


Abstract
The canalicular multispecific organic anion transporter (cMOAT), also termed MRP2, is a recently identified ATP-binding cassette transporter. We previously established stable human cMOAT cDNA-transfected cells, LLC/cMOAT-1 from LLC-PK1 cells, and LLC/CMV cells that were transfected with an empty vector. We found that LLC/cMOAT-1 cells have increased resistance to vincristine (VCR), 7-ethyl-10-hydroxy-camptothecin, and cisplatin but not to etoposide. The multidrug resistance-reversing agents cyclosporin A (CsA) and 2-[4-(diphenylmethyl)-1-piperazinyl]-5-(trans-4,6-dimethyl-1,3,2-dioxaphosphorinan-2-yl)-2,6-dimethyl-4-(3-nitrophenyl)-3-pyridinecarboxylate P-oxide (PAK-104P) almost completely reversed the resistance to VCR, 7-ethyl-10-hydroxy-camptothecin, and cisplatin of LLC/cMOAT-1 cells; anddl-buthionine-(S,R)-sulfoximine, (3′-oxo-4-butenyl-4-methyl-threonine1, (valine2) cyclosporin (PSC833), and 3-([{3-(2-[7-chloro-2-quinolinyl]ethenyl)phenyl}-{(3-dimethylamino-3-oxopropyl)-thio}-methyl]thio)propanoic acid (MK571) partially reversed the resistance to these drugs. CsA and PAK-104P at 10 μM enhanced the accumulation of VCR in LLC/cMOAT-1 cells almost to the level in LLC/CMV cells without the agents. The efflux of VCR from LLC/cMOAT-1 cells was enhanced compared with LLC/CMV cells and inhibited by CsA and PAK-104P. Transport of leukotriene C4 (LTC4) and S-(2, 4-dinitrophenyl)glutathione also was studied with membrane vesicles prepared from these cells. LTC4 and S-(2, 4-dinitrophenyl)glutathione were actively transported into membrane vesicles prepared from LLC/cMOAT-1 cells. TheK m and V maxvalues for the uptake of LTC4 by the LLC/cMOAT-1 membrane vesicles were 0.26 ± 0.05 μM and 7.48 ± 0.67 pmol/min/mg protein, respectively. LTC4 transport was competitively inhibited by PAK-104P, CsA, MK571, and PSC833, withK i values of 3.7, 4.7, 13.1, and 28.9 μM, respectively. These findings demonstrate that cMOAT confers a novel drug-resistance phenotype. CsA and PAK-104P may be useful for reversing cMOAT-mediated drug resistance in tumors.
After selection for resistance to a single cytotoxic drug, cells sometimes become cross-resistant to a wide range of drugs having different structures and cellular targets, a phenomenon called multidrug resistance (MDR). MDR results from overexpression of P-glycoprotein (Pgp), a 170-kDa product of the MDR1 gene. Pgp, an ATP-binding cassette transporter, functions as an ATP-dependent efflux pump that reduces the intracellular accumulation of chemotherapeutic agents by transporting them across the plasma membrane (Endicott and Ling, 1989; Gottesman and Pastan, 1993). The 190-kDa MDR protein (MRP or MRP1), another ATP-binding cassette transporter (Cole et al., 1992), was found in many non-Pgp-mediated MDR cells, and it confers MDR by transporting anticancer agents outside the cells. MRP is able to transport reduced glutathione (GSH) conjugates, and it has been suggested that it is an ATP-dependent glutathione S-conjugate export pump (GS-X pump) (Jedlitschky et al., 1994; Leier et al., 1994). The discovery that MRP is a GS-X pump raised the question whether other GS-X pumps that exist in human tissues may be involved in drug resistance. The canalicular multispecific organic anion transporter (cMOAT), also called MRP2, transports organic anions from hepatocytes into bile (Ishikawa, 1992; Mayer et al., 1995; Oude Elferink et al., 1995). Transport-deficient mutant rat strains (Jansen et al., 1985) and Eisai hyperbilirubinemic rats (Takikawa et al., 1991) contributed to our understanding of the substrate specificity of this transporter (Oude Elferink et al., 1995). The substrate specificity of cMOAT is similar to that of MRP. The biochemical abnormalities in transport-deficient mutant rats mimic those of human patients with the inborn error of metabolism known as the Dubin-Johson syndrome. Rat cMOAT cDNA was isolated and sequenced, and cMOAT was shown to be expressed in the canalicular membrane of hepatocytes. It has a 47.6% overall identity with human MRP (Paulusma et al., 1996), and human cMOAT (MRP2) has 46% identity with human MRP (MRP1; Taniguchi et al., 1996). The membrane vesicles isolated from cells stably expressing rat cMOAT or human cMOAT can transport GSH conjugates in an ATP-dependent manner (Evers et al., 1998; Ito et al., 1998). These findings suggested that cMOAT is a member of the MRP gene family. Koike et al. (1997) demonstrated that transfection of antisense cMOAT cDNA into human hepatic cancer HepG2 cells that stably express cMOAT increased their sensitivity to cisplatin, vincristine (VCR), doxorubicin (Adriamycin; ADM), and camptothecin derivatives 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin (CPT-11) and 7-ethyl-10-hydroxy-camptothecin (SN-38), but not to etoposide. However, the resistance phenotypes of cell lines transfected with cMOAT have not been reported. Therefore, the ability of this transporter to confer drug resistance has not been confirmed. In the present study, we used an LLC-PK1 cell line that was transfected with an expression vector containing human cMOAT cDNA (Kawabe et al., 1999) and examined the drug resistance phenotype of the transfectants. Transport activity of cMOAT and its inhibition by agents that reverse MDR also were examined. We show that human cMOAT is involved in drug resistance. MDR-reversing agents, such as cyclosporin A (CsA), 2-[4-(diphenylmethyl)-1-piperazinyl]-5-(trans-4,6-dimethyl-1,3,2-dioxaphosphorinan-2-yl)-2,6-dimethyl-4-(3-nitrophenyl)-3-pyridinecarboxylate P-oxide (PAK-104P), 3-([{3-(2-[7-chloro-2-quinolinyl]ethenyl)phenyl}-{(3-dimethylamino-3-oxopropyl)-thio}-methyl]thio)propanoic acid (MK571), and (3′-oxo-4-butenyl-4-methyl-threonine1, (valine2) cyclosporin (PSC833) reverse the drug resistance conferred by cMOAT by interacting with its substrate-binding site.
Experimental Procedures
Materials.
