


Abstract
The mammalian CYP2C subfamily is one of the largest and most complicated in the cytochrome P450 superfamily. In this report, we describe the organization of the mouse Cyp2c locus, which contains 15 genes and four pseudogenes, all of which are located in a 5.5-megabase region on chromosome 19. We cloned three novel mouse CYP2C cDNAs (designated CYP2C50, CYP2C54, and CYP2C55) from mouse heart, liver, and colon, respectively. All three cDNAs contain open reading frames that encode 490 amino acid polypeptides that are 57 to 95% identical to other CYP2Cs. The recombinant CYP2C proteins were expressed in Escherichia coli after N-terminal modification, partially purified, and shown to be active in the metabolism of both arachidonic acid (AA) and linoleic acid, albeit with different catalytic efficiencies and profiles. CYP2C50 and CYP2C54 metabolize AA to epoxyeicosatrienoic acids (EETs) primarily, and linoleic acid to epoxyoctadecenoic acids (EOAs) primarily, whereas CYP2C55 metabolizes AA to EETs and hydroxyeicosatetraenoic acids and linoleic acid to EOAs and hydroxyoctadecadienoic acids. Northern blotting and reverse transcription-polymerase chain reaction analysis reveal that CYP2C50 transcripts are abundant in liver and heart; CYP2C54 transcripts are present in liver, kidney, and stomach; and CYP2C55 transcripts are abundant in liver, colon, and kidney. Immunoblotting studies demonstrate that CYP2C50 protein is expressed in liver and heart, CYP2C54 protein is detected primarily in liver, and CYP2C55 protein is present primarily in colon. Immunohistochemistry reveals that CYP2C55 is most abundant in surface columnar epithelium in the cecum. We conclude that these new CYP2C enzymes are probably involved in AA and linoleic acid metabolism in mouse hepatic and extrahepatic tissues.
Cytochrome P450s (P450s) have been the subject of intense investigation by toxicologists and pharmacologists because they catalyze the metabolism of a wide range of exogenous compounds, including drugs, industrial chemicals, environmental pollutants, and carcinogens (Guengerich, 1992; Nelson et al., 1996; Nebert and Russell, 2002). Many of the enzymes are also active in the NADPH-dependent oxidation of endogenous compounds such as arachidonic acid (AA) and linoleic acid (Capdevila et al., 2000; Zeldin, 2001; Nebert and Russell, 2002). The P450-derived AA metabolites include cis-epoxyeicosatrienoic acids (5,6-, 8,9-, 11,12-, 14,15-EET), midchain hydroxyeicosatetraenoic acids (5-, 8-, 9-, 11-, 12-, and 15-HETE), and ω-terminal alcohols of AA (16-, 17-, 18-, 19-, and 20-HETE) (Capdevila et al., 2000; Zeldin, 2001). These metabolites possess potent biological functions in a variety of different tissues. For example, the EETs and their hydration products, the dihydroxyeicosatrienoic acids, have been shown to control peptide hormone secretion in the pancreas (Falck et al., 1983), regulate vascular tone in the heart (Campbell and Harder, 1999; Fisslthaler et al., 1999), affect ion transport in the kidney (Zou et al., 1996), and possess antiinflammatory properties in the vasculature (Node et al., 1999). Similarly, 19- and 20-HETE have been reported to affect the vascular tone and ion transport in the kidney and brain (Carroll et al., 1996; Imig et al., 1996; Gebremedhin et al., 2000).
The CYP2C subfamily is one of the largest and most complicated in the cytochrome P450 superfamily. Multiple members of the CYP2C subfamily have been identified in many species, including nine in rabbits, nine in rats, and four in humans (for update, see http://drnelson.utmem.edu/CytochromeP450.html). The four human CYP2C subfamily members are CYP2C8, CYP2C9, CYP2C18, and CYP2C19. The human CYP2Cs account for ∼20% of total human liver P450 content and metabolize many clinically used drugs, including diazepam, omeprazole, mephenytoin, tolbutamide, warfarin, ibuprofen, and indomethacin (Goldstein and de Morais, 1994; Pelkonen et al., 1998). The human CYP2Cs also metabolize endogenous compounds, such as AA. For example, CYP2C8 catalyzes the regio- and stereoselective epoxidation of AA to 11R,12S- and 14R,15S-EET, whereas CYP2C9 produces a mixture of 8S,9R-EET, 11S,12R-EET, and 14S,15R-EET (Daikh et al., 1994; Zeldin et al., 1995).
