


Abstract
Rat canalicular membranes contain microdomains enriched in cholesterol and ATP-binding cassette transporters. Cholesterol is known to regulate the activity of transporters. Here, we investigated the effect of membrane cholesterol on the transport kinetics of multidrug resistance–associated protein 2 (MRP2) and of bile salt export pump (BSEP) variants and mutants. MRP2 and BSEP were expressed with baculoviruses in insect cells, followed by vesicle isolation from control and cholesterol-loaded cells (1 mM cholesterol@randomly methylated-β-cyclodextrin) for transport assays. We found that cholesterol stimulates MRP2 transport activity for substrates of different molecular weights: estradiol-17-β-glucuronide (E17βG), prostaglandin E2 (PGE2), cholecystokinin 8 (CCK8), and vasopressin displayed an increase of Vmax and a variable decrease of Km. Kinetics of E17βG showed a sigmoidal shape and a mild cooperativity in Hanes–Woolf plots in control membranes. High cholesterol content shifted E17βG to Michaelis–Menten kinetics. PGE2/glutathione transport followed Michaelis–Menten kinetics irrespective of cholesterol. The MRP2 substrates CCK8 and vasopressin exhibited Michaelis–Menten kinetics independent of membrane cholesterol content. Transport of ochratoxin A was ATP-dependent but was neither mediated by MRP2 nor stimulated by cholesterol. Transport of the two most common BSEP variants p.444V/A showed Michaelis–Menten kinetics irrespective of membrane cholesterol, whereby cholesterol leads to an increased Vmax while Km remains unchanged. The transport activity of the BSEP mutants p.E297G and p.R432T increased at high cholesterol content but did not reach the capacity of normal BSEP. Hence, changing membrane cholesterol content modulates BSEP and MRP2 transport kinetics differently. Cholesterol increases the transport rates of BSEP and MRP2, but with the latter, may also modify the binding site as for E17βG.
Introduction
Bile is constantly produced by the liver, contains a high concentration of bile salts, and provides a route for hepatocellular elimination of poorly water-soluble substances (Small, 2003). Bile salts and biliary phospholipids form mixed micelles, which protect the biliary system from the detergent action of bile salts (Trauner et al., 2008). Despite the partitioning of bile salts into mixed micelles, monomeric bile salt concentration in bile remains high (Roda et al., 1995). Since bile salts have detergent properties (Perez and Briz, 2009) and are cytotoxic (Utanohara et al., 2005), their high concentration can potentially damage the canalicular plasma membrane (cLPM). Furthermore, elevated concentrations of bile salts in hepatocytes impair hepatocellular functions.
Bile formation works against steep concentration gradients of solutes, and therefore involves several ATP-binding cassette transporters (ABC transporters) (Small, 2003): bile salts are secreted by the bile salt export pump (BSEP; ABCB11), which drives bile salt–dependent bile flow and has a rather narrow substrate specificity (Stieger, 2011). The multidrug resistance–associated protein 2 (MRP2; ABCC2) transports a wide range of organic anions (mostly conjugates), which are the driving force for bile salt–independent bile flow (Nies and Keppler, 2007). Phosphatidylcholine is translocated to the outer leaflet of the cLPM by multidrug resistance protein 3 (MDR3; ABCB4) (Oude Elferink and Paulusma, 2007), and cholesterol release from the cLPM is facilitated by the heterodimeric transporter ABCG5/ABCG8 (ABCG5, ABCG8) (Hazard and Patel, 2007).
We have recently demonstrated that cLPM contains detergent- and bile salt–resistant microdomains enriched in sphingomyelin and cholesterol, in which several ABC transporters, such as BSEP, MRP2, and MDR1, are located (Ismair et al., 2009; Guyot and Stieger, 2011). These microdomains may help protect the cLPM from solubilization by bile salts. Cholesterol, which is enriched in microdomains, has been shown to modulate the activity of several ABC transporters such as BSEP, MRP2, ABCG2, or MDR1 (Kimura et al., 2007a; Telbisz et al., 2007; Ito et al., 2008; Kis et al., 2009). Partitioning of ABC transporters in and out of cholesterol-rich microdomains could passively regulate the canalicular secretion of the respective substrates of these ABC transporters.
The 3.8 Å structure of mouse Mdr1a reveals a large substrate-binding pocket, explaining how Mdr1a is able to cope with a vast variety of compounds having a wide range of molecular mass (MM) (Aller et al., 2009). It was demonstrated that cholesterol stimulates MDR1 ATPase activity induced by small drugs (<500 Da), has no effect on ATPase activity stimulated by large drugs (800–900 Da), and suppresses ATPase activity induced by larger drugs (>1000 Da) (Kimura et al., 2007a). These findings suggest that the drug-binding site of MDR1 fits to drugs with a size of 800–900 Da, and that cholesterol might support the recognition of smaller drugs by tightening the drug-binding site (Kimura et al., 2007b).
Similar to MDR1, MRP2 transports a large variety of substrates with largely different MM (Hirono et al., 2005). Hence, membrane cholesterol content may also differentially stimulate transport of small and large MRP2 substrates. On the other hand, BSEP has a much more restricted substrate specificity (Stieger et al., 2007; Stieger, 2011), which may lead to a different effect of membrane cholesterol content on its transport properties. Several dozen mutations in the ABCB11 gene have been characterized in terms of expression and function. Often, BSEP function was studied in insect cells (Stieger, 2011), known for their very low cholesterol content (Gimpl et al., 1995), but the impact of membrane cholesterol on BSEP mutants awaits investigation. BSEP function may be inhibited by drugs (Morgan et al., 2010; Stieger, 2010; Dawson et al., 2012). We have presented evidence that the p.444A variant of BSEP, which constitutes the major polymorphic variant, is a susceptibility factor for drug- and estrogen-induced cholestasis but is kinetically indistinguishable from the p.444V variant (Lang et al., 2007; Meier et al., 2008). Although the two variants show similar kinetic properties, the p.444A variant tends toward lower protein levels in human liver (Meier et al., 2006). These two BSEP variants could potentially interact differently with membrane cholesterol. Therefore, here we studied the effect of cholesterol on transport activity and kinetic parameters of the two most common variants of BSEP and on two mutant forms of BSEP with decreased transport activity (Noe et al., 2005) and compared the effect of cholesterol on kinetic parameters of MRP2 assessed with low- and high-MM substrates.
Materials and Methods
Radiolabeled [3H]estradiol-17-β-D-glucuronide ([3H]E17βG; 45 Ci/mmol), [3H]taurocholic acid (4.6 Ci/mmol), [3H]cholecystokinin 8 ([3H]CCK8; 70–80.2 Ci/mmol), [5,6,8,11,12,14,15-3H(N)]prostaglandin E2 ([5,6,8,11,12,14,15-3H(N)]PGE2; 181.7–200.0 Ci/mmol), and [phenlylalanyl-3,4,5-3H(N)]vasopressin (8-L-arginine) (55.9 Ci/mmol) were purchased from PerkinElmer (Schwerzenbach, Switzerland). [3H]Ochratoxin A ([3H]OTA; 10.6–26.0 Ci/mol) was obtained from PerkinElmer or from Moravek Biochemicals (Brea, CA). CCK8 was purchased from Bachem (Bubendorf, Switzerland), and taurocholic acid sodium salt, E17βG, OTA, and vasopressin were purchased from Sigma-Aldrich (St. Louis, MO). PGE2 was supplied by Cayman Chemical (Ann Arbor, MI). Randomly methylated-β-cyclodextrin (RAMEB) and a cholesterol complex of RAMEB (cholesterol@RAMEB) were purchased from Cyclolab (Budapest, Hungary). Purified polyclonal rabbit antibody against BSEP was described in Noe et al. (2002), and mouse monoclonal antibody against MRP2 was purchased from Enzo Life Sciences (Lausen, Switzerland). Horseradish peroxidase–conjugated secondary antibodies were purchased from GE Healthcare (Little Chalfont, UK). Hanks’ balanced saline solution (HBSS) was purchased from Gibco/Invitrogen (Carlsbad, CA).
