular AHR status, we confirmed AHR dependence of ABCG2 gene regulation. By gene promoter analysis, chromatin immunoprecipitation, and electrophoretic mobility shift assays, an active, proximal dioxin-response element at −194/−190 base pairs upstream of the transcription start site of the human ABCG2 gene was identified. Despite a common observation in human-derived cells, our in vitro and in vivo studies supported by phylogenetic footprinting analysis did not find that mouse Abcg2 is subject to AHR regulation. We conclude that AHR is a direct transcriptional regulator of human BCRP and provide an unprecedented role of AHR in cellular adaptive response and cytoprotection by up-regulating an important ATP-binding cassette efflux transporter.
Breast cancer resistance protein (BCRP) is an ATP-binding cassette (ABC) transporter encoded by the ABCG2 gene, which attracts growing research efforts directed at its involvement in toxicity and elimination of various drugs and xenobiotics (Doyle et al., 1998; Ishikawa, 2009). BCRP is a promiscuous cellular efflux pump of a broad spectrum of drugs (e.g., topotecan, acyclovir, mitoxantrone), carcinogens [e.g., aflatoxin, benzo(a)pyrene, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine], nutrients and phytochemicals (e.g., riboflavin, folate, daidzein, flavonoids), and metabolites (e.g., sulfated estrogens, eicosanoids). The apical domain of intestinal mucosa, mammary ductal epithelia, liver, kidney, and sanctuary barrier tissues (blood-brain barrier, blood-testis barrier, and blood-placental barrier) is an important site of BCRP expression (Ishikawa, 2009; Robey et al., 2009). Thus, BCRP has important pharmacological and toxicological implications in systemic pharmacokinetics, tissue distribution, and disposition of various drugs and toxins. Because it limits intracellular accumulation of numerous chemotherapeutic agents, BCRP overexpression may be a key factor of multidrug resistance phenotype in some cancers (Doyle and Ross, 2003). BCRP expression is also a known marker of certain stem cell populations with a function ascribed to promoting cell survival during hypoxia by reducing toxic heme accumulation (Krishnamurthy et al., 2004).
An increasing number of studies have focused recently on unraveling the molecular regulation of BCRP because BCRP/ABCG2 expression is highly sensitive to various developmental and environmental stimuli (Doyle and Ross, 2003; Jonker et al., 2005). To date, four major transcription pathways [hypoxia-inducible factor 1α (HIF-1α), estrogen receptor-α (ERα), peroxisome proliferator-activated receptor-γ, and progesterone receptor (PgR)] have been identified to regulate BCRP mRNA expression (Ee et al., 2004; Krishnamurthy et al., 2004; Szatmari et al., 2006; Wang et al., 2008). Of these, HIF-1α has been shown to regulate the ABCG2 gene in both species, humans and mice. These transcription factors constitute part of the physiological and endocrine control network for BCRP mRNA expression.
Despite having critical roles in cellular defense against toxic xenobiotics, the molecular regulation of ABCG2, with respect to this adaptive-response role, is poorly understood. Using human colon cancer cells, Ebert et al. (2005, 2007) showed that BCRP mRNA and/or protein is increased by inducers of the aryl hydrocarbon receptor (AHR) and the nuclear factor (erythroid 2-like) factor 2 (Nrf2). Given that many AHR agonists, such as benzo(a)pyrene, α-naphthoflavone, and oltipraz, are potential oxidative stress inducers and/or estrogenic compounds capable of activating Nrf2 or hormone receptors (Ma et al., 2004; Abdelrahim et al., 2006), direct involvement of AHR in BCRP transcriptional control remains uncertain. Furthermore, the observation of blocking ABCG2 induction by 2′-Amino-3′-methoxyflavone (PD98059), an AHR antagonist (Ebert et al., 2005), cannot be confidently attributed to an AHR-mediated mechanism, because this inhibitor is also a potent inhibitor of the mitogen-activated protein kinase kinase 1-mitogen-activated protein kinase pathway, which plays a central role in gene regulation of Nrf2 and other hormone receptors (McKenna and O'Malley, 2002; Tan et al., 2008). Hence, it is currently unknown whether the induction of ABCG2 occurs via direct transcriptional regulation by AHR through its cognate DNA motif, dioxin-response element (DRE), and whether such control occurs in various tissues across species.
AHR is a member of the basic helix-loop-helix/PER-AHR nuclear translocator (ARNT)-SIM superfamily of nuclear receptors. It governs a wide range of developmental and toxicological processes, such as development of vascular and immune systems, cell proliferation, and xenobiotic metabolism (Kawajiri and Fujii-Kuriyama, 2007). AHR has long been touted as a major mediator of undesirable toxic responses, and its contributory role in cancer has been recognized (Nebert et al., 2004). Ligands/agonists of AHR are diverse and include poly- and halogenated aromatic hydrocarbon (PAH) such as dioxin [2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)], dimethyl-benzo(a)pyrene (DBA), and 3-methylcholanthrene (3MC). Upon ligand activation, cytoplasmic AHR with its chaperone proteins (namely, a 90-kDa heat shock protein dimer and hepatitis B virus X-associated protein 2) translocates to the nucleus and associates with ARNT, thereby binding to the cognate DRE to transactivate target genes, particularly the phase I and II drug-metabolizing enzymes, such as CYP1A (Nebert et al., 2004; Kawajiri and Fujii-Kuriyama, 2007).
In this study, we provide evidence of a direct transcriptional regulation of the human ABCG2 gene by a DRE located in the proximal region of the ABCG2 gene promoter. We further show that whereas the human ABCG2 5′-flanking region contains an active proximal DRE and many putative DREs, none of these DREs is conserved in the mouse Abcg2 gene. This discrepancy may explain why mouse Abcg2 is not under AHR regulation in the present study and others (Han and Sugiyama, 2006; Tijet et al., 2006; Boutros et al., 2009).
Materials and Methods
Cell Cultures and Chemicals.