CYP2C29 was the first mouse CYP2C member identified (Matsunaga et al., 1994), followed by CYP2C37, CYP2C38, CYP2C39, and CYP2C40 (Luo et al., 1998). The mouse CYP2Cs differ in their tissue distribution and catalytic function (Luo et al., 1998; Tsao et al., 2000, 2001). Thus, CYP2C29 transcripts are detected mainly in liver, brain, kidney, lung, heart, intestine, adrenals, aorta, seminal vesicles, testis, and ovary. CYP2C37 is most abundant in liver, white blood cells, and female adrenals. CYP2C38 is found in liver, intestine, kidney, and brain. CYP2C39 is present primarily in liver but also found in epididymis. CYP2C40 is highly expressed in liver, colon, heart, kidney, and skin. The mouse CYP2Cs metabolize AA to different regio- and stereospecific products, including EETs and HETEs. For example, CYP2C29 produces mainly 14R,15S-EET, whereas CYP2C39 produces mainly 14S,15R-EET. CYP2C29, CYP2C37, and CYP2C40 are stereoselective for production of 8S,9R-EET, whereas CYP2C38 and CYP2C39 are stereoselective for production of 8R,9S-EET. CYP2C40 produces primarily 16-HETE, but also produces 14R,15S-EET and a small amount of 8S,9R-EET. CYP2C37, CYP2C38, CYP2C39 also generate HETEs (Luo et al., 1998; Tsao et al., 2000, 2001).
The mouse is being used increasingly as a model for human physiology and disease. Indeed, targeted disruption to elucidate the functional relevance of individual genes has become relatively routine. Identification and enzymatic characterization of the mouse CYP2C enzymes will be an important first step to further progress in this area. Using exonic sequences of known human, rat and mouse CYP2Cs and the BLAST software program, we characterized the mouse Cyp2c gene cluster in the Celera Discovery System. We located all five known mouse Cyp2c genes and ten potentially new mouse Cyp2c genes on a single contig on chromosome 19. We then cloned cDNAs for three of the new members designated CYP2C50, CYP2C54, and CYP2C55 and heterologously expressed these in Escherichia coli. Enzymatic assays showed that each was active in metabolism of both AA and linoleic acid; however, their catalytic efficiencies and metabolic profiles were different. We also examined the tissue distribution and relative abundance of these three new CYP2C enzymes in various mouse tissues using specific probes at both the mRNA and protein levels.
Materials and Methods
Materials. [1-14C]Arachidonic acid and [1-14C]linoleic acid were purchased from Amersham Biosciences (Little Chalfont, Buckinghamshire, UK). Restriction enzymes were purchased from New England Biolabs (Beverly, MA). Human NADPH-cytochrome P450 oxidoreductase (POR) was purchased from Oxford Biomedical Research, Inc. (Oxford, MI). Oligonucleotides were synthesized by BioBserve Biotechnologies (Laurel, MD). All other chemicals and reagents were purchased from Sigma unless otherwise specified.
Database Searching. The BLAST software program was used to search the entire mouse genome in the Celera Discovery System (assembly R26) to identify exons of potentially new members of this P450 subfamily. Exonic sequences of known mouse, rat, and human CYP2C members were entered into the program, and an identity cutoff of 55% was used for this analysis (Nelson et al., 1996). The nine exons for all five of the known mouse Cyp2c genes were found on a single contig on chromosome 19 (GA_x6K02T2RE5P:29000001.34500000). We also found 10 new Cyp2c genes and four new Cyp2c pseudogenes on this contig. A physical map of the mouse Cyp2c locus was then assembled using this information (Fig. 1) and putative cDNA sequences of the new mouse CYP2Cs were derived by combining the exonic sequences. Each of the new mouse Cyp