Transporter Expression in Sf9 and Sf21 Insect Cells and Membrane Isolation.
Human BSEP was expressed in Sf9 insect cells using the baculovirus system as described in Noe et al. (2002). Human MRP2 was expressed in Sf21 insect cells using the baculovirus system as described in de Waart et al. (2006). Membranes from insect cells expressing the different ABC transporters were isolated as described in Gerloff et al. (1998), resuspended in 50 mM sucrose, 100 mM KNO3, 10 mM HEPES/Tris pH 7.4, and stored in liquid nitrogen until use. Protein content was determined with the bicinchoninic acid method using a kit from Interchim (Montlucon, France).
Cholesterol Loading.
For cholesterol loading of infected Sf9 cells, we compared two different protocols. In the first protocol, the loading was performed with intact cells as described in Kis et al. (2009). In brief, cells were collected and treated for 30 minutes at 37°C with 10 times their volume of HBSS containing 1 mM cholesterol@RAMEB. After 30 minutes, cells were washed in HBSS (in 20 times the initial volume). Membrane vesicle isolation from cholesterol-loaded and mock-treated virus-infected cells was performed as described earlier. The second loading protocol was performed with vesicles isolated as described in Telbisz et al. (2007). In brief, isolated vesicles were resuspended in cholesterol@RAMEB in 50 mM sucrose, 100 mM KNO3, and 10 mM HEPES/Tris pH 7.4 at a final cholesterol concentration of 2 mM and incubated at 37°C for 45 minutes. After the incubation, cold buffer was added and vesicles were recovered by centrifugation and used for transport experiments. The efficiencies of the cholesterol loading and taurocholate transport activity of wild-type BSEP expressing Sf9 cell vesicles are given in Table 1.
Comparison of different cholesterol-loading protocols
Taurocholate transport activity of wild-type BSEP-expressing Sf9 cell vesicles was determined after cholesterol loading with two different protocols. For each protocol, the cholesterol enrichment was calculated after lipid extraction and determination of cholesterol amount by thin-layer chromatography. Transport experiments were realized as described in Materials and Methods. Cholesterol enrichment and transport studies were reproduced at least two times independently.
Transport Studies.
ATP-dependent transport was determined by a rapid filtration technique as described in Gerloff et al. (1998). In brief, BSEP and MRP2 membrane vesicles (70 µg protein) were incubated with 50 mM sucrose, 100 mM KNO3, 12.5 mM Mg(NO3)2, and 10 mM HEPES/Tris pH 7.4, supplemented with [3H]-labeled and cold substrate at concentrations given in the figure legends in the presence or absence of 5 mM ATP. For CCK8 and vasopressin transport experiments, BSA was added to the uptake solution (to prevent proteolysis of the substrate) at 1 mg/ml and to the cold stop solution at 10 mg/ml to prevent nonspecific filter binding. For the transport experiments with PGE2, glutathione (GSH) and dithiothreitol (5 mM each) were added to the incubation solutions (de Waart et al., 2006), unless stated otherwise. For kinetic experiments, linearity of initial uptake rates as a function of time was determined in preliminary experiments (data not shown). All data were obtained from at least two independent experiments (except for PGE2 kinetics) and are presented as the mean ± S.D. for individual experiments. As the transporter expression levels varied to some extent between different membrane batches, only representative individual experiments are presented. Kinetic parameters were obtained by fitting initial uptake rates to the Michaelis–Menten equation by nonlinear regression analysis with Prism 5 (GraphPad Software, San Diego, CA). The same program was used to transform and plot the data for the Hanes–Woolf plots (Neet, 1980).
Lipid Determination and Western Blotting.
To assess cholesterol enrichment, lipids were extracted from isolated membrane vesicles as described in Bligh and Dyer (1959). Extracted lipids were analyzed by thin-layer chromatography for cholesterol amount as described in Ismair et al. (2009). The amount of total phospholipids was determined as described in Rouser et al. (1970). The lipid amounts were normalized to the amount of protein of the vesicles used for lipid extractions to be able to compare vesicles obtained from different isolations. To assess BSEP and MRP2 expression levels in cholesterol-loaded and mock-treated vesicles, 30 µg of vesicles was processed by SDS–polyacrylamide electrophoresis and transferred to nitrocellulose membranes as described in Stieger et al. (1994). For detection of proteins, nitrocellulose membranes were blocked for 1 hour with 5% (w/v) nonfat dry milk dissolved in Tris-buffered saline/Tween 20 (TBST) [10 mM Tris-HCl pH 7.6, 150 mM NaCl, 0.1% (v/v) Tween 20] and incubated with primary antibodies in TBST for 2 hours at room temperature by three 10-minute washes in TBST. The blots were then incubated for 1 hour at room temperature with the appropriate secondary antibodies diluted in TBST/5% (w/v) dry milk, washed three times in TBST, and developed with the UptiLight chemiluminescence reagent (Interchim).
Results
Since several protocols for membrane loading with cholesterol were available from the literature, we decided to compare two of them. The cholesterol enrichment of membranes after loading and the effect of cholesterol loading on BSEP function were studied on Sf9 cells infected with BSEP (Table 1). Cholesterol loading of intact cells or of isolated vesicles leads to an 8-fold increase in cholesterol content normalized to protein content. We observed a 2.2- and 1.5-fold increase in transport activity when the cholesterol loading was performed with intact cells (protocol 1) or isolated vesicles (protocol 2), respectively. The ATP-dependent transport after loading with protocol 1 is considerably larger than after cholesterol loading with protocol 2. This may be explained by slightly different BSEP expression levels between different infections, and more importantly by the stress put on the vesicles in protocol 2, which, after their isolation, went through additional incubations and high-speed centrifugation. As a consequence, we decided to use loading protocol 1 for further experiments. To test a potential impact of cholesterol loading on membrane lipid composition, total phospholipid content and cholesterol content of Sf9 and Sf21 vesicles were determined in three independent experiments. Control Sf9 cell vesicles contain 0.22 ± 0.021 μmol/mg total phospholipids and 0.020 ± 0.017 μmol/mg protein cholesterol, resulting in a phospholipid-to-cholesterol ratio of 16.0 ± 9.3. In cholesterol-loaded vesicles, these values were 0.33 ± 0.046 μmol/mg protein total phospholipids and 0.194 ± 0.187 μmol/mg protein cholesterol with a ratio of 1.9 ± 1.2. Sf21 cell vesicles displayed a higher total phospholipid content—namely, 0.493 ± 0.071 μmol/mg protein before and 0.529 ± 0.034 μmol/mg protein after cholesterol loading. The cholesterol content was 0.023 ± 0.012 μmol/mg protein before and 0.145 ± 0.033 μmol/mg protein after loading, resulting in a phospholipid-to-cholesterol ratio of 25.1 ± 11.1 before and 3.8 ± 0.7 after cholesterol loading. Hence, the total phospholipid content of the insect cell vesicles was not affected by cholesterol loading. Comparison of the total phospholipid content of insect cell vesicles to published values for highly purified rat cLPM vesicles [0.83 μmol/mg protein (Meier et al., 1984), 0.67 μmol/mg protein (Bossard et al., 1993)] reveals that, in rat cLPM, the total phospholipid content is about twice the amount determined here in insect cell vesicles. Nevertheless, the total phospholipid-to-cholesterol ratio is comparable in insect cells after cholesterol loading to reported rat cLPM membrane vesicles [1.8 (Meier et al., 1984), 3.9 (Bossard et al., 1993)].