The human carcinoma cell lines (C2bbe1, LS180, LS174T, HepG2, and MCF7) and mouse-derived cell lines (EMT6, Hepa1c1c7, and CMT93) were purchased from the American Type Culture Collection (Manassas, VA), and grown as recommended. Cryopreserved primary colonocytes (Celprogen, San Pedro, CA), and fresh primary hepatocytes (Celsis In Vitro Technologies, Chicago, IL; and BD Biosciences, San Jose, CA) were thrived in media and conditions recommended by respective suppliers. MCF7 AHR100 cells, which are an AHR-deficient subclone of MCF7 cells, were kindly provided by Dr. Ciolino (National Cancer Institute-Frederick Cancer Research and Development Center, Frederick, MD). All cells were maintained at 37°C with 5% CO2/95% air. TCDD was purchased from Cerilliant (Round Rock, TX), 3,4-dimethoxyflavone (DMF) was from Lancaster (Pelham, NH), fumitremorgin C (FTC) was from Alexis Biochemicals (Farmingdale, NY), and most other chemicals were from Sigma-Aldrich (St. Louis, MO), unless otherwise indicated. Radiochemicals [3H]mitoxantrone was obtained from Moravek Biochemicals (Brea, CA) and [14C]mannitol from GE Healthcare (Chalfont St. Giles, Buckinghamshire, UK). Oligonucleotides, including biotinylated DNA, were synthesized at Integrated DNA Technologies (Coralville, IA). Antibodies used were the following: mouse anti-BCRP BXP-21 (Millipore, Billerica, MA), fluorescein isothiocyanate-conjugated mouse anti-ZO1 (Invitrogen, Carlsbad, CA), and mouse monoclonal antibody against AHR (Thermo Fisher Scientific, Waltham, MA).
Real-Time RT-PCR.
Total RNA isolated with RNeasy Kit (QIAGEN, Valencia, CA) was reverse-transcribed by Superscript II reverse transcriptase (Invitrogen), and PCR reaction was carried out on Prism 7700 Sequence Detection System or 7500 Real-Time PCR system (Applied Biosystems, Foster City, CA) using inventoried, preoptimized Assay-on-Demand Taqman's gene expression probes and primer sets (ABI). The amplification-fold difference of gene transcripts between treatments and the vehicle control was analyzed after adjustment for the endogenous control gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Tan et al., 2007). Analysis was based on triplicate determinations, and similar experiments were repeated at least three times.
Immunoblots and Immunohistochemistry.
Cells were lysed in hypotonic lysis buffer (10 mM KCl, 1.5 mM MgCl2, and 10 mM Tris-HCl, pH 7.4) with protease inhibitors, homogenized, and centrifuged at 4000g for 10 min. The supernatant was centrifuged at 100,000g for 1 h, and the precipitated, membrane protein-rich pellets were dissolved in sucrose buffer (250 mM sucrose, 10 mM Tris-HCl, and 150 mM NaCl, pH 7.4). Immunoblot analysis was performed on the NuPAGE Novex gel system (Invitrogen) using 4 to 12% bis-Tris gel. Primary antibodies used were the following: mouse monoclonal BXP-21 (Millipore Bioscience Research Reagents, Temecula, CA) against human BCRP (1:1000) and rabbit antiserum against calnexin α (1:1000) (a gift from Dr. David Williams, University of Toronto, Toronto, ON, Canada). For immunohistochemistry, cells were exposed to 10 nM TCDD or vehicle for 24 h, fixed with 4% paraformaldehyde and permeabilized with 0.25% Triton X-100. The fixed cells were first incubated with blocking solution (1% bovine serum albumin in phosphate-buffered saline/Tween 20) for 30 min and anti-BCRP (1:1000) for 2 h followed by incubation with the Cy3-conjugated anti-rat IgG secondary antibody (1:1500) in blocking solution for 1 h and finally the fluorescein isothiocyanate-conjugated anti-ZO1 antibody (1:200) for 1 h at room temperature in the dark. The primary antibody was replaced with nonimmune IgG in negative controls, and specificity of the secondary antibody was examined in the absence of the primary antibody. Images were acquired using an LSM510 META laser-scanning confocal microscope (Carl Zeiss, Inc., Thornwood, NY).
Mitoxantrone Uptake Assay.
C2bbe1 cells pretreated for 24 h with vehicle dimethyl sulfoxide (DMSO) or 10 nM TCDD were incubated with 1 μM mitoxantrone (with or without 1 or 10 μM FTC, a BCRP inhibitor) spiked with 0.334 μCi/ml [3H]mitoxantrone and 0.0344 μCi/ml [14C]mannitol at 37°C for 1 h. The cell membrane impermeant mannitol was used as an extracellular compartment marker to ensure minimal media entrapment in cell lysate. Accumulation of cellular mitoxantrone determined by the tritium radioactivity was adjusted against cellular total protein.
Plasmid Constructs.
We used the luciferase-based ABCG2 reporter constructs reported by Bailey-Dell et al. (2001). The gene reporter constructs harboring five sequentially deleted fragments of the human ABCG2 5′-regulatory regions (−1285/+362, −628/+362, −312/+362, −243/+362, −115/+362) cloned into Mlu1/BglII restriction enzyme sites of basic pGL3 vector (Promega, Madison, WI) were synthesized. To test the enhancer activity of individual DREs, DNA fragments (25 bp) containing a core DRE motif or mutant DRE (two point mutations on the DRE core sequence, i.e., GCGTG → ATGTG, mutations shown in italic) and its adjacent nucleotides were chemically synthesized. They were inserted into similar restriction enzyme sites of pGL3 vector with simian virus 40 (SV40) promoter (Promega). Serving as positive control, a 25-bp DNA fragment of the DRE3 of mouse Cyp1a1 promoter region (Harper et al., 1992) or its mutant (two point mutations as described above) and its adjacent nucleotides were synthesized and inserted into a similar reporter vector. Sequence information of these individual DRE motifs is provided in Supplementary Table S1.
Transient Transfection and Reporter Luciferase Assays.
MCF7 AHR100 or C2bbe1 cells at ∼50% confluence were transfected overnight with 0.25 μg of the reporter firefly (Photinus pyralis) luciferase plasmid, 0.02 μg of Renilla reniformis luciferase control reporter pRL-TK with or without cotransfection with 0.25 μg of human AHR expression vector, or its empty vector using the liposome carrier Lipofectamine 2000 (Invitrogen). Treatment with TCDD or vehicle followed for 4 or 24 h. Luciferase activities of cell lysates were analyzed with the Dual-Luciferase Reporter Assay System (Promega). Reporter activity was derived from the luciferase signal ratio (firefly to R. reniformis), from which fold differences between TCDD and vehicle treatments were calculated. For AHR ectopic expression studies, the AHR-deficient MCF7 AHR100 cells at ∼50% confluence were transfected with 1-μg AHR expression vector or respective empty vector for 24 h, followed by TCDD or vehicle treatment for another 24 h. All experiments were done in triplicate and repeated at least three times.
RNA Interference.