Lipofectamine and geneticin (G418) were purchased from Life Technologies, Inc. (Grand Island, NY). Monoclonal antibody against cMOAT, M2III-6, was kindly provided by Drs. Marcel Kool and Piet Borst (The Netherlands Cancer Institute, Amsterdam, the Netherlands). [3H]VCR sulfate (5.70 Ci/mmol) was obtained from Amersham International Ltd. (Buckinghamshire, UK). [14,15,19,20-3H(N)]leukotriene C4 (LTC4; 146 Ci/mmol) was obtained from DuPont NEN (Boston, MA), and antimony potassium tartrate, sodium arsenite, and 1-chloro-2,4-dinitrobenzene were obtained from Wako Pure Chemical Industries Ltd. (Osaka, Japan). UnlabeledS-(2, 4-dinitrophenyl)glutathione (DNP-SG) and [3H]DNP-SG were synthesized from 1-chloro-2,4-dinitrobenzene and unlabeled or [Gly-2-3H]GSH (Daiichikagaku yakuhin Co. Ltd., Tokyo, Japan) in the presence of bovine liver glutathioneS-transferase as described by Awasthi et al. (1981). Etoposide was obtained from Nippon Kayaku Co. (Tokyo, Japan) and unlabeled LTC4 was obtained from Calbiochem (La Jolla, CA). The leukotriene D4 receptor antagonist MK571 (Jones et al., 1989) was kindly provided by Dr. A. W. Ford-Hutchinson (Merck-Frosst Center for Therapeutic Research, Pointe Claire-Dorval, Quebec, Canada). CsA and its analog PSC833 were kindly provided by Sandoz (Tsukuba, Japan), and PAK-104P was supplied by Nissan Chemical Industries (Chiba, Japan). CPT-11 and SN-38 were kindly donated by Daiichi Seiyaku (Tokyo, Japan). Nucleotides, cisplatin, VCR, ADM, mitoxantrone (MX), paclitaxel (Taxol), glutathione S-transferase,dl-buthionine-(S,R)-sulfoximine (BSO), and other drugs were obtained from Sigma Chemical Co. (St. Louis, MO).
Construction of Human cMOAT Expression Vector.
Human cMOAT expression vector was constructed previously (Kawabe et al., 1999). Briefly, cDNA-overlapping clones (L3, AL28–33, 4–1, 1–1, AK1–8) (Taniguchi et al., 1996) were subcloned into pBluescript II SK(−) (Stratagene, Inc., La Jolla, CA) and reconstructed into a full-length cDNA with NotI linker. The human cMOAT cDNA in pBluescript II SK(−) was inserted into the NotI site of pCI-neo mammalian expression vector (Promega Biotec, Madison, WI).
Cell Culture and Transfection with Human cMOAT Expression Vector.
LLC-PK1 cells were cultured in M199 medium (Life Technologies, Inc.) containing 10% fetal bovine serum (Cell Culture Laboratories, Cleveland, OH), 100 μg/ml kanamycin, and 100 U/ml penicillin in a humidified atmosphere of 5% CO2at 37°C. Exponentially growing cells (2 × 106) were washed with PBS and placed in serum-free medium. A mixture of 50 μg of lipofectamine and 5 μg of human cMOAT expression vector was added for 12 h, after which fresh medium was added. The cells were incubated in selection medium containing 800 μg/ml G418 for 3 to 4 weeks. As described elsewhere (Kawabe et al., 1999), LLC/cMOAT-1, one of the stable transfectants among the G418 resistant clones expressing human cMOAT, was selected by immunoblot analysis. As a control, we also established a G418-resistant empty vector transfected subline, LLC/CMV (Kawabe et al., 1999). In addition, KB-3–1, a human epidermoid KB carcinoma cell line and its Pgp-mediated MDR mutant KB-C2 (Akiyama et al., 1985), as well as KB/MRP (Taguchi et al., 1997), KB-3–1 cells transfected with MRP cDNA, were grown in minimal essential medium (Nissui Seiyaku Co., Tokyo, Japan) with 10% newborn calf serum in a humidified atmosphere of 5% CO2 at 37°C. The human hepatic cancer cell line, HepG2, was cultured as described previously (Koike et al., 1997).
Immunoblot Analysis.
Immunoblot analyses of human cMOAT and MRP were performed as described in Chen et al. (1998). Monoclonal antibody M2III-6, generated against amino acids 1340 to 1541 of rat cMOAT protein (Paulusma et al., 1996), was used for cMOAT immunoblots and the monoclonal antibody C219 was used to detect Pgp. Membrane vesicles (10 μg of protein) were electrophoresed on 7.5% (w/v) SDS-polyacrylamide minigels and transferred onto Immobilon-P membranes (Millipore Corp., Bedford, MA). The membranes were incubated with 10-fold diluted monoclonal antibody against cMOAT for 1 h at room temperature. Membranes were developed by chemiluminescence according to the enhanced chemiluminescence protocol (Amersham International).
Cell Survival by 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) Assay.
The MTT colorimetric assay was used as described in Chen et al. (1998)to assess the sensitivity of the cells to agents in vitro. Exponentially growing cells were trypsinized and harvested, and 6 × 103 cells in 180 μl of culture medium were inoculated into each well. After overnight incubation, 20 μl of various drugs was added and the cells were incubated for 4 days. Thereafter, 50 μl of MTT (1 mg/ml in PBS) was added to each well and the cells were incubated for an additional 3 h. After aspiration of the culture medium, the resulting formazan was dissolved with 100 μl of dimethyl sulfoxide. Plates were placed on a shaker for 5 min and read immediately at 570 nm with a microplate reader, MRP-A4i (Tosoh, Tokyo, Japan). To examine the effects of CsA, PSC833, MK571, PAK-104P, BSO, or cephalanthine on drug sensitivity, cells were preincubated without or with CsA (10 μM), PSC833 (10 μM), MK571 (5 μM), PAK-104P (5 μM), or cephalanthine (5 μM) for 1 h or BSO (10 μM) for 24 h and then incubated with various concentrations of drugs.
Drug Accumulation and Efflux.
The accumulation and efflux of VCR was determined as described in Sumizawa et al. (1997). To measure drug accumulation, confluent monolayers of LLC/CMV and LLC/cMOAT-1 cells in 24-well plates were incubated in Hanks' balanced salt solution for 1 h at 37°C and then with 1 μM [3H]VCR in Hanks' balanced salt solution with or without 10 μM CsA or PAK-104P for 2 h at 37°C. After being washed with ice-cold PBS three times, the cells were solubilized in 1% Triton X-100 and 0.2% SDS in 10 mM phosphate buffer, pH 7.4, and the radioactivity was determined. To measure drug efflux, cells were incubated overnight and then further incubated in the presence or absence of 10 μM CsA or PAK-104P for 1 h. Medium was then changed to fresh medium with or without CsA or PAK-104P, and cells were incubated with 1 μM [3H]VCR for 1 h at 37°C. Each dish was washed once with PBS, and fresh medium without [3H]VCR and with or without 10 μM CsA or PAK-104P was added. Then the cells were incubated for the indicated times at 37°C and harvested, and radioactivity was determined.