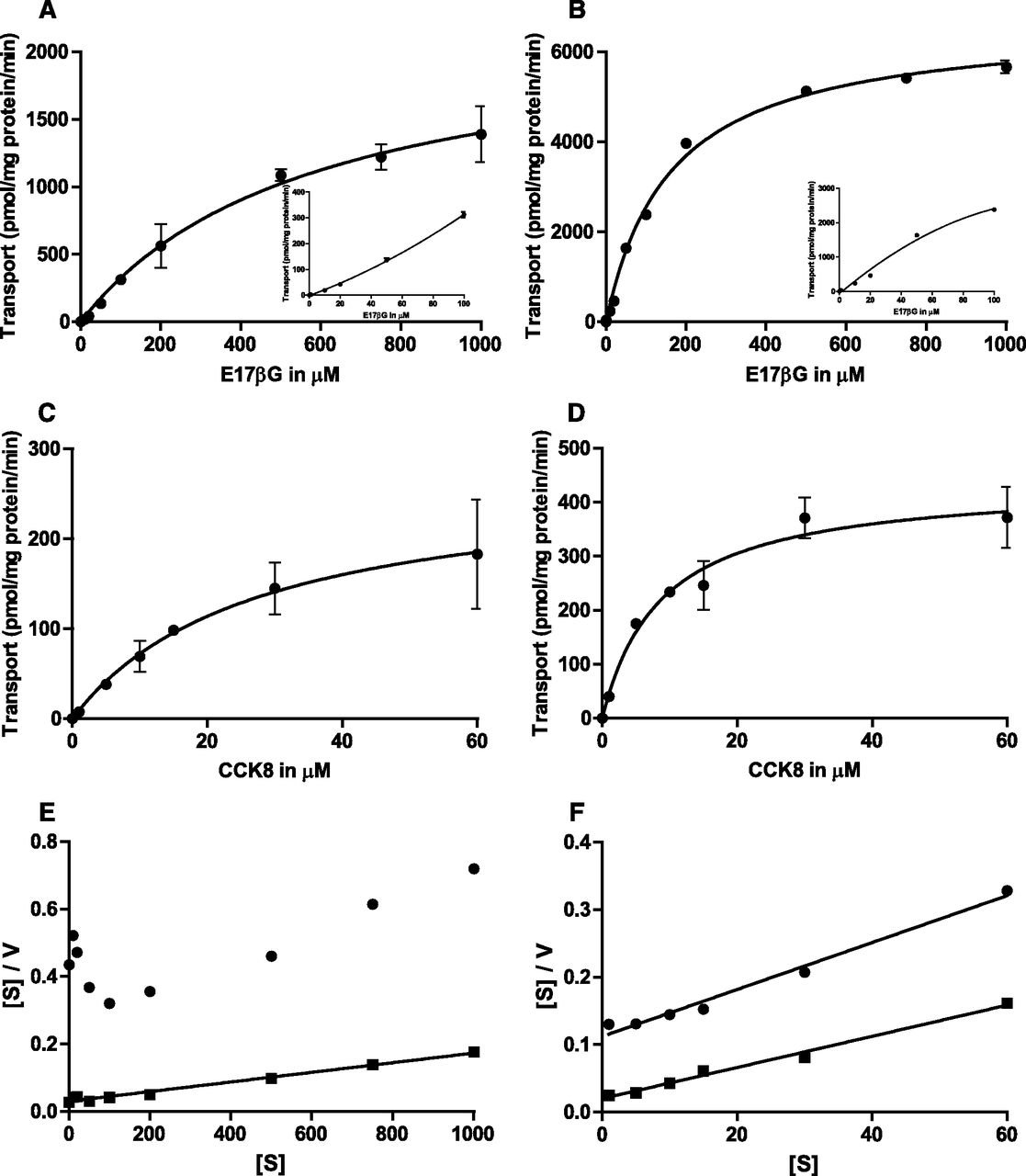
To study the effect of cholesterol on MRP2 transport activity, Sf21 cells infected with MRP2 were loaded with cholesterol as described in Materials and Methods. Vesicles were analyzed for MRP2 expression by Western blotting. Figure 1A shows a representative Western blot of the cholesterol-loaded and control vesicles, demonstrating that cholesterol treatment had no effect on MRP2 expression level. Figure 1B demonstrates that the cholesterol content is massively increased in membrane vesicles after cholesterol loading (0.413 ± 0.012 μmol cholesterol per mg of protein; n = 8) compared with mock-treated cells (0.038 ± 0.0017 μmol cholesterol per mg of protein; n = 8), corresponding to an 11-fold increase in the specific cholesterol content. As MRP2 transports numerous substrates spanning a wide range of MM, we studied the effect of cholesterol on the transport of E17βG as a small substrate (MM 470) and of CCK8 as a large substrate (MM 1143). In contrast to others (Kopplow et al., 2005), we were not able to demonstrate MRP2-mediated estrone-3-sulfate (MM 372) transport in mock- or cholesterol-loaded Sf21 membrane vesicles (data not shown), confirming an earlier study (Sasaki et al., 2002). Figure 1 shows that cholesterol loading has a marked effect on ATP-dependent transport of MRP2-expressing Sf21 cell vesicles: E17βG uptake was increased about 8-fold in the presence of cholesterol (Fig. 1C), whereas CCK8 transport was increased about 6-fold in the presence of cholesterol (Fig. 1D). It is interesting to note that E17βG uptake, but not CCK8 uptake, was somewhat increased also in the absence of ATP after cholesterol loading (Fig. 1, C versus D).

Effect of cholesterol loading on MRP2 expression and transport activity. (A) Western blot analysis of MRP2-expressing vesicles with and without cholesterol loading (30 µg protein per lane). (B) Cholesterol content in MRP2-expressing vesicles detected by thin-layer chromatography; 50 and 5 µl of extracted lipids were loaded for nonloaded vesicles and vesicles loaded with cholesterol, respectively. (C) Initial rate of E17βG uptake (10 µM) for 5 minutes by membrane vesicles isolated from MRP2-expressing Sf21 cells with and without cholesterol loading. (D) Initial rate of CCK8 uptake (0.2 µM) for 10 minutes by membrane vesicles isolated from MRP2-expressing Sf21 cells with and without cholesterol loading. Uptake was measured in the presence (white bars) or absence of ATP (black bars), as described in Materials and Methods; the difference in the presence and absence of ATP is represented with gray bars. Values given are the mean ± S.D. of triplicate measurements of a representative experiment.
To distinguish between an effect of membrane cholesterol on the substrate affinity of MRP2 or on the turnover number, we next determined the kinetic parameters of E17βG and CCK8 transport by MRP2 (Fig. 2, A–D) in the presence and absence of cholesterol loading. In the absence of cholesterol loading, the uptake of E17βG as a function of substrate concentration was not consistent with classic Michaelis–Menten kinetics, but the plot showed the well known sigmoidal shape (Bodo et al., 2003; Zelcer et al., 2003) (Fig. 2A, insert). This sigmoidal shape indicates a mild cooperativity, which is clearly displayed in the U-shaped Hanes–Woolf plot (Fig. 2E). Very interestingly, after loading with cholesterol, E17βG uptake versus substrate concentration shifted to classic Michaelis–Menten kinetics (Fig. 2B), leading to a linear Hanes–Woolf plot (Neet, 1980) (Fig. 2E). The loading with cholesterol leads to an increase in Vmax (from 2214 to 6673 pmol/mg protein/min calculated for Fig. 2, A and B), whereas the estimated low-affinity Km (581 to 161 µM) is decreased. As we encountered solubility problems for E17βG at concentrations higher than 1 mM, this was the highest concentration used for the kinetic experiments. In contrast to E17βG, CCK8 transport by MRP2 showed a classic Michaelis–Menten behavior with linear Hanes–Woolf plots at low and high membrane cholesterol content (Fig. 2, C, D, and F). Loading with cholesterol leads to an increase in Vmax (from 269 to 438 pmol/mg protein/min calculated for Fig. 2, C and D), whereas the Km value (27 to 9 µM) is decreased. Hence, cholesterol loading affects the kinetic parameters of MRP2 similarly for low- and high-MM substrates.