A cocktail of four gene-specific small interfering RNA (siRNA) against human AHR (NM_001621) was procured (Dharmacon ON-TARGET plus SMARTpool siRNA Reagent; Thermo Fisher Scientific). Overnight-seeded C2bbe1 cells, which are characterized by their relative abundance of endogenous AHR (Supplementary Figure S1), at ∼50% confluence were transfected for 48 h with 50 nM siRNA against AHR or equal molar mismatched siRNA controls. Treatment with TCDD or vehicle followed for 24 h. These siRNAs were complexed in advance with liposome carrier DharmaFect I (Dharmacon RNA Technologies, Lafayette, CO) at 0.2 μl/nM siRNA concentration in serum-free Opti-MEM medium (Invitrogen). For gene reporter luciferase assays, transfection with reporter plasmids was carried out after 24 h of transfection with siRNA followed by 24 h of TCDD treatment. Successful functional knockdown of AHR was confirmed by ∼70% reduction of the AHR gene transcript with significantly lower TCDD-induced CYP1A1 gene expression and DRE reporter activity compared with cells treated with siRNA controls and TCDD.
Electrophoretic Mobility Shift Assay.
Electrophoretic mobility shift assay (EMSA) was conducted as described previously (Kosuge et al., 2007). C2bbe1 cells were treated with vehicle or 10 nM TCDD for 4 h, at which point their nuclear protein was extracted. To detect AHR-DRE interaction, 0.25 pmol concentration of 25-bp biotinylated DNA segments of human ABCG2 containing the −194/−190 DRE (GCTTGTCCCTGCGTGTCACGGCAGG; the core DRE is underlined) were incubated with 1.5 μg of nuclear extracts with or without excess amounts (100–400 times) of similar or mutant (GCTTGTCCCTATGTGTCACGGCAGG; mutations shown in italic) unbiotinylated DNA oligonucleotides. A 25-bp biotinylated DNA motif containing a mouse Cyp1a1 DRE (Supplementary Table S1) was similarly examined as a positive control. The mouse monoclonal antibody against AHR (Thermo Fisher Scientific, Waltham, MA) was used to probe the AHR-DNA complexes. As a nonspecific negative control, an irrelevant mouse monoclonal antibody against phophoinositide-3 kinase was used.
Chromatin Immunoprecipitation.
The protocol of chromatin immunoprecipitation (ChIP) and quantitative PCR detection has been detailed elsewhere (Tan et al., 2007). After 4-h treatment with TCDD or vehicle, C2bbe1 cells were fixed and subjected to sonication. AHR-chromatin DNA complexes were then coimmunoprecipitated with a mouse monoclonal AHR antibody (Affinity BioReagents) or equal amounts of a nonspecific mouse monoclonal antibody (negative control) at a concentration of 1:100 dilution. Quantitation of AHR occupancy to the proximal DRE region of the ABCG2 gene promoter by SYBR Green real-time PCR was carried out using the following primer set: forward strand, 5′-GTCCCTGCGTGTCACGGCAGGGTG-3′; and reverse strand, 5′-GTGAGGCGTGGCCCCGACTGCCGG-3′, which amplifies a product of 150 bp (−200/−51). The detection of DREs of CYP1A1, which serves as positive control, was included in similar PCR reactions using the following primer set: forward strand, 5′-ACCCGCCACCCTTCGACAGTTC-3′; and reverse strand, 5′-TGCCCAGGCGTTGCGTGAGAAG-3′.
Phylogenetic Footprinting.
A “Hidden Markov Model” phyloHMM analysis was used to identify the degree of conservation between mouse and human ABCG2 promoter sequences (Siepel and Haussler, 2004). A total of 20 kilobase pairs upstream of the ABCG2 gene were extracted, along with the entire ABCG2 gene sequence for human genome build hg17, from the University of California Santa Cruz genome database and for mouse genome build mm5. The phyloHMM data were retrieved from the same source and associated with the raw sequence using custom-designed Perl scripts in an Oracle database. The sequence was then searched for core DRE motifs (GCGTG), as defined previously (Boutros et al., 2004). For display, moving averages in a 7-bp window were calculated. Scores are reported as the average phyloHMM score for the putative transcription factor binding sites, with bases missing phyloHMM scores ignored. This region of the mouse and human genomes has not been significantly updated because builds hg17 and mm5, with more than 99.9% identity between current builds (hg19 and mm9) and the builds originally used in this analysis.
Mouse Experiments.
Because of the significance of placental BCRP, pregnant mice were used in this study. Timed-pregnant C57BL/6N (Charles River Laboratories, Montreal, QC, Canada) mice were given a single intraperitoneal injection of either 30 μg/kg TCDD or equal volume vehicle corn oil on gestational day 16. After 24 h, the mice were killed by cervical dislocation and the maternal liver, small intestine (divided into three parts to approximately reflect duodenum, jejunum, and ileum), colon, and placenta were collected and stored frozen in RNAlater (QIAGEN). Total RNA was extracted using TRIzol reagent (Invitrogen) followed by the QIAGEN RNeasy Mini protocol for RNA cleanup according to the manufacturer's instructions. All mice were housed in a 12-h light/dark cycle and given standard animal chow and water ad libitum. Experimental protocols were approved by the Hospital for Sick Children Animal Care Committee (Toronto, ON, Canada).
Statistical Analyses.
Data were presented as mean ± S.E.M. Normality and equal variance check was first carried out to guide the selection of statistical tests. Comparisons between groups were designed and conducted using Student's t test or one- or two-way ANOVA, where appropriate. Pair-wise post hoc comparisons in multiple test settings were conducted using the Holm-Sidak method. P values lower than 0.05 were considered statistically significant.
Results
ABCG2 Gene Induction by AHR Ligands.
In the human intestinal C2bbe1 (a subclone of Caco2) cells, ABCG2 mRNA was increased by three different AHR ligands (TCDD, DBA, and 3MC) in a dose-dependent manner starting as early as 4 h (Fig. 1, A and B). Significant induction of ABCG2 (2- to 100-fold) and CYP1A1 (200- to 2000-fold) was also noted in other human secondary carcinoma cells of the colon, liver, and mammary glands exposed to TCDD treatment (Fig. 1C). The difference in basal AHR and ABCG2 gene expression in different cell models seem to influence the magnitude of ABCG2 induction by TCDD. A higher basal AHR but lower ABCG2 expression level was associated with a much higher induction of ABCG2. Likewise, primary human colon and liver cells treated with TCDD showed significant induction of ABCG2, although the magnitude was relatively modest in both ABCG2 (1.5- to 2-fold) and CYP1A (10- to 50-fold) compared with those of the secondary cell lines (Fig. 1D). The enriched growth factors and steroids in the media used for these cells might have mitigated the effects of AHR agonist treatment because these factors are capable of activating hormone receptors and multiple signaling pathways potentially modulating ABCG2 expression (Freyschuss et al., 1993). It is noteworthy that the gene induction by TCDD was fully prevented with coincubation with DMF, an AHR inhibitor with partial agonist function, suggesting a possible AHR dependence of ABCG2 induction (Fig. 1, C and D).