Membrane Vesicle Preparation.
Membrane vesicles were prepared by the nitrogen cavitation method (Cornwell et al., 1986) from LLC/CMV, LLC/cMOAT-1, KB-3–1, and KB/MRP cells grown in 24 × 24 cm dishes (Nunc, Inc., Naperville, IL) under standard growth conditions (Akiyama et al., 1985). Cell monolayers (109) were washed once and scraped into PBS containing 1% aprotinin (Sigma Chemical Co.). The cells were washed by centrifugation (4000g for 10 min) at 4°C in PBS and then suspended in buffer A (10 mM Tris-HCl, pH 7.4, 0.25 M sucrose, and 0.2 mM CaCl2) and equilibrated at 4°C under a nitrogen pressure of 25 kg/cm2 for 30 min. Ethylenediaminetetraacetic acid at a final concentration of 1 mM was added to the suspension of lysed cells, which was then diluted one-fourth with buffer B (10 mM Tris-HCl, pH 7.4, and 0.25 M sucrose) and centrifuged at 1000g for 10 min at 4°C to remove nuclei and unlysed cells. The supernatant was layered onto a 35% sucrose cushion (10 mM Tris-HCl, pH 7.4, 35% sucrose, and 1 mM ethylenediaminetetraacetic) and centrifuged for 30 min at 16,000g at 4°C. The interface was collected and diluted one-fifth in buffer B, and then centrifuged for 45 min at 100,000g. The vesicle pellet was resuspended in buffer B by repeated passages through a 25 gauge needle. Sialidase accessibility for the determination of inside-out vesicles was examined as described in Ishikawa (1989). The percentage of inside-out membrane vesicles from the cells was ∼50%. Protein concentration in the vesicles was determined by the method of Bradford (1976). Vesicles were stored at −80°C.
Membrane Vesicle Transport Studies.
ATP-dependent transport of LTC4 into the membrane vesicles and its inhibition by some MDR-reversing agents was measured by filtration essentially as described by Ishikawa and Ali-Osman (1993). The standard incubation medium contained membrane vesicles (25 μg of protein), 1.37 nM [3H]LTC4, 0.25 M sucrose, 10 mM Tris-HCl, pH7.4, 10 mM MgCl2, 2 mM ATP, 10 mM phosphocreatine, and 100 μg/ml creatine phosphokinase with or without unlabeled LTC4 in a final volume of 50 μl. The inhibitor concentrations are given in the figure legends. The reactions were carried out at 37°C and stopped with 3 ml of ice-cold stop solution (0.25 M sucrose, 100 mM NaCl, and 10 mM Tris-HCl, pH 7.4). The diluted samples were passed through Millipore filters (GVWP, 0.22-μm pore size; Millipore Corp.) under light vaccum to measure the [3H]LTC4 trapped in the vesicles. The filters were washed with 3 ml of ice-cold stop solution three times and then oven-dried at 50°C for 10 min. Each filter was placed in scintillation fluid, and the level of radioactivity was measured with a liquid scintillation counter. In control experiments ATP was replaced by an equal concentration of 5′-AMP. Rates of net ATP-dependent transport were calculated by subtracting the values obtained in the presence of 5′-AMP from those obtained in the presence of ATP. To examine whether [3H]LTC4 was actually transported into intravesicular spaces or was bound to vesicle membranes, we measured the uptake of [3H]LTC4 with different concentrations of sucrose in the assay mixtures.
ATP-dependent transport of [3H]DNP-SG into membrane vesicles was measured as described above, with a substrate concentration of 20 μM [3H]DNP-SG and with or without unlabeled DNP-SG.
Statistical Analysis.
Differences between groups were tested by one-way ANOVA or Student's t test. Significance levels given are those for the two-tailed Student's paired t test. Data are presented as means ± S.E. Differences were considered significant when P < .05.
Results
Generation of LLC-PK1 Cells Expressing Human cMOAT.
A cMOAT transfectant of LLC-PK1 cells, LLC/cMOAT-1, expressed a 180-kDa human cMOAT, whereas the cells transfected with empty vector (LLC/CMV) did not (Fig. 1). The molecular mass of cMOAT (180 kDa) expressed in the transfected LLC-PK1 cells was smaller than that (190 kDa) in human hepatic cancer HepG2 (Mayer et al., 1995), KB-3–1, KB/MRP, and KB-C2 cells. MRP was detected in the MRP-transfectant KB/MRP cells (Taguchi et al., 1997) but not in LLC/CMV or LLC/cMOAT-1 cells (Fig. 1), even after prolonged exposure (data not shown). Pgp was readily detected in KB-C2 cells by incubating the same blot with monoclonal antibody C219, and a low level of Pgp was expressed in LLC/CMV but not in LLC/cMOAT-1 cells (Fig. 1).

Immunoblot analyses for cMOAT, MRP, and Pgp. Membrane vesicles (10 μg of protein) from HepG2, LLC/CMV, LLC/cMOAT-1, KB-3–1, KB/MRP, and KB-C2 cells were separated by SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membrane. Immunoblot analyses of the transferred proteins with antibodies against cMOAT (M2III-6), MRP, or Pgp were performed as described in Experimental Procedures.
Drug Resistance in LLC-PK1 Cells Expressing Human cMOAT.
We examined the drug sensitivity of each cell line by the MTT assay. The spectrum of drug resistance in the cMOAT-transfected LLC/cMOAT-1 cells was determined and the results are summarized in Table1. LLC/cMOAT-1 cells were 8.02-, 6.28-, 3.04-, 2.03-, and 1.55-fold more resistant to VCR, SN-38, cisplatin, ADM, and CPT-11, respectively, than LLC/CMV. LLC/cMOAT-1 cells were not resistant to etoposide, MX, Taxol, antimony potassium tartrate, and sodium arsenite.
Resistance of cMOAT transfected LLC/cMOAT-1 cells to anticancer agents and heavy metals
Effect of MDR-Reversing Agents on Sensitivity to Anticancer Agents.
We examined the cytotoxic effects of the MDR-reversing agents by the MTT assay. CsA (≤10 μM), PSC833 (≤10 μM), PAK-104P (≤5 μM), MK571 (≤5 μM), cephalanthine (≤5 μM), and BSO (≤10 μM) had no cytotoxic effect on LLC/CMV and LLC/cMOAT-1 cells (data not shown). Table 2 summarizes the data from dose-response curves of anticancer agents with or without reversing agents. CsA (10 μM) and PAK-104P (5 μM) almost completely reversed the resistance to VCR, SN-38, and cisplatin, and MK571 (5 μM), PSC833 (10 μM), and BSO (10 μM) moderately reversed the resistance of LLC/cMOAT-1 cells to these drugs. In contrast, cephalanthine had no effect (data not shown).