Kinetics of ATP-dependent uptake of E17βG (A, B, and E) and CCK8 (C, D, and F) by MRP2 in membrane vesicles isolated from Sf21 cells. Initial uptake rates are given as the difference of uptake in the presence and absence of ATP. (A) Uptake in the presence of increasing concentrations of E17βG without cholesterol loading. (B) Uptake in the presence of increasing concentrations of E17βG after cholesterol loading. (C) Uptake in the presence of increasing concentrations of CCK8 without cholesterol loading. (D) Uptake in the presence of increasing concentrations of CCK8 after cholesterol loading. (E) Corresponding Hanes–Woolf plots for E17βG transport with (▪) and without cholesterol loading (●). (F) Corresponding Hanes–Woolf plots for CCK8 transport with (▪) and without cholesterol loading (●). (A–D) Values given are the mean ± S.D. of triplicate measurements, but in some cases, error bars are smaller than the size of the symbol.
Next, we determined the kinetic parameters for MRP2-mediated vasopressin transport (Madon et al., 2000). As for CCK8, cholesterol consistently stimulated ATP-dependent vasopressin transport (data not shown). Kinetic analysis revealed a low affinity of vasopressin to MRP2, both in the absence and presence of cholesterol, with high Km values (Fig. 3A): 315 μM in the absence of cholesterol and 103 μM in the presence of cholesterol. For technical reasons, no concentrations considerably higher than Km could be used. Therefore, the estimation of the Vmax value was subject to large uncertainty, but cholesterol loading increased this value (data not shown). The corresponding Hanes–Woolf plots are shown in Fig. 3B. Again, for this rather large MRP2 substrate (MM 1084), we found no evidence for cooperative kinetics in the absence or presence of cholesterol.
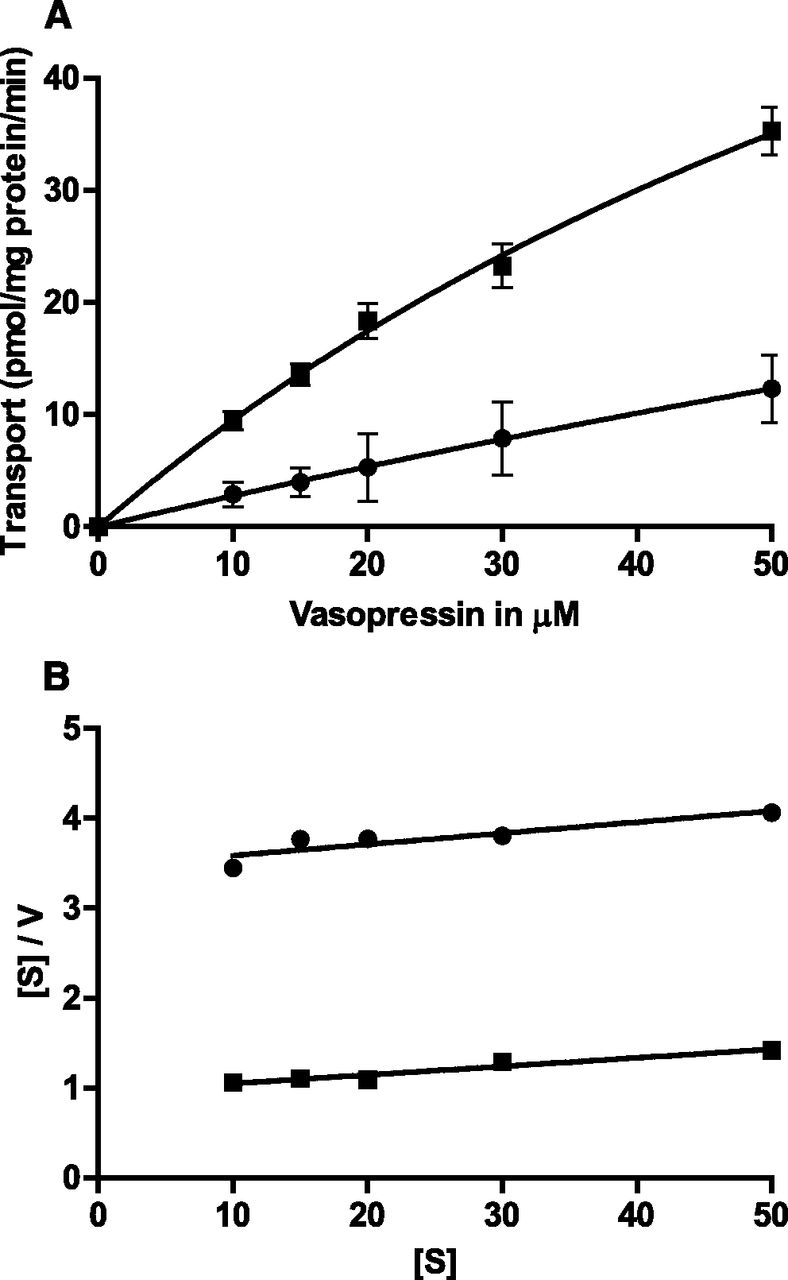
Kinetics of ATP-dependent uptake of vasopressin by MRP2 in membrane vesicles isolated from Sf21 cells. (A) Initial uptake rates in the presence of increasing vasopressin concentrations without (●) and with (▪) cholesterol loading. (B) Corresponding Hanes–Woolf plots without (●) and with (▪) cholesterol loading. (A) Values are the mean ± S.D. of quadruplicate determinations, but in some cases, error bars are smaller than the size of the symbols.