BCRP (ABCG2) induction by AHR ligands. mRNA levels were normalized to endogenous control GAPDH and are expressed as fold change to vehicle DMSO control. A, time course of ABCG2 and CYP1A1 mRNAs treated with the indicated AHR ligands in the C2bbe1 cells. B, dose-dependent response of C2bbe1 cells on ABCG2 and CYP1A1 mRNAs after 24-h treatment with the indicated AHR ligands. C, ABCG2 mRNA levels in human secondary cell lines after 24-h exposure to 10 nM TCDD in the presence or absence of 10 μM DMF, an AHR inhibitor with partial agonist function. The data were analyzed within each cell line using ANOVA followed by the post hoc Holm-Sidak test (∗, p < 0.01). D, ABCG2 mRNA levels in human primary colon cells (top) and liver cells (bottom) after 24-h exposure to the indicated chemicals. Data of the hepatocytes are representative of two independent hepatocyte preparations from two suppliers showing comparable results. Note that CYP1A2 mRNA is shown for the primary liver cells. ABCG2, CYP1A1, and CYP1A2 mRNA levels were measured using real-time RT-PCR and normalized to endogenous control GAPDH and expressed as fold change to vehicle DMSO control. ∗, p = 0.001 (one-way ANOVA: F3,8 = 14.32); p < 0.001 (post hoc Holm-Sidak test).
Concurrent Increase of BCRP Protein and Function by AHR Ligands.
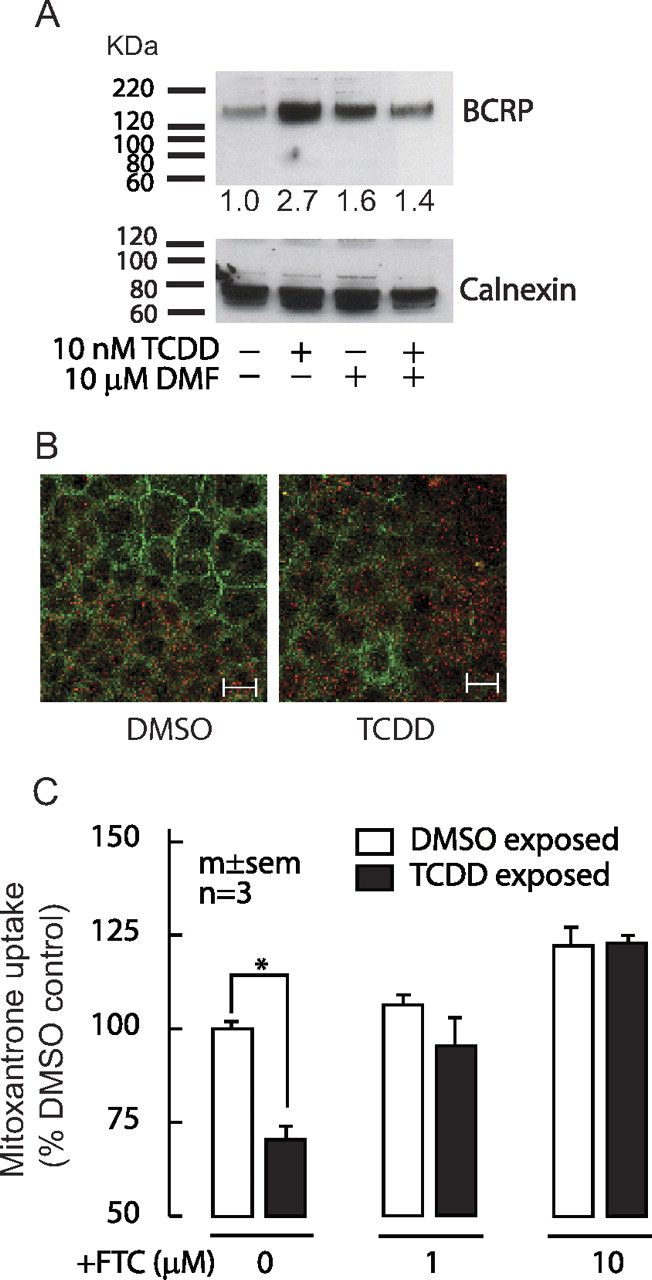
The colonic C2bbe1 cells were used in most analyses because they possess functional BCRP activity and relatively higher gene expression of AHR compared with the other test cell lines (Supplementary Fig S1). Immunoblotting of membrane fractions of C2bbe1 cells showed an increase of BCRP protein detected as homodimer (∼140 kDa), a known functional unit of this transporter, after 24-h exposure to TCDD, and this increase was abolished by DMF cotreatment (Fig. 2A). Immunostaining analyses also exhibited an increased diffuse apical and cytoplasmic BCRP staining in TCDD-exposed C2bbe1 cells, compared with that of vehicle-treated control cells (Fig. 2B). Consistent with these findings, cellular accumulation of a selective BCRP substrate, mitoxantrone, was significantly decreased in TCDD-treated cells (Fig. 2C). This TCDD-induced efflux activity was dose-dependently reversed by cotreatment with a specific BCRP inhibitor, FTC. ABCG2 mRNA decay in the presence of a potent transcription inhibitor (5 μg/ml actinomycin D) did not differ significantly between DMSO- and TCDD-treated C2bbe1 cells (TCDD: 100% at time 0, 54.5% at 5 h, 35.4% at 7.5 h, and 12.5% at 24 h of treatment; DMSO: 46.5% at 5 h, 27.5% at 7.5 h, and 14.6% at 24 h of treatment), indicating no major contribution of ABCG2 mRNA-stability effect by TCDD treatment.

Increased BCRP (ABCG2) protein and transporter activity after exposure to TCDD. A, immunoblots of BCRP (top) and a membrane marker, calnexin, (bottom, arrow) of C2bbe1 cells after 24-h exposure to 10 nM TCDD, 10 μM DMF, or TCDD + DMF. Densitometric measurements by ImageJ software (http://rsbweb.nih.gov/ij/) of BCRP adjusted for individual calnexin and expressed as fold difference to DMSO are indicated on the blot. Blot images shown are representative of three independent experiments. B, representative confocal immunofluorescence images (1.5-μm depth horizontal slices from the coverslip-attached basal side) of BCRP (red) and tight-junction marker ZO-1 (green) proteins in C2bbe1 cells exposed for 24 h to vehicle DMSO or 10 nM TCDD. Similar experiments were repeated three times with similar results. Scale bar, 20 μM. C, C2bbe1 cells treated for 24 h with 10 nM TCDD or DMSO were incubated for 1 h with 1 μM mitoxantrone containing spiked [3H]mitoxantrone with or without FTC. Total uptake of mitoxantrone adjusted for total protein was measured. Results were expressed as the percentage of DMSO control without FTC. Two-way ANOVA showed significant effects of TCDD (F1,12 = 9.87; p = 0.009), FTC (F2,12 = 23.79; p < 0.001), and TCDD × FTC treatment interaction (F2,12 = 4.88; p = 0.028); post hoc pair-wise comparison with Holm-Sidak test (∗, p = 0.001 from DMSO-exposed control without FTC; p < 0.01 from TCDD/ FTC-exposed cells).