Effect of MDR-reversing agents on sensitivity of LLC/CMV and LLC/cMOAT-1 cells to anticancer agents
Effect of MDR-Reversing Agents on Cellular Accumulation and Efflux of [3H]VCR.
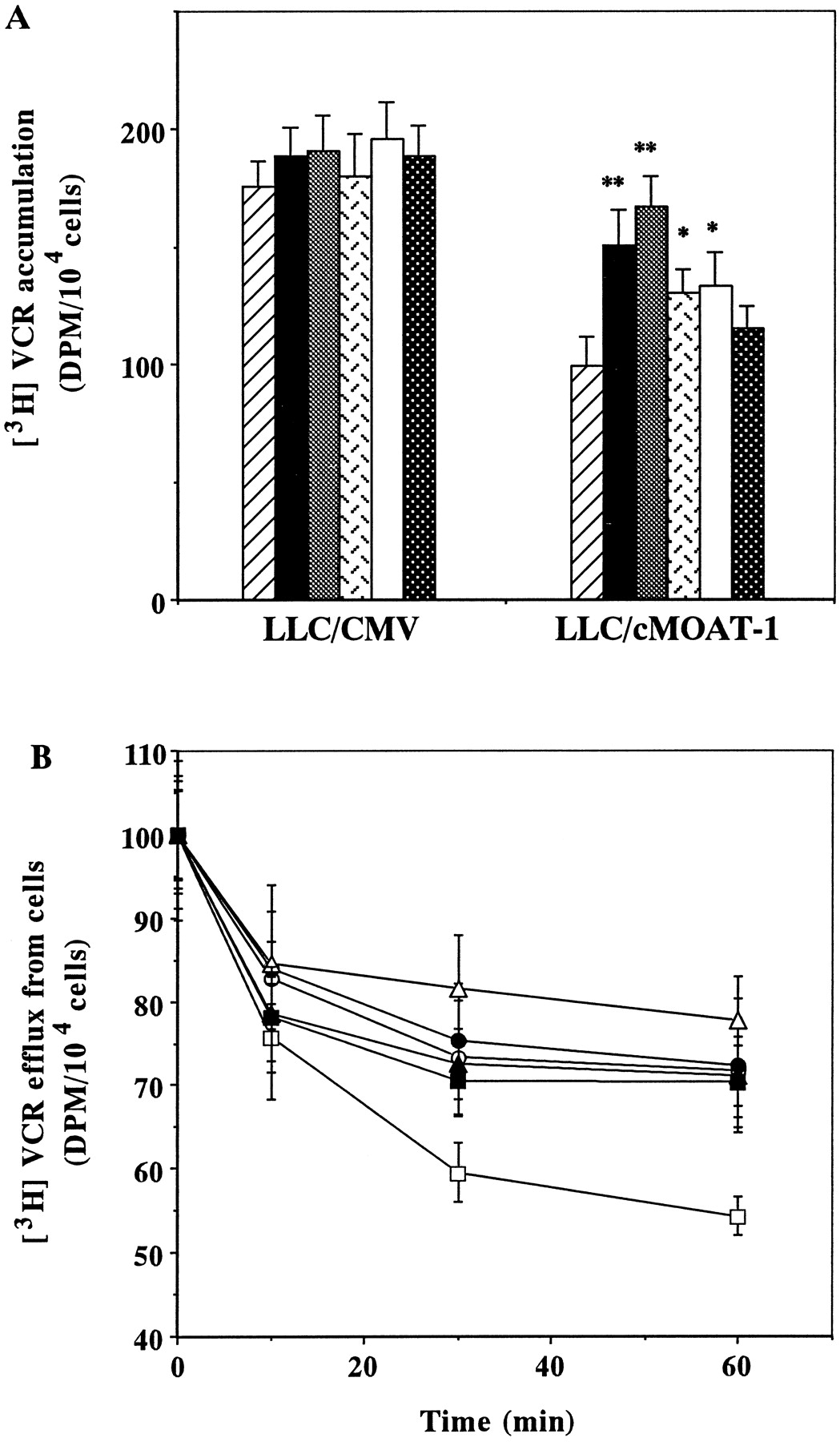
To investigate how MDR-reversing agents overcome resistance to VCR in LLC/cMOAT-1 cells, we examined their effect on the accumulation of VCR in LLC/CMV and LLC/cMOAT-1 cells. As shown in Fig. 2, the intracellular concentration of VCR in LLC/cMOAT-1 cells was ∼65% of that in LLC/CMV cells. The addition of 10 μM CsA and PAK-104P enhanced the accumulation of VCR in LLC/cMOAT-1 cells to a level similar to that in LLC/CMV cells without MDR-reversing agents (Fig.2A). Although MK571 and PSC833 significantly enhanced the accumulation of VCR in LLC/cMOAT-1 cells, the effect was less than that of CsA and PAK-104P at the same concentrations. Cephalanthine at 10 μM had no effect (Fig. 2A).

Effects of MDR-reversing agents on accumulation and efflux of VCR in LLC/CMV and LLC/cMOAT-1 cells. A, VCR accumulation in LLC/CMV and LLC/cMOAT-1 cells was measured after incubation with 1 μM [3H]VCR for 2 h without (▨) or with 10 μM CsA (▪), PAK-104P (▩), MK571 (), PSC833 (■), or cephalanthine (▩). Bars represent means ± S.E. of triplicate determinations;* P < .05 compared with control. B, efflux of VCR. VCR retained after 1 h of VCR accumulation by LLC/CMV (○, ●, ▵) and LLC/cMOAT-1 cells (■, ▪, ▴) in the absence (○, ■) or presence of CsA (●, ▪) or PAK-104P (▵, ▴). Values represent means ± S.E. of triplicate determinations.
We examined whether the increased accumulation of VCR in LLC/cMOAT-1 cells caused by CsA and PAK-104P was due to inhibition of VCR efflux. The time course of release of VCR after 1 h of accumulation is shown in Fig. 2B. LLC/cMOAT-1 cells released a higher percentage of accumulated VCR than KB-3–1 cells. At 60 min, 45% of accumulated VCR was lost from LLC/cMOAT-1 cells, whereas <25–30% was lost from LLC/CMV cells. CsA and PAK-104P at 10 μM apparently inhibited the efflux of VCR from the LLC/cMOAT-1 cells, and less so from LLC/CMV cells.
ATP-Dependent Uptake of [3H]LTC4 and [3H]DNP-SG by Membrane Vesicles Prepared from LLC/cMOAT-1 Cells.