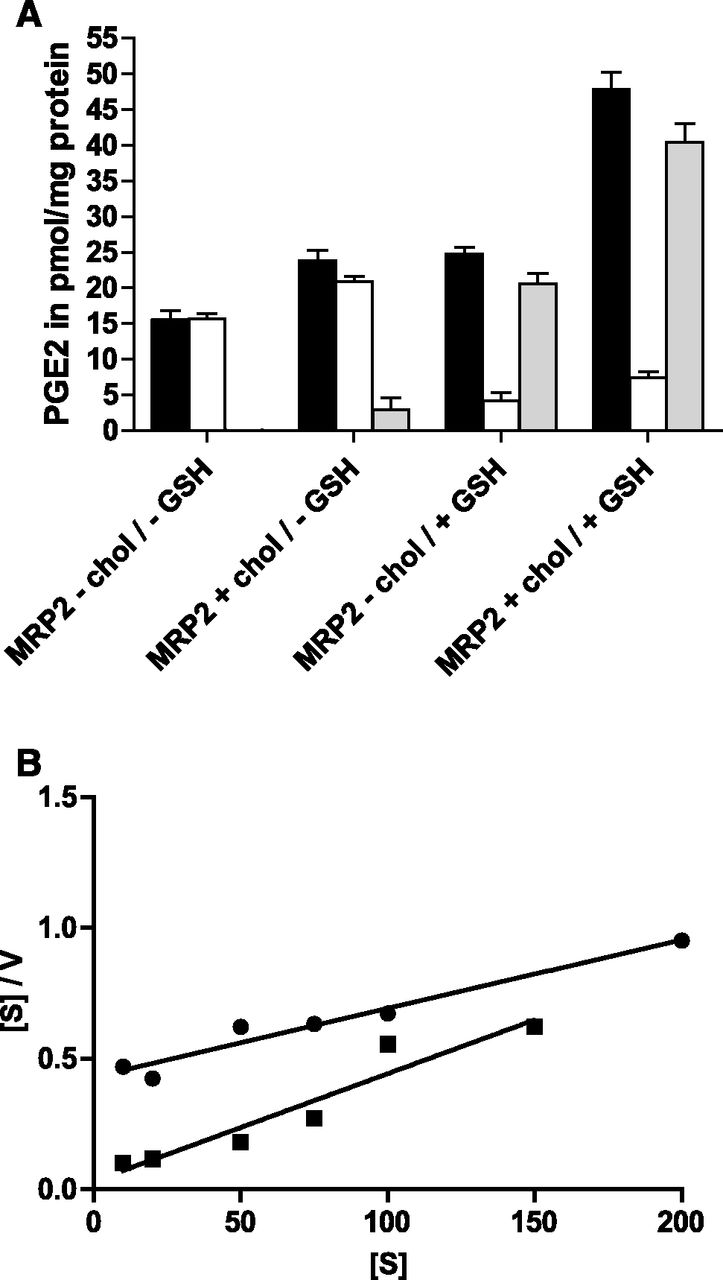
PGE2 is a substrate of MRP2 (de Waart et al., 2006) and has a MM of 353, which is comparable to the small substrate E17βG. Figure 4A shows that, in the absence of cholesterol, PGE2 transport depends on the presence of GSH. If dithiothreitol was added instead of GSH, only a marginal MRP2-dependent PGE2 transport was observed (data not shown), indicating that GSH is needed as a cosubstrate for MRP2. In vesicles containing cholesterol, a marginal stimulation of transport was observed in the absence of GSH (Fig. 4A), which, however, was not observed in all experiments (data not shown). We conclude from this that cholesterol cannot replace GSH as a cosubstrate. Interestingly, however, cholesterol was able to stimulate PGE2 transport in the presence of GSH. Initial kinetic experiments indicated no saturation of PGE2 transport in the nanomolar range (de Waart et al., 2006), irrespective of the presence of cholesterol. Consequently, we performed the kinetic analysis in the micromolar substrate range and found a large decrease of the Km value from 174 μM in the absence of cholesterol to 11.9 μM. The Hanes–Woolf plot in Fig. 4B shows that PGE2 transport did not display cooperativity either in the absence or in the presence of cholesterol. Due to the low affinity of this substrate for MRP2, the interpretation of the Vmax values was not feasible.
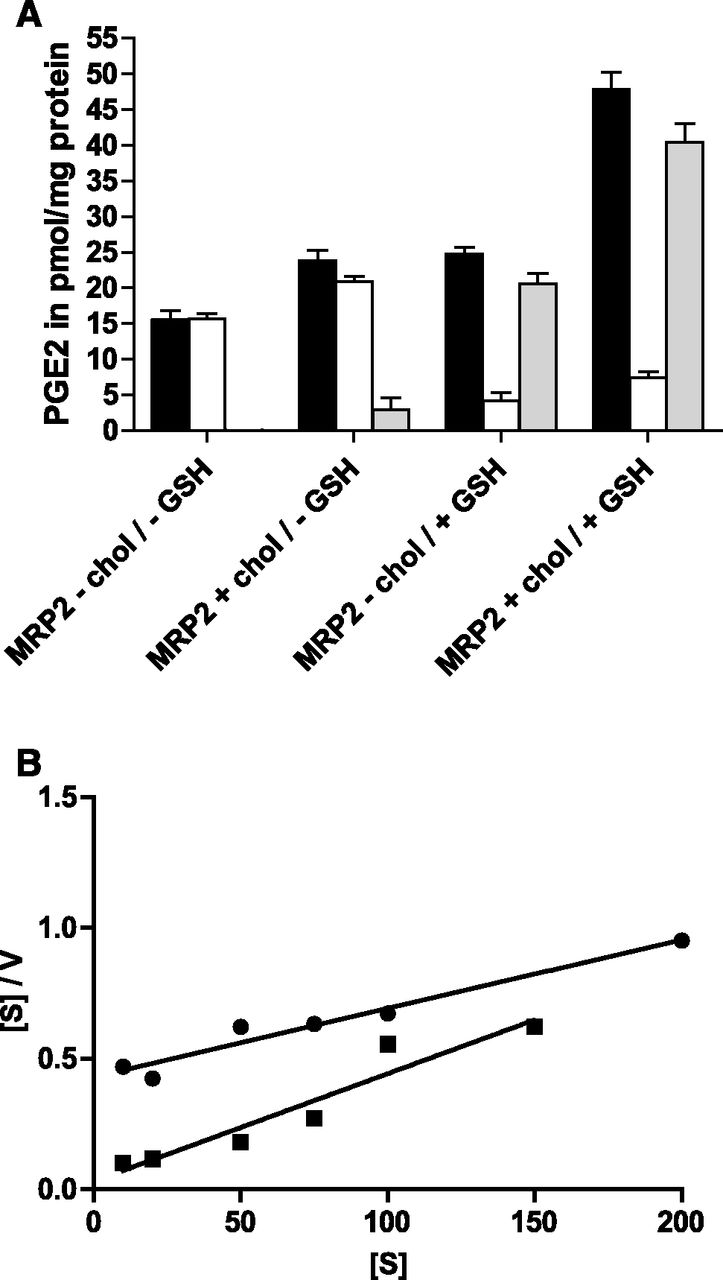
Effect of cholesterol loading on MRP2-mediated PGE2 transport in the presence and absence of GSH. (A) Uptake of 1 μM PGE2 was determined for 30 minutes in the absence or presence of 5 mM GSH in control and cholesterol (chol)-loaded vesicles. Uptake was measured in the presence (white bars) or absence of ATP (black bars) as described in Materials and Methods; the difference in the presence and absence of ATP is represented with gray bars. Values given are the mean ± S.D. of quadruplicate measurements of a representative experiment. (B) Hanes–Woolf plot for ATP-dependent PGE2 transport without (●) and with cholesterol loading (▪) at increasing concentrations of PGE2 under initial uptake rates. Values are means of quadruplicate determinations.
OTA has been reported to be an MRP2 substrate (Leier et al., 2000), has a MM of 404, and was therefore considered to be an additional small MRP2 substrate. Interestingly, for this substrate, we were not able to see a stimulation of transport activity by cholesterol (data not shown). Kinetic experiments pointed to OTA being a very low-affinity substrate for MRP2 with no evidence for cooperativity in the corresponding Hanes–Woolf plot (Fig. 5A). Furthermore, the addition of GSH did not change its transport (Fig. 5B). We therefore also tested wild-type Sf21 vesicles and found practically the same OTA transport activity as for vesicles isolated from MRP2-expressing cells, indicating that the observed ATP-dependent OTA transport is due to an endogenous transporter in insect cells but is not mediated by MRP2 (data not shown). In addition, we tested the effect of MK571 (3-[[[3-[(1E)-2-(7-chloro-2-quinolinyl)ethenyl]phenyl][[3-(dimethylamino)-3-oxopropyl]thio]methyl]thio]propanoic acid) [an inhibitor of MRP2 and other MRPs (Schrickx et al., 2006)] and PSC833 (6-[(2S,4R,6E)-4-methyl-2-(methylamino)-3-oxo-6-octenoic acid]-7-L-valine-cyclosporin A) [an inhibitor for MDR1 (Schrickx et al., 2006)] on OTA transport. In MRP2-expressing Sf21 cell vesicles (and in control vesicles; data not shown), we observed a weak inhibition of OTA transport by MK571, but no inhibition by PSC833 (Fig. 5B), irrespective of the presence of GSH. This indicates that the observed endogenous OTA transport may be mediated by an MRP-like transporter in insect cells.