AHR-Dependence of ABCG2 Induction.
We performed gain-of-function assays by ectopically restoring AHR status in an AHR-deficient human mammary cell line, MCF7 AHR100, and loss-of-function assays by knocking down AHR in C2bbe1 cells using siRNA techniques. Transfection of AHR expression vector to MCF7 AHR100 cells resulted in significant induction of ABCG2 and CYP1A1 mRNAs upon TCDD treatment (Fig. 3A). In contrast, AHR knockdown with siRNA in C2bbe1 cells (70% suppression of AHR transcript levels compared with control siRNA treatment) attenuated ABCG2 and CYP1A1 induction by TCDD (Fig. 3B). In support of the experiments of AHR inhibitor cotreatment, these data suggest that AHR activation is required for induction of human ABCG2 by TCDD.

AHR-dependence of ABCG2 induction. A, mRNA levels of ABCG2 and CYP1A1 of the AHR-deficient MCF7 AHR100 cells transfected with AHR expression vector followed by 24 h of 10 nM TCDD treatment. B, mRNA levels of ABCG2 and CYP1A1 in C2bbe1 cells treated with 10 nM TCDD after AHR knockdown by siRNA. mRNA was measured by real-time RT-PCR. Results were normalized to GAPDH and expressed as a percentage of DMSO-treated controls transfected with the empty vector (A) or with the control siRNA (B). Mean and S.E.M. values are shown for ABCG2 and are representative results of two three independent experiments for CYP1A1. One-way ANOVA was used to test statistical significance: A, F2,9 = 12.95, p = 0.002 (post hoc pair-wise comparison, ∗, p = 0.0024 from −AHR/+TCDD, and p = 0.0013 from +AHR/−TCDD); B, F2,6 = 46.27; p < 0.001 (post hoc pair-wise comparison: ∗, p < 0.001 from +siRNA/−TCDD and from +siRNA/+TCDD).
Lack of Induction of Mouse Abcg2 by TCDD.
In contrast to human cell lines, mouse-derived hepatic (hepa1c1c7), mammary (EMT 6), and intestinal (CMT93) cell lines showed no significant Abcg2 induction by TCDD (Fig. 4A). In vivo administration of TCDD to mice also failed to induce Abcg2 gene transcripts in various organs, including liver, small intestine, and colon, as opposed to Cyp1a1 mRNA, which was markedly increased by more than 100-fold compared with vehicle control (Fig. 4B). Similar observations were noticed in the placenta (J. MacAulay and P. A. Harper, unpublished data).

No effects of TCDD treatment on mouse Abcg2 in vitro and in vivo. A, Abcg2 and Cyp1a1 mRNAs of mouse-derived liver (hepa1c1c7), mammary (EMT 6), and intestinal (CMT93) carcinoma cell lines were treated with 10 nM TCDD or DMSO for 24 h. B, timed pregnant C57BL/6N mice at gestational day 16 were injected (intraperitoneally) a single dose of 30 μg/kg TCDD or corn-oil vehicle for 24 h. mRNA levels of Abcg2 and Cyp1a1 of maternal liver, small intestine (p, proximal third; m, middle third; d, distal third, corresponding to duodenum, jejunum, and ileum, respectively), and colon were determined. Real-time RT-PCR was used to quantify mRNA levels. Significant difference (p < 0.05) for Cyp1a1 but not Abcg2 was observed between the corn-oil control and treatment groups.
Lack of Conserved DREs between Human and Mouse ABCG2.
The apparent disparities in AHR regulation of ABCG2 between human and mouse may suggest differences in genomic organizations of their AHR-binding DREs. To take a broader look at the ABCG2 loci across human and mouse species, we initiated phyloHMM assessments of interspecies conservation (Supplemental Figure S2). A systematic scan of human and mouse ABCG2 found that most exons are well conserved (i.e., phyloHMM conservation scores of nearly 1.0), but the majority of the noncoding regions, including the 5′-flanking and intronic regions, were not (i.e., phyloHMM conservation scores of <0.5) between the two species. In particular, none of the putative core DREs found on the 10-kb 5′-flanking region of human ABCG2 is conserved in the aligned mouse region, with the phyloHMM conservation scores of less than 0.25. This suggests that AHR regulation of ABCG2 is probably human-specific.
AHR Recruitment to the Proximal DREs of Human ABCG2.