To examine the substrate specificity of cMOAT, inside-out membrane vesicles were prepared from LLC/cMOAT-1 and LLC/CMV cells. Figure 3 shows the time course of LTC4 and DNP-SG uptake into membrane vesicles from LLC/CMV and LLC/cMOAT-1 cells in the presence of ATP or 5′-AMP. ATP-dependent [3H]LTC4uptake by membrane vesicles from LLC/cMOAT-1 cells was linear for ∼2 min and reached a plateau in 10 min (Fig. 3A). In contrast, no ATP-dependent [3H]LTC4uptake by LLC/CMV vesicles was observed. ATP-dependent DNP-SG uptake also was observed only in LLC/cMOAT-1 vesicles (Fig. 3B).

Time course of [3H]LTC4 and [3H]DNP-SG accumulation by membrane vesicles. Membrane vesicles (25 μg of protein) prepared from LLC/CMV (●) and LLC/cMOAT-1 (▪) cells were incubated at 37°C in 50 μl of transport buffer (0.25 M sucrose, 10 mM Tris-HCl, pH7.4, 10 mM MgCl2, 2 mM ATP, 10 mM phosphocreatine, 100 μg/ml creatine phosphokinase) containing 1.37 nM [3H]LTC4 (A) or 20 μM [3H]DNP-SG (B). In control experiments, ATP was replaced by an equal concentration of 5′-AMP (○, ■). [3H]LTC4 or [3H]DNP-SG transport by membrane vesicles was determined as described inExperimental Procedures. Data represent means ± S.E. of three separate experiments.
Effect of Osmolarity on [3H]LTC4 and [3H]DNP-SG Uptake.
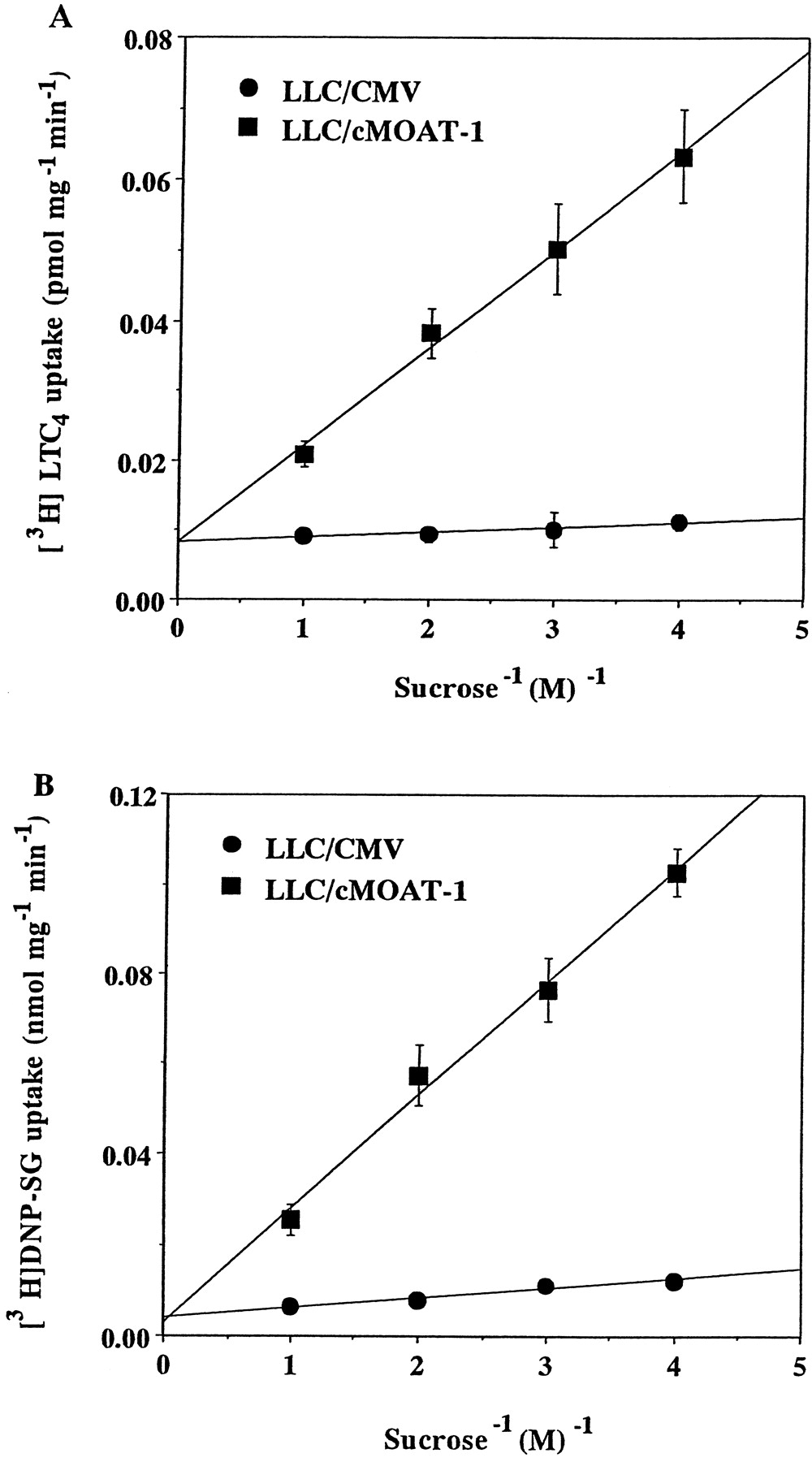
To determine whether the uptake of [3H]LTC4 and [3H]DNP-SG is due to transport of the substrates into the membrane vesicles or to binding to the vesicles, the osmotic sensitivity of [3H]LTC4 and [3H]DNP-SG uptake was analyzed. A major biochemical characteristic of transport into membrane vesicles, as opposed to nonspecific binding, is its inhibition at high osmotic pressures (Kamimoto et al., 1989). High osmotic pressure reduces the intravesicular space by shrinking the membrane vesicles, resulting in reduced uptake capacity. The uptake of [3H]LTC4 and [3H]DNP-SG by LLC/cMOAT-1 vesicles was osmotically sensitive and decreased linearly with increasing extravesicular sucrose concentrations from 0.25 to 1.0 M (Fig.4), whereas the uptake of [3H]LTC4 and [3H]DNP-SG by LLC/CMV vesicles was not significantly changed by these treatments.

Osmotic sensitivity of ATP-dependent [3H]LTC4 and [3H]DNP-SG uptake by LLC/CMV and LLC/cMOAT-1 membrane vesicles. Membrane vesicles (25 μg of protein) prepared from LLC/CMV (●) and LLC/cMOAT-1 (▪) cells were incubated at 37°C for 2 min in increasing concentrations of sucrose (0.25–1.0 M) in transport buffer containing 1.37 nM [3H]LTC4 (A) or 20 μM [3H]DNP-SG (B) in the presence of 2 mM ATP. Data represent the means ± S.E. of three separate experiments.