Kinetics of ATP-dependent uptake of OTA by MRP2 in membrane vesicles isolated from Sf21 cells. (A) Initial uptake rates in the presence of increasing OTA concentrations without cholesterol loading (top). The initial uptake rates of increasing OTA concentrations with cholesterol loading are omitted for clarity of the figure, as the values are too close to the values without cholesterol loading. (Bottom) Corresponding Hanes–Woolf plots without (●) and with (▪) cholesterol loading. (B) Effect of the inhibitors (inh) MK571 (25 μM) and PSC833 (5 μM) in the presence and absence of 5 mM GSH on transport of 10 μM OTA after 1-minute incubation. Black bars, uptake in the absence of ATP; white bars, uptake in the presence of 5 mM ATP as described in Materials and Methods; gray bars, ATP-dependent transport. Values are the mean ± S.D. of quadruplicate determinations, but in some cases, error bars are smaller than the size of the symbols.
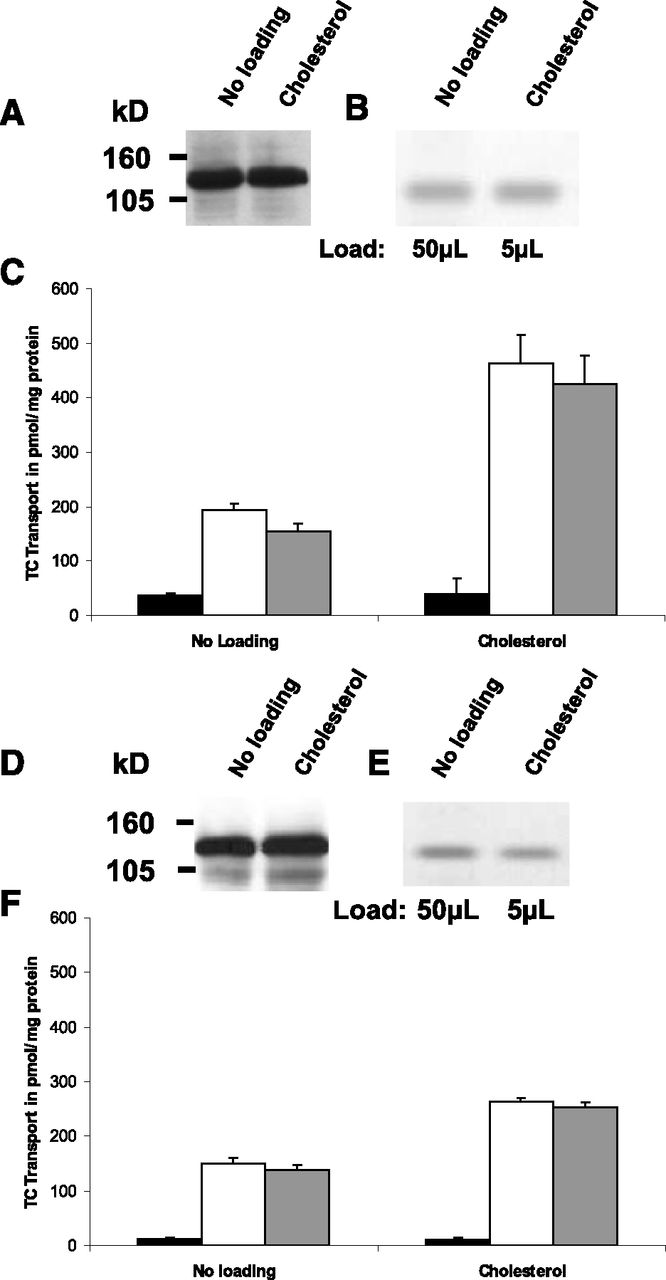
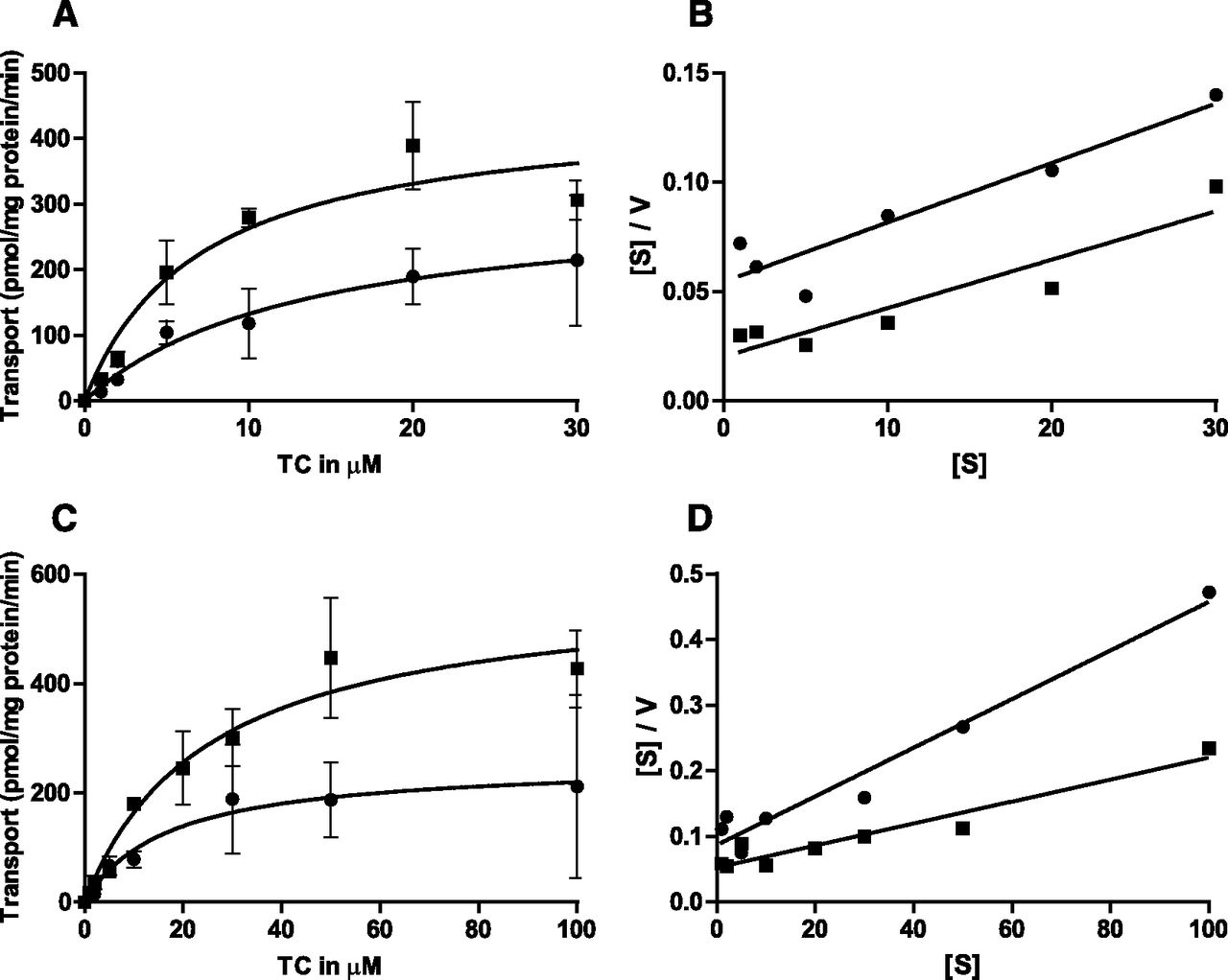
We next studied the effect of cholesterol on the transport activity of BSEP, which has a narrow substrate and size specificity (Stieger, 2011). Sf9 cells were infected with the two major BSEP isoforms (Pauli-Magnus et al., 2004)—namely, the p.444A and the p.444V variants—to determine the impact of membrane cholesterol on taurocholate transport activity. The conditions for infection of Sf9 cells with the two BSEP isoforms were adjusted such that comparable protein expression levels as shown by Western blotting were achieved in mock- and cholesterol-treated cells (Fig. 6, A and D). The cholesterol loading of the membranes was comparable (Fig. 6B) for the two isoforms, with an enrichment of about 11-fold (0.0292 μmol cholesterol per mg of protein in mock-treated vesicles compared with 0.307 μmol cholesterol per mg of protein in cholesterol-loaded vesicles). Figure 6, C and F, shows a comparable stimulation of the taurocholate transport activity of the p.444A variant and p.444V variant after loading with cholesterol by 2.8- and 1.8-fold, respectively. Both BSEP isoforms show classic Michaelis–Menten kinetics and linear Hanes–Woolf plots (Fig. 7, A and C) in the presence and absence of cholesterol, indicating the absence of cooperativity. This absence of cooperativity of taurocholate transport by BSEP in the absence of cholesterol loading is in accordance with previous results obtained in our laboratory (Noe et al., 2002, 2005; Lang et al., 2007). The membrane loading with cholesterol leads to a marked increase in Vmax (from 256 to 579 pmol/mg protein/min for p.444V and from 311 to 447 pmol/mg protein/min for p.444A, calculated for Fig. 7, A and C), whereas the Km values (17–25.3 µM for p.444V and 13.5–7 µM for p.444A, calculated for Fig. 7, A and C) remain unchanged in all experiments, irrespective of the isoform. The analysis of the kinetic parameters of the two BSEP variants by Hanes–Woolf transformations of the respective data at high and low cholesterol shows lines of comparable slopes and y-axis intercepts (see example in Fig. 7, B and D) corresponding to a classic Michaelis–Menten behavior.