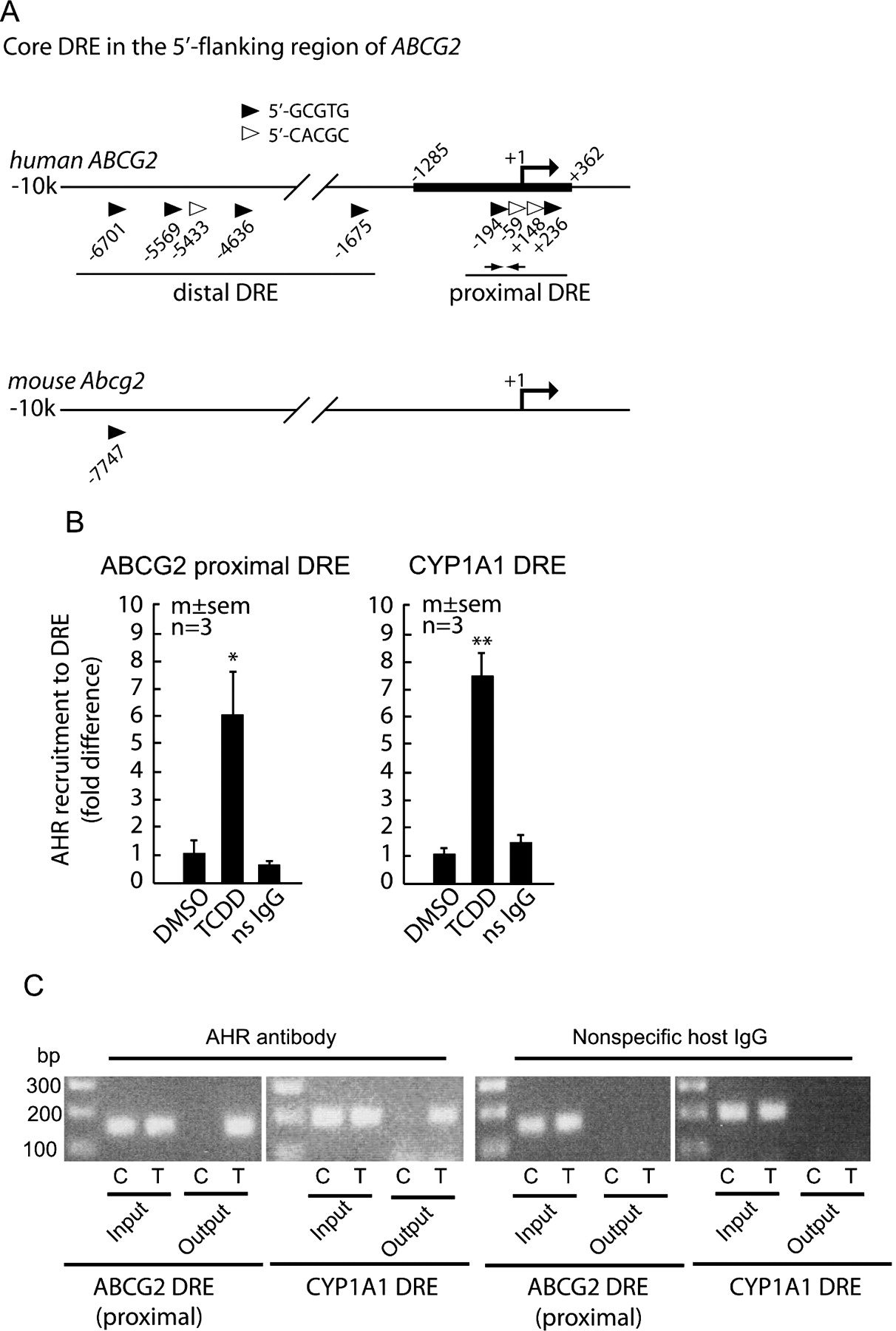
In silico analysis of the 10-kb 5′-flanking region revealed that human ABCG2 has nine putative core DREs, in contrast to that of mouse, which has only one (Fig. 5A). The four putative DREs on the proximal region (within ∼200 bp from the transcription start site and 5′-untranslated region) was of particular interest to us because within this region reside the binding regions of HIF1α, ERα, and PgR (Krishnamurthi et al., 2004; Ee et al., 2006; Wang et al., 2008). Because physical interactions between estrogen receptors and AHR in gene regulation have been reported (Rüegg et al., 2008), we hypothesized that AHR binds to any of these proximal DREs to mediate the induction of ABCG2. To test this hypothesis, we examined AHR recruitment to this region in the native chromatin context by ChIP assays. As shown in Fig. 5, B and C, an increased AHR occupancy to the proximal promoter region of ABCG2 was evident in C2bbe1 exposed for 4 h to TCDD. Similar observations were made with another AHR ligand, DBA (data not shown). Because of the proximity of these four DREs to one another (within ∼400 bp), individual contribution of these proximal DREs cannot be determined with the ChIP assays.

AHR recruitment to proximal DREs. A, schematic representation of the 5′-flanking regions of human ABCG2 and mouse Bcrp1 genes. Core DREs are shown as triangles with respective locations of the 5′-end base from the reported transcription start sites. The thick line (−1285/+362) on the human ABCG2 indicates the DNA segment used in the reporter assays. The approximate positions of the ChIP probes are shown as small arrows. B and C, C2bbe1 cells were treated with 10 nM TCDD or DMSO for 4 h. ChIP shows AHR recruitment to the proximal DRE region of ABCG2. The known CYP1A1 DRE region was amplified and used as positive control. Nonspecific host antibody (ns IgG) was used as negative control. B, quantitation of DREs was done by real-time PCR. Data were normalized to input DNA and expressed as the fold difference from the DMSO-treated control (mean and S.E. of three experiments). Statistical tests were one-way ANOVA: F2,6 = 5.97, p = 0.037; post hoc pair-wise comparison: ∗, p = 0.032 from DMSO. CYP1A1 DRE as positive control also showed similar results (one-way ANOVA: F2,6 = 32.16, p < 0.001; post hoc pair-wise comparison: ∗∗, p < 0.001 from DMSO). C, conventional PCR analysis (35 cycles) for proximal DREs of ABCG2 and CYP1A1 was also carried out to confirm the specificity of PCR amplification and quantitation in SybGREEN real-time PCR. Representative agarose gel images are shown. C, vehicle control treatment; T, 10 nM TCDD treatment.
TCDD-Induced Reporter Activity of the Proximal DREs Is AHR-Dependent.
We then used luciferase-based reporter assays to ascertain whether any of the proximal DREs are functional. The ABCG2 gene promoter reporter construct (−1285/+362) contains two core DREs (−194/−190 and −59/−55) upstream and two DREs (+148/+152 and +236/+240) downstream of the transcriptional start site (Fig. 5A). The −1285/+362 reporter construct transfected into the AHR-deficient MCF7 AHR100 cells responded to TCDD treatment after coexpression with an AHR expression vector (Fig. 6A). Consistent with this, AHR knockdown in the C2bbe1 cells caused a significant decrease in TCDD response in similar reporter construct (Fig. 6B). These findings convincingly suggest that the −1285/+362 ABCG2 promoter construct contains active DRE(s), which is AHR-driven.

Active DRE in the human ABCG2 5′-flanking region. A, activity of the ABCG2 reporter construct (−1285/+362) in the AHR-deficient MCF7 AHR100 cells transfected with AHR expression vector and/or 24-h 10 nM TCDD treatment. B, activity of the ABCG2 reporter construct (−1285/+362) in C2bbe1 cells transfected with siRNA against AHR with or without 24-h TCDD treatment. Results were fold changes to DMSO-exposed, empty vector-transfected control (A) or to DMSO-exposed, control siRNA-treated cells (B). One-way ANOVA (A, F2,6 = 25.32, p = 0.001; B, F2,6 = 24.19, p = 0.001); post hoc pair-wise comparisons (A, ∗, p < 0.01 from +AHR/−TCDD and −AHR/+TCDD cells; B, ∗∗, p < 0.005 from −siRNA/+TCDD cells). C, MCF7 AHR100 cells were cotransfected with the reporter constructs and the AHR expression vector or the empty vector and exposed to 10 nM TCDD or DMSO for 24 h. Each DRE (A, −194/−190; B, −59/−55; C, +148/+152; D, +236/+240) is schematically shown as a triangle (▶, 5′-GCGTG; ▷, 5′-CACGC). Results were fold changes from DMSO-treated, empty vector-transfected control for each construct. One-way ANOVA (p < 0.01 for the −1285, −628, −312, and −243 reporters; not significant for the −115 construct); post hoc pair-wise comparisons: ∗, p < 0.025; ∗∗, p < 0.01; ∗∗∗, p < 0.001 from DMSO-treated AHR-transfected cells (ns, not significant). D, individual −194/−190 ABCG2 DRE, its mutant, Cyp1a1 DRE, and its mutant reporter constructs were examined (two mutations were introduced to DRE: ▼) in an SV40 promoter-driven luciferase (pGL3-promoter). Protocols and results are as described in C. One-way ANOVA (F4,10 = 15.90, p < 0.001); post hoc pair-wise comparisons (significant results, ∗, p = 0.014; ∗∗, p < 0.01).