Kinetic Parameters and Dependence on Concentration of ATP on [3H]LTC4 Uptake by LLC/cMOAT-1 Vesicles.
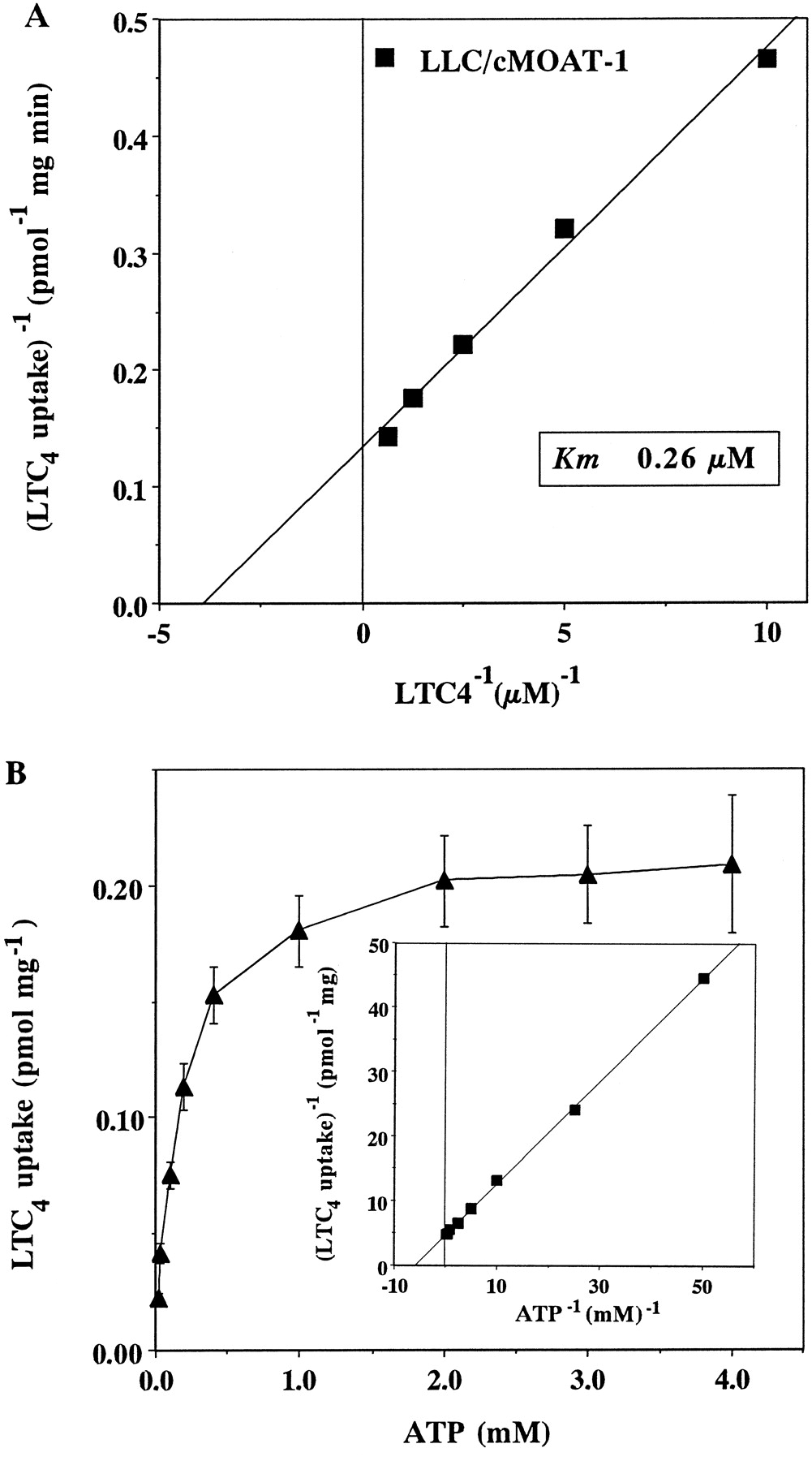
ATP-dependent LTC4 uptake by LLC/cMOAT-1 vesicles was examined at various concentrations of LTC4(10 to 1600 nM) and K m andV max values for the transport by cMOAT were determined. A Lineweaver-Burk plot of the data yielded an apparentK m value of 0.26 ± 0.05 μM for LTC4 and a V max value of 7.48 ± 0.67 pmol/mg protein/min (Fig.5A). If uptake is energy-dependent, transport should increase with increasing concentrations of ATP in the incubation mixture. As shown in Fig. 5B, [3H]LTC4 uptake by vesicles prepared from LLC/cMOAT-1 cells increased with increasing ATP concentrations. [3H]LTC4uptake was saturable with respect to ATP concentration, and the apparent K m value for ATP was 170 μM (Fig. 5B, inset).

Effects of LTC4 and ATP concentrations on [3H]LTC4 uptake by vesicles prepared from LLC/cMOAT-1 cells. A, membrane vesicles (25 μg of protein) prepared from LLC/cMOAT-1 cells were incubated in 50 μl of transport buffer containing various concentrations of LTC4 (100–1600 nM) in the presence of 2 mM ATP or 5′-AMP at 37°C for 2 min. ATP-dependent [3H]LTC4 uptake in the vesicles was determined as described in Experimental Procedures. Kinetic parameters (K m = 0.26 μM andV max = 7.48 pmol/mg protein min) were determined from double reciprocal plots of the ATP-dependent LTC4 uptake. B, ATP-dependent uptake of [3H]LTC4 by membrane vesicles (25 μg of protein) from LLC/cMOAT-1 cells was measured at various concentrations of ATP (0.02–4 mM) in the presence of 1.37 nM [3H]LTC4 for 2 min. Kinetic parameters were determined from double reciprocal plots of the ATP-dependent LTC4 uptake (inset).
Competitive Inhibition by MDR-Reversing Agents of ATP-Dependent LTC4 Transport.
ATP-dependent [3H]LTC4 transport by membrane vesicles from LLC/cMOAT-1 cells was studied in the presence of MDR-reversing agents. We found that four of six MDR-reversing agents, i.e., CsA, PAK-104P, MK571, and PSC833, inhibited [3H]LTC4 uptake competitively with K i values of 4.7, 3.7, 13.1, and 28.9 μM, respectively (Fig.6). However, [3H]LTC4 transport by vesicles prepared from KB/MRP cells was only slightly, if at all, inhibited by CsA even at 100 μM (data not shown). Cephalanthine and BSO even at 100 μM had no significant effects on [3H]LTC4 uptake by vesicles prepared from LLC/cMOAT-1 cells (data not shown). DNP-SG transport by vesicles prepared from LLC/cMOAT-1 cells also was inhibited by CsA, PSC833, MK571, and PAK-104P and not by cephalanthine and BSO (data not shown).