Effect of cholesterol loading on BSEP expression and transport activity. (A and D) Western blot analysis of p.444A variant–expressing (A) and p.444V variant–expressing (D) vesicles with and without cholesterol loading (30 µg protein per lane). (B and E) Cholesterol content in p.444A variant–expressing (B) and p.444V variant–expressing (E) vesicles detected by thin-layer chromatography; 50 and 5 µl of extracted lipids were loaded for nonloaded vesicles and vesicles loaded with cholesterol, respectively. (C and F) initial rate of taurocholate (TC) uptake (2.5 µM) for 10 minutes by membrane vesicles isolated from p.444A variant–expressing (C) and p.444V variant–expressing (F) Sf9 cells with and without cholesterol loading. Uptake was measured in the presence (white bars) or absence of ATP (black bars) as described in Materials and Methods; the difference of uptake in the presence and absence of ATP is represented with gray bars. Values given are the mean ±S.D. of triplicate measurements of a representative experiment.

Kinetics of ATP-dependent uptake of taurocholate (TC) by BSEP p.444A variant (A and B) and p.444V variant (C and D) in membrane vesicles isolated from Sf9 cells. Initial uptake rates were determined in the presence and absence of ATP with and without cholesterol loading (▪ and ●, respectively). ATP-dependent uptake values represent the difference between uptake in the absence and presence of ATP. (A) p.444A variant uptake in the presence of increasing concentration of taurocholate. (B) Corresponding Hanes–Woolf plot. (C) p.444V variant uptake in the presence of increasing concentration of taurocholate. (D) Corresponding Hanes–Woolf plot. (A and C) Values given are the mean ± S.D. of triplicate measurements, but in some cases, error bars are smaller than the size of the symbol.
Finally, we studied the effect of cholesterol on two mutant forms of BSEP, p.E297G and p.R432T, found in patients with BSEP deficiency syndromes. These two mutants were previously shown to have a dramatically decreased transport activity (Noe et al., 2005). Western blot analysis detected comparable protein amounts for p.444A, p.297G, and p.432T expressed in Sf9 vesicles (Fig. 8, A, D, and G) and a comparable amount of cholesterol after loading (Fig. 8, B, E, and H). As expected, in a low cholesterol environment, the two mutants display a clearly reduced transport activity as compared with the p.444A variant (Fig. 8, C, F, and I), which considerably increased after cholesterol loading (6-fold increase for p.297G and 3-fold increase for p.432T). However, this increase was not sufficient to reach the transport activity of the p.444A BSEP variant.

Comparison of the effect of cholesterol loading on p.297G and p.432T on transport activity with p.444A. (A, D, and G) Western blot analysis of p.444A variant (A), p.E297G (D), and p.R432T (G) mutant BSEP-expressing vesicles with and without cholesterol loading (30 µg protein per lane). (B, E, and H) Cholesterol content in p.444A variant (B), p.E297 (E), and p.432T (H) mutant BSEP-expressing vesicles detected by thin-layer chromatography; 50 and 5 µl of extracted lipids were loaded for nonloaded vesicles and vesicles loaded with cholesterol, respectively. (C, F, and I) Initial rate of taurocholate (TC) uptake (2.5 µM) for 10 minutes by membrane vesicles isolated from p.444A variant (C), p.297G (F), and p.432T (I) mutant BSEP-expressing Sf9 cells with and without cholesterol loading. Uptake was measured in the presence (white bars) or absence of ATP (black bars) as described in Materials and Methods; the difference between values in the presence and absence of ATP is represented with gray bars. Values of a representative experiment are given as the mean ± S.D. of triplicate measurements.
Discussion
In the present work, we have investigated the role of membrane cholesterol content in the kinetics of the two ABC transporters MRP2 and BSEP. The main finding is that membrane cholesterol content modifies transport kinetics of BSEP and MRP2 in different ways. The established protocol for cholesterol loading of insect cells increases the cholesterol content of isolated vesicles about 10-fold. Sf9 cell membranes are known for having much lower cholesterol contents than mammalian plasma membranes (Gimpl et al., 1995). Hence, they are a good model for studying the effect of increasing membrane cholesterol levels without the prior need for cholesterol depletion. In cholesterol-loaded insect membrane vesicles, the ratio of total phospholipid to cholesterol is comparable to rat cLPM membrane vesicles, allowing a cautious extrapolation of the present findings to transporters in cLPM.