Localization of Active DRE.
Sequential deletion analyses of the −1285/+362 reporter construct showed that all reporter constructs, except that of −115/+362, had significantly increased activities in response to AHR coexpression, and the activity could be further augmented by 24-h TCDD treatment (Fig. 6C). TCDD response of the −115/+362 reporter construct was marginal if not completely absent. Comparable findings were seen when similar experiments were done in C2bbe1 cells (data not shown). These findings indicate that the −194/−190 DRE, which is absent in the −115/+362 reporter construct, is a key motif for AHR-mediated induction of ABCG2. Because the −115/+362 reporter construct retained noticeable enhanced activity with AHR coexpression alone, contribution of the other DREs and/or indirect effects of AHR through alternative transcription factor(s) that bind to respective enhancer site present in the construct cannot be ruled out.
We then examined the gene-transactivation activity of individual proximal DREs with special consideration to minimize indirect effects of AHR through alternative pathways. A 25-bp DNA sequence containing each of the four proximal DREs and its obligatory flanking nucleotides was subcloned into a similar luciferase-based reporter driven by a SV40 promoter. Consistent with earlier findings, the −194/−190 DRE was the only motif showing significantly higher activity upon AHR coexpression and TCDD treatment (Fig. 6D). With similar treatments, the activity of the other three proximal DREs was subtle (∼1.2-fold) and did not differ significantly from that of the empty vector-transfected control (data not shown). Moreover, introducing mutations into the core sequence of the −194/−190 DRE and positive control Cyp1a1 DRE completely abolished their reporter activity (Fig. 6D). These findings suggest that the −194/−190 DRE is an AHR-driven enhancer of human ABCG2.
Specific Binding of AHR to the −194/−190 DRE.
EMSA was conducted to examine the ability of the −194/−190 DRE to bind AHR. Nuclear extracts of C2bbe1 cells exposed to TCDD caused a more intense band, indicative of a DRE-AHR protein complex, retarded on the gel compared with those of vehicle treatment (Fig. 7). Excess amounts of unlabeled DRE oligonucleotides were able to reduce the complex formation, as evidenced by reduced band intensity. This, however, was not noted with excess mutant DRE, suggesting the requirement of an intact core DRE sequence to form this DNA-protein complex. The fact that coincubation with an AHR antibody reduced the intensity of this band confirms the presence of AHR in this DNA-protein complex. Instead of a supershifted band, studies have shown a reduction of AHR/ARNT-DRE complex formation in the presence of an AHR antibody (Rivera et al., 2007). The Cyp1a1 DRE used as positive control also showed similar results (Fig. 7). It is worth mentioning that the reported palindromic estrogen response element (ERE) in the human ABCG2 promoter (Ee et al., 2004) is in the immediate neighboring region of the −194/−190 DRE. The oligonucleotide DRE probe used in our EMSA analysis, however, does not include the full palindromic ERE sequence necessary for binding of the dimer estrogen receptors, rendering contribution of ERE to AHR-DRE binding unlikely. Taken altogether, our data indicate that AHR-mediated transcriptional induction of ABCG2 is, at least partly, through this −194/−190 DRE.

Direct AHR binding to −194/−190 ABCG2 DRE. Biotinylated DNA probes (25 bp) containing the −194/−190 DRE or Cyp1a1 DRE were incubated with nuclear extracts of C2bbe1 cells treated with DMSO or 10 nM TCDD in the presence or absence of excess unlabeled probe or the mutant DRE. A mouse monoclonal antibody against AHR was used to show antibody-induced reduction of intensity of the AHR-protein complex band (arrow). Incubation with equal amounts of a nonspecific mouse monoclonal antibody did not reduce the band intensity.
Discussion
In this study, we show that induction of human ABCG2 by AHR ligands is accomplished, at least in part, by direct AHR binding to an active, proximal DRE located at the −194/−190 bp upstream from the reported transcription start site. Our findings expand the roles of AHR into adaptive regulation of the BCRP transporter. This is the first report linking the AHR/ARNT-DRE transcription system directly to the regulation of an important ABC efflux transporter in humans. Our studies provide evidence that AHR-induced ABCG2 is common in various carcinoma and primary cell lines, particularly those of the liver and intestine, which are important sites of xenobiotic detoxification.
We further show that the proximal DREs in the human ABCG2 promoter, which include the −194/−190 DRE, do not have conserved counterparts in that of the mouse Abcg2 gene. Our phyloHMM-based analysis revealed a general paucity of conserved DREs in the regulatory regions of the ABCG2 gene between humans and mice. In addition, the 5′-flanking region of mouse Abcg2 contains much less putative DREs than that of humans. All of this serves to explain our in vitro and in vivo observations that mouse Abcg2 did not respond to TCDD treatment. Similar negative findings were reported in mice exposed to another AHR ligand, 3MC (Han and Sugiyama, 2006). Gene-expression profiling analysis of both Ahr-null and wild-type mice exposed to TCDD showed no increase of Abcg2 mRNA despite decreased constitutive or basal Abcg2 gene transcripts in the Ahr-null mice (Tijet et al., 2006; Boutros et al., 2009). Our findings further shed light on the possible widespread species divergence of AHR-regulated genes. Recent data from the transcriptome profiling analysis have witnessed different target genes in response to TCDD treatment across species (Boutros et al., 2008; Carlson et al., 2009). At present, it is still unclear whether the difference in the AHR-mediated adaptive induction of ABCG2 between humans and mice constitutes an underlying factor to the species-dependent spectrum of AHR-mediated toxicities.