Competitive inhibition by CsA (A), PAK-104P (B), MK571 (C), and PSC833 (D) of ATP-dependent [3H]LTC4 transport. Double reciprocal Lineweaver-Burk plots are shown in the absence (○) or presence of 3 (▴), 10 (▪), and 30 μM (●) CsA, PAK-104P, MK571, and PSC833. Data are means ± S.E. of triplicate determinations.
Discussion
Pgp is expressed in various tumors and is involved in MDR (Gottesman and Pastan, 1993). MRP also confers MDR (Grant et al., 1994;Zaman et al., 1994) and is a transporter of glutathione conjugates (Jedlitschky et al., 1994; Leier et al., 1994). Another ATP-dependent transporter named cMOAT (Oude Elferink et al., 1995) is the glutathione S-conjugate export pump (Ishikawa, 1992) expressed in canalicular membranes of hepatocytes (Mayer et al., 1995). Recently, cDNAs for rat and human cMOAT have been cloned, and the substrate specificity of cMOAT was found to be similar to that of MRP (Taniguchi et al., 1996; Evers et al., 1998; Ito et al., 1998). Because the substrate specificity and molecular structure of cMOAT are similar to those of MRP, cMOAT also is referred to as MRP2 (Keppler and König, 1997). However, the differences in substrate specificity between MRP and cMOAT have not been fully understood and the contribution of cMOAT to drug resistance is also unclear.
In the present study, we demonstrated that human cMOAT was overexpressed in LLC/cMOAT-1, a subline of LLC-PK1 pig kidney epithelial cells transfected with cMOAT cDNA (Fig. 1; Kawabe et al., 1999). The molecular mass of cMOAT expressed in LLC/cMOAT-1 cells was smaller than that in human hepatic cancer HepG2 and KB cell lines. This difference may be attributed to a difference in glycosylation between the two species. LLC/cMOAT-1 cells were less sensitive to vincristine, SN-38, and cisplatin than LLC/CMV cells (Table 1). Koike et al. (1997)transfected an expression vector containing cMOAT antisense cDNA into HepG2 human hepatic cancer cells that express cMOAT. cMOAT was reduced in the antisense transfectants and their sensitivity to VCR, cisplatin, ADM, and camptothecin derivatives was increased (Koike et al., 1997).Kool et al. (1997) analyzed the characteristics of cell lines selected with ADM and cisplatin and found a positive association between cMOAT overexpression and cisplatin resistance. Recently, Chu et al. (1998)reported that cMOAT may be involved in the ATP-dependent transport of the carboxylate forms of SN-38 and SN-38 glucuronide in humans. Our results are consistent with the results of these previous articles.Evers et al. (1998) reported that polarized kidney epithelial MDCK cells transfected with cMOAT cDNA expressed cMOAT in plasma membranes and showed that cMOAT causes transport of vinblastine. These findings suggested that cMOAT may be involved in drug resistance in mammalian cells. However, the clinical significance of cMOAT-mediated MDR is unclear. Matsunaga et al. (1998) recently demonstrated enhanced expression of cMOAT and MRP1 in childhood malignant liver tumors and suggested that these increases are related to their clinical chemoresistance. We demonstrated in this study that cMOAT is involved in resistance to VCR, SN-38, cisplatin, ADM, and CPT-11, but not to etoposide, MX, Taxol, and two heavy metals. KB/MRP cells that overexpress MRP were moderately resistant to etoposide and MX (>5-fold), and also resistant to heavy metals and Taxol (data not shown; Taguchi et al., 1997) at low (≤3-fold) levels. The spectrum of drug resistance in LLC/cMOAT-1 cells is different from that in KB/MRP cells. Ishikawa et al. (1996) speculated that overexpression of MRP and the increased level of GSH in cisplatin-resistant human leukemia HL-60 cells are responsible for cisplatin resistance. However, an MRP-transfected cell line that expressed MRP showed no cross-resistance to cisplatin (Grant et al., 1994). When the GSH levels in C-A120 cells that overexpressed MRP were increased to nearly the same levels as those in cisplatin-resistant KCP-4 cells by transfection with γ-glutamylcysteine synthetase cDNA, the resistance to and the accumulation of cisplatin were not significantly changed (Chen et al., 1998). These results indicated that MRP is not involved in cisplatin resistance. Collectively, these findings suggest that the substrate specificities of cMOAT and MRP are different.
Kinetic analysis revealed that human cMOAT had aK m value of 0.26 μM for LTC4. Human MRP also transports LTC4 and had K mvalues of 97 to 105 nM for LTC4 (Leier et al., 1994; Loe et al., 1996). The K m values for LTC4 of human cMOAT appear to be ∼2.5- to 2.7-fold higher than that of human MRP. Rat cMOAT in membrane vesicles from apical site hepatic cells shows K mvalues of 0.25 to 0.32 μM for LTC4 (Ishikawa et al., 1990; Böhme et al., 1993), which appear to be comparable to that of human cMOAT. LTC4 and DNP-SG were transported by human cMOAT in LLC-PK1 cells, indicating that human cMOAT functions as a transporter of a number of GSH conjugates, consistent with the data from a Dubin-Johnson syndrome model rat defective in cMOAT function (Chu et al., 1997).
The agents that reverse drug resistance mediated by cMOAT are unknown. The transport of DNP-SG by cMOAT was inhibited only slightly by compounds known to block MRP, such as sulfinpyrazone, indomethacin, probenecid, and bromosulfophthalein (Evers et al., 1998). However, CsA and PSC833 inhibited ATP-dependent LTC4 transport in hepatocyte canalicular membranes (Böhme et al., 1993). CsA and PSC833, which are very potent reversing agents of Pgp (Boesch et al., 1991), usually show no or only small effects on the drug sensitivity of MRP-overexpressing MDR cells (Barrand et al., 1993). We found that CsA and PSC833 increase the sensitivity of LLC/cMOAT-1 cells to VCR, SN-38, and cisplatin (Table 2). The pyridine analog PAK-104P has been reported to reverse both MRP- and Pgp-mediated drug resistance (Chen et al., 1997; Sumizawa et al., 1997), and the leukotriene D4 receptor antagonist MK571 has been reported to modulate drug resistance mediated by MRP (Versantvoort et al., 1994). Both of these compounds also reversed the cMOAT-mediated drug resistance (Table 2). PAK-104P and PSC833 also increased the sensitivity of LLC/CMV cells to VCR significantly, but did not increase the sensitivity of LLC/CMV cells to SN-38 and cisplatin (Table 2), which are not substrates of Pgp. A low level of Pgp was expressed in LLC/CMV cells but not in LLC/cMOAT-1 cells (Fig. 1). These findings indicate that endogenous Pgp in LLC-PK1 cells is one of the causes of the low relative resistance to VCR in LLC/cMOAT-1 cells. In addition,Kawabe et al. (1999) found that human cMOAT is mainly expressed in the plasma membrane in polarized LLC/cMOAT-1 cells, although some intracellular staining also was observed in some cells. The fact that some cMOAT is localized in intracellular membrane may explain why high cMOAT expression conferred a low level of drug resistance on LLC/cMOAT-1 cells. VCR accumulation was decreased in LLC/cMOAT-1 cells that overexpress cMOAT, and 10 μM CsA, PAK-104P, MK571, or PSC833 increased the level of [3H]VCR accumulation in LLC/cMOAT-1 cells. Furthermore, CsA and PAK-104P inhibited efflux of [3H]VCR from LLC/cMOAT-1 cells. These findings suggest that MDR-reversing agents inhibit VCR transport by cMOAT expressed in LLC/cMOAT-1 cells.