We took advantage of the broad substrate specificity of MRP2 accepting low-MM (e.g., E17βG) and high-MM (e.g., CCK8) substrates. At low membrane cholesterol, the kinetics of E17βG transport by MRP2 is sigmoidal, yielding a U-shaped Hanes–Woolf plot. With cholesterol-loaded membranes, the kinetics of E17βG transport follows Michaelis–Menten. This suggests a dependence of the allosteric properties of MRP2 for E17βG on membrane cholesterol. We tested PGE2 as another low-MM substrate and found its transport activity to depend on the presence of GSH, as previously published (de Waart et al., 2006). An earlier publication reported no PGE2 transport by MRP2 expressed in insect cells (Reid et al., 2003). As the latter study was performed without GSH, our data on GSH dependence of PGE2 transport confirm these earlier findings. Although cholesterol loading stimulated PGE2 transport by MRP2, we did not observe cooperativity for this small substrate irrespective of membrane cholesterol. Previously, an increased affinity of MRP2 for E17βG (without major modification of Vmax) together with a shift from cooperative to Michaelis–Menten kinetics in the presence of indomethacin and sulfanitran was reported (Zelcer et al., 2003). Our findings therefore confirm and extend this study, as cholesterol or GSH also abolishes cooperativity of MRP2 for small substrates. Here, membrane cholesterol increases the Vmax for two substrates but only mildly changes the Km. In contrast, when transporting the large substrates CCK8 and vasopressin, MRP2 does not display cooperativity irrespective of membrane cholesterol content. Cooperativity of MRP2 for E17βG kinetics was reported previously (Bodo et al., 2003; Zelcer et al., 2003) and was interpreted as two drug-binding sites interacting cooperatively (Borst et al., 2006). The data with GSH/PGE2 transport support a model with two binding sites for small substrates. Taking together small and large substrates, the data suggest that the binding site of MRP2 may be rather large. In the case of small substrates, two substrate molecules may bind sequentially to or close to the binding site, leading to an allosteric behavior of MRP2. The exact mechanism of the cholesterol-mediated activation of MRP2 transport activity is not evident. Currently, two possible mechanisms can be envisioned. The crystal structure of the Na,K-ATPase from the shark rectal gland displays a cholesterol molecule, which is tightly bound to the membrane helix M7 and carried along from the native tissue (Shinoda et al., 2009). The activity of Na,K-ATPase strongly depends on cholesterol (Cornelius et al., 2001). Cholesterol could similarly activate MRP2. This concept is supported by recent findings on ABCG2, where mutations of Arg482, residing in or very close to the third transmembrane helix, abolished cholesterol stimulation (Telbisz et al., 2014). Alternately, cholesterol could interact with the binding site(s) of MRP2. This concept was put forward for the ABC transporter MDR1 with a very broad specificity for substrates of largely differing MM. The affinity of MDR1 substrates with low MM is increased by cholesterol, whereas the affinity for high-MM substrates is cholesterol-independent (Kimura et al., 2007a). To explain this difference, a cholesterol fill-in model for substrate recognition of MDR1 and other ABC transporters such as ABCG1, ABCA1, or ABCB4 was developed: part of the binding pocket is, if space allows, occupied by cholesterol (Kimura et al., 2007b). Indeed, a crystal structure of mouse P-glycoprotein at 3.8 Å reveals a large substrate-binding area compatible with this concept (Aller et al., 2009). From our results, it is tempting to speculate that a similar mechanism may apply for some substrates of MRP2. In further support of this concept, bile salts have been shown to stimulate the transport activity of MRP2 (Bodo et al., 2003). Bile salts are synthesized from cholesterol and retain the four-ring structure of cholesterol. Hence, they could bind to MRP2 at the same site as cholesterol and stimulate its transport activity. Also, E17βG is derived from cholesterol, retaining the four-ring element of cholesterol. Taken together, the binding site for bile salts, cholesterol, or the second E17βG molecule could adopt molecules with the four-ring element of cholesterol. Importantly, our data together with literature data demonstrate that the transport mechanism of MRP2 for small substrates depends on the individual substrate, thus precluding a general transport mechanism for small MRP2 substrates, whereas larger substrates, based on our two examples, may share a common transport mechanism.
We also tested transport of OTA, previously reported to be a substrate of MRP2 (Leier et al., 2000), but found no MRP2-mediated OTA transport. The observed endogenous OTA transport in insect cell vesicles was subject to weak inhibition by MK571 but not by PSC833, suggesting that the endogenous transporter may belong to the ABCC family. Indeed, arthropods have genes belonging to the ABCC family (Dermauw and Van Leeuwen, 2014) and the Drosophila MRP (dMRP), which is the Drosophila melanogaster ortholog of the so-called long human MRPs displays typical transport characteristics of human MRPs (Szeri et al., 2009). As we were not able to stimulate the OTA transport activity by cholesterol, it is tempting to speculate that invertebrate ABC transporters may be cholesterol insensitive.
Cholesterol is known to stimulate the transport activity of BSEP (Kis et al., 2009; Paulusma et al., 2009). BSEP displays a considerably narrower substrate specificity than MRP2, and hence, we investigated the effect of cholesterol on BSEP variants and mutants. The ABCB11 gene coding for BSEP occurs with various polymorphic variants (Pauli-Magnus et al., 2004; Stieger, 2011). The most common single-nucleotide polymorphism is c.1331T>C (p.444A) tending toward lower protein expression levels (Pauli-Magnus et al., 2004; Meier et al., 2006). The c.1331C variant is a susceptibility factor for drug-induced cholestasis and intrahepatic cholestasis of pregnancy (Pauli-Magnus et al., 2010), whereas the taurocholate transport kinetics of both variants are indistinguishable (Lang et al., 2007). Here, p.444V and p.444A showed an increased transport activity and a classic Michaelis–Menten behavior after cholesterol loading but lacked cooperativity, irrespective of cholesterol content (Fig. 4). We therefore conclude that cholesterol dependence of the two major BSEP variants does not constitute an additional susceptibility factor for acquired forms of cholestasis. The two investigated BSEP mutants were responsive to cholesterol loading. Compared with MRP2, kinetic experiments with BSEP are more challenging due to the toxic action of high bile salt concentrations, which impair the integrity of membranes (Guyot and Stieger, 2011). The mutant forms of BSEP also showed an increase in Vmax after cholesterol loading (data not shown). However, this increase was not sufficient to restore the transport activity to levels comparable to the wild-type isoforms. Consequently, data obtained from the in vitro characterization of BSEP mutants in the insect cells are valid to extrapolate to phenotypes of BSEP in mammalian membranes. This is further supported by the observation that inhibition of ATP-dependent taurocholate transport into rat cLPM vesicles and rat Bsep expressed in Sf9 cells without cholesterol loading yields comparable inhibition constants (Stieger et al., 2000). Although BSEP is a member of the B family of ABC transporters and a close relative of ABCB1 and transports low-MM substrates, it does not display cooperativity as does ABCB1. The exact mechanism of the cholesterol effect(s) on transport activity will require the determination of the crystal structure of MRP2 and BSEP, which to date has not yet been accomplished. The cLPM of rats (and likely of humans) contains microdomains, which are enriched in cholesterol and contain rat MRP2 and rat BSEP (Ismair et al., 2009; Guyot and Stieger, 2011). Partitioning (modulated by canalicular bile salt concentration) of ABC transporters into microdomains may enhance their activity and constitute a regulatory mechanism in cLPM. Comparison of the extent of stimulation of MRP2 and BSEP reveals that the latter is much less stimulated by increased membrane cholesterol. Consequently, the stimulation of these two transporters after partitioning into microdomains may differ.
In conclusion, we have shown that susceptibility of MRP2 and BSEP to cholesterol stimulation differs. Cholesterol increases the transport rates of BSEP and MRP2. However, for MRP2 but not BSEP, cholesterol may modify the binding of individual substrates. Therefore, transport properties of BSEP, such as potential inhibition by drugs or drug metabolites, are most likely less dependent on the expression system than the transport properties of MRP2. Hence, characterization of MRP2 may warrant the utilization of more than one expression system.
Authorship Contributions
Participated in research design: Guyot, Stieger.
Conducted experiments: Guyot, Hofstetter.
Performed data analysis: Guyot, Hofstetter, Stieger.
Wrote or contributed to the writing of the manuscript: Guyot, Hofstetter, Stieger.
Footnotes
- Received February 14, 2014.
- Accepted April 7, 2014.
This work was supported by the Swiss National Science Foundation (SNF) [Grant 310030_144195] (to B.S.), the SNF National Center of Competence in Research TransCure (University of Berne, Berne, Switzerland) (to B.S.), and the Novartis Foundation for Biomedical Research (Basel, Switzerland) [07C75] (to B.S.).
Abbreviations
- ABC transporter
- ATP-binding cassette transporter
- BSEP
- bile salt export pump
- CCK8
- cholecystokinin 8
- cLPM
- canalicular plasma membrane
- E17βG
- estradiol-17-β-glucuronide
- GSH
- glutathione
- HBSS
- Hanks’ balanced saline solution
- MDR
- multidrug resistance protein
- MK571
- 3-[[[3-[(1E)-2-(7-chloro-2-quinolinyl)ethenyl]phenyl][[3-(dimethylamino)-3-oxopropyl]thio]methyl]thio]propanoic acid
- MRP2
- multidrug resistance–associated protein 2
- MM
- molecular mass
- OTA
- ochratoxin A
- PGE2
- prostaglandin E2
- PSC833
- 6-[(2S,4R,6E)-4-methyl-2-(methylamino)-3-oxo-6-octenoic acid]-7-L-valine-cyclosporin A
- RAMEB
- randomly methylated-β-cyclodextrin
- TBST
- Tris-buffered saline/Tween 20
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}