Krishnamurthy et al. (2004) reported that mouse and human Abcg2/ABCG2 expression is increased upon exposure to hypoxia in a manner dependent on HIF-1α. Using a similar reporter construct (−312/+362) of the human ABCG2 promoter, two hypoxia response elements (HREs) were identified. The core HRE motif (5′-[A/G]CGTG) is similar to the core DRE (5′-GCGTG). It is intriguing that one of the two HREs (−194/−190) identified by this group exactly overlaps the active DRE shown to bind AHR in the present study. Although this HRE was shown to have much weaker transactivation activity than the downstream motif (−115/−111), it is possible that the −194/−190 motif, depending on predominant cellular milieu and exogenous stimuli, is dually controlled by AHR and HIF-1α. Similar findings of a binding site on the ABCG2 promoter shared by two transcription factors were also reported for ERα and PgR (Ee et al., 2004; Wang et al., 2008).
The observation that these enhancer motifs are in such close proximity (∼15 bp) to each other may denote that the neighborhood region of these motifs is a crucial recruitment site for transcriptional activation machinery on the human ABCG2 gene. Whether there exists competition or collaboration of the above transcription factors in ABCG2 regulation is a subject for future studies. For example, cross-talk between ER and AHR has been a focus of intense research because, depending on experimental conditions and cell context, AHR ligands can lead to paradoxical dysregulation or enhancement/synergism of ER-mediated pathways (Ohtake et al., 2003; Abdelrahim et al., 2006). In our studies, it is nevertheless unlikely that the ER/ERE pathways contribute to the effects of AHR ligands on the induction of ABCG2. It was noticed that the ER-deficient cell lines, notably C2bbe1 and HepG2, exerted a similar response in inducing ABCG2 upon AHR ligand exposure, whereas the AHR-deficient, ER-expressing MCF7 AHR100 cells are deprived of this ability.
In addition to the −194/−190 DRE, it is possible that other active DRE(s) in regulation of ABCG2 exist. Our preliminary studies suggested that some distal putative DREs (Fig. 5A) may have gene-transactivation activities that are much lower than the proximal −194/−190 DRE (data not shown). We also believe that AHR-mediated induction of BCRP may as well be operated indirectly by activating other transcription factors such as Nrf2 (K. P. Tan and S. Ito, unpublished data) whose gene regulation is reportedly under AHR regulation (Miao et al., 2005). There are many putative Nrf2 binding motifs known as the antioxidant response elements along the 5′-regulatory region of human ABCG2 (data not shown). Studies have shown that many ABC transporters such as MRP 2–4 (ABCC 2–4) are regulated by Nrf2 (Maher et al., 2007). In addition, AHR-mediated gene induction may be pursued without AHR's binding to a DRE. Studies have revealed an AHR-mediated p-38 mitogen-activated protein kinase signaling pathway in the up-regulation of c-Jun expression (Weiss et al., 2005).
Because most AHR agonists are highly carcinogenic and toxic, human intervention studies on the effects of AHR ligands are ethically impossible. Nonetheless, there are observational studies that may provide possible effects of AHR activation on BCRP in humans exposed to substances that contain numerous AHR inducers such as those in tobacco smoke. In two studies, moderate increases of placental BCRP expression and/or activity were detected in mothers who smoked compared with those of nonsmokers (Kolwankar et al., 2005; Huuskonen et al., 2008). Because of a small sample size (n = 5 and 10 for each group) and substantial interindividual variability, however, statistical significance of these results was not reached. In a clinical study, irinotecan and its active metabolite, 7-ethyl-10-hydroxycamptothecin (SN-38), were found to be eliminated significantly faster in smokers than in nonsmokers, indicating a potential smoking-induced risk of therapeutic failure (van der Bol et al., 2007). Given that irinotecan and SN-38 are preferential substrates of BCRP (Houghton et al., 2004), the authors suggest that enhanced BCRP-mediated elimination of these drugs might have occurred among the smokers. Despite the absence of direct evidence, indirect data from human studies in subjects exposed to tobacco smoke seem to suggest a potential effect of AHR-mediated induction of ABCG2 (BCRP) that actually occurs in the human population.
In conclusion, this is the first report to illustrate that an important phase III efflux transporter, ABCG2/BCRP, is transcriptionally regulated by AHR. The resultant enhancement of BCRP function represents an adaptive response coordinated by AHR to eliminate toxicants and carcinogens that are directly sensed by AHR or are consequently produced by its activated phase I/II enzymes, like CYP1As. Studies have shown that AHR agonists poly- and halogenated aromatic hydrocarbons and benzo(a)pyrene are substrates of BCRP (Kondo et al., 2004; Ebert et al., 2005). This human-specific adaptive response regulation of AHR sheds light on species-specific toxicity profile and may indicate a possible resistance of humans compared with rodents to AHR ligand-induced carcinogenesis and health adversities.
Acknowledgments
We sincerely thank Dr. Ciolino (Cellular Defense and Carcinogenesis Section, National Cancer Institute-Frederick Cancer Research and Development Center, Frederick, MD) for providing MCF7 AHR100 cells. We greatly appreciate Dr. David Williams (University of Toronto) for providing the calnexin antibody, Dr. Allan B. Okey for insightful discussions, and Song Gao and Derrick Tam for technical assistance.
Footnotes
-
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material. -
This work was supported by the Canadian Institute of Health Research [Grant MT13747] and the VA Merit Review.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.110.065078.
-
ABBREVIATIONS:
- BCRP
- breast cancer resistance protein
- ABC
- ATP-binding cassette
- ABCG2
- ATP-binding cassette G2 protein
- AHR
- aryl hydrocarbon receptor
- ChIP
- chromatin immunoprecipitation
- DBA
- dimethyl-benzo(a)pyrene
- DMF
- 3,4-dimethoxyflavone
- DMSO
- dimethyl sulfoxide
- DRE
- dioxin response element
- EMSA
- electrophoretic mobility shift assay
- ERα
- estrogen receptor-α
- FTC
- fumitremorgin C
- HIF-1α
- hypoxia-inducible factor 1α
- 3MC
- 3-methylcholanthrene
- Nrf2
- nuclear factor (erythroid 2-like) factor 2
- PgR
- progesterone receptor
- PCR
- polymerase chain reaction
- siRNA
- small interfering RNA
- TCDD
- 2,3,7,8-tetrachlorodibenzo-p-dioxin
- bp
- base pair
- RT-PCR
- reverse-transcriptase polymerase chain reaction
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- SV40
- simian virus 40
- ANOVA
- analysis of variance
- ERE
- estrogen response element
- HRE
- hypoxia response element
- ARNT
- aryl hydrocarbon receptor nuclear translocator
- PD98059
- 2′-amino-3′-methoxyflavone
- SN-38
- 7-ethyl-10-hydroxycamptothecin.

- Received March 25, 2010.
- Accepted May 5, 2010.
- U.S. Government work not protected by U.S. copyright





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}