Four of the six MDR-reversing agents examined, CsA, PAK-104P, MK571, and PSC833, competitively inhibited ATP-dependent LTC4 transport in LLC/cMOAT-1 vesicles, and their inhibiting activities correlated with their drug resistance-reversing activities. LTC4 transport was inhibited by PAK-104P, CsA, MK571, and PSC833 with K ivalues of 3.7, 4.7, 13.1, and 28.9 μM, respectively. These data suggest that the MDR-reversing agents may reverse cMOAT-mediated drug resistance by interacting with cMOAT. Böhme et al. (1993) have shown that PSC833 is a more potent inhibitor of Pgp-mediated daunorubicin transport than CsA. In the present study, we found that PCS833 is less effective than CsA in reversing cMOAT-mediated drug resistance (Table 2) and inhibiting cMOAT-mediated LTC4 transport. TheK i value (13.1 μM; Fig. 6) for MK571 inhibition of LTC4 transport in LLC/cMOAT-1 membrane vesicles was higher than that (0.6 μM) in membrane vesicles from cells expressing MRP (Leier et al., 1994). These results suggest that MK571 inhibits MRP much more effectively than does cMOAT. Probenecide and sulfinpyrazone have been reported to inhibit MRP-mediated transport. We found that these two agents inhibited MRP-mediated LTC4 transport at 1 mM. However, cMOAT-mediated LTC4 transport was not inhibited by these compounds at 1 mM (Z.-S.C. and S.-I.A, unpublished data). It seems that CsA, an inhibitor of Pgp, is more effective for reversing cMOAT-mediated drug resistance than inhibitors of MRP, such as MK571, probenecide, and sulfinpyrazone.
BSO partially reversed the cMOAT-mediated drug resistance (Table 2), and cMOAT-mediated LTC4 transport was more effectively inhibited by VCR or cisplatin in the presence of GSH than in its absence (Kawabe et al., 1999). These data suggest that GSH is needed for cMOAT-mediated drug resistance.
When a high dose of CsA was used in combination with anticancer agents in the clinical inhibition of Pgp-mediated drug resistance, hyperbilirubinemia was seen in 24 or 39% of cases (Stiff et al., 1995;Samuels et al., 1997). Our finding that CsA inhibits the transporting activity of cMOAT suggests that CsA inhibits the transport of bilirubin glucuronide by cMOAT expressed in the liver and increases the serum level of bilirubin glucuronide. However, the hyperbilirubinemia appeared to be of no clinical significance in these trials.
In conclusion, our results indicate that human cMOAT overexpression confers a novel drug resistance phenotype and suggest that the substrate specificity of cMOAT overlaps with but is distinct from that of MRP. CsA and PAK-104P almost completely reverse drug resistance by interacting with the substrate-binding site of cMOAT and may be useful for reversing cMOAT-mediated drug resistance in tumors.
Note Added in Proof—After submission of the manuscript, Cui et al. (Mol Pharmacol 55:929–937, 1999) reported that drug resistance and ATP-dependent conjugate transport are mediated by MRP2 expressed in human and canine cells.
Acknowledgments
We thank Drs. Marcel Kool and Piet Borst (The Netherlands Cancer Institute, Amsterdam, the Netherlands) for the monoclonal antibody against cMOAT used in this study, Dr. A. W. Ford-Hutchinson (Merck-Frosst Center for Therapeutic Research, Pointe Claire-Dorval, Quebec, Canada) for the gift of MK571, Dr. Kazumitsu Ueda (Kyoto University, Kyoto, Japan) for the gift of KB/MRP cells, and Dr. Kiyotomo Seto (Nissan Chemical Industries, Chiba, Japan) for the gift of PAK-104P. Thanks also are due to Sandoz (Tsukuba, Japan) for kindly providing CsA and PSC833, to Daiichi Seiyaku (Tokyo, Japan) for kindly providing CPT-11 and SN-38, and to Hiromi Kakura for her excellent secretarial assistance. Z.-S.C. appreciates the postdoctoral fellowship from the Japan Society for the Promotion of Science.
Footnotes
- Received May 17, 1999.
- Accepted August 24, 1999.
-
Send reprint requests to: Dr. Shin-ichi Akiyama, Department of Cancer Chemotherapy, Institute for Cancer Research, Faculty of Medicine, Kagoshima University, 8–35-1 Sakuragaoka Kagoshima 890-8520, Japan. E-mail:akiyamas{at}khosp2.kufm.kagoshima-u.ac.jp
-
This work was supported by grants from the Ministry of Education, Science, and Culture; the Ministry of Health and Welfare, Japan; and Japan Society for the Promotion of Science.
Abbreviations
- MDR
- multidrug resistance
- Pgp
- P-glycoprotein
- MRP
- multidrug resistance protein
- GSH
- reduced glutathione
- GS-X pump
- ATP-dependent glutathione-S conjugate export pump
- cMOAT
- canalicular multispecific organic anion transporter
- VCR
- vincristine
- ADM
- doxorubicin (Adriamycin)
- CPT-11
- 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin
- SN-38
- 7-ethyl-10-hydroxy-camptothecin
- MX
- mitoxantrone
- CsA
- cyclosporin A
- PSC833
- (3′-oxo-4-butenyl-4-methyl-threonine1,(valine2)cyclosporin
- PAK-104P
- 2-[4-(diphenylmethyl)-1-piperazinyl]-5-(trans-4,6-dimethyl-1,3,2-dioxaphosphorinan-2-yl)-2,6-dimethyl-4-(3-nitrophenyl)-3-pyridinecarboxylate P-oxide
- MK571
- 3-([{3(2-[7-chloro-2-quinolinyl]ethenyl)phenyl}-{(3-dimethylamino-3-oxopropyl)-thio}-methyl]thio)propanoic acid
- BSO
- dl-buthionine-(S,R)-sulfoximine
- LTC4
- leukotriene C4
- DNP-SG
- S-(2, 4-dinitrophenyl)glutathione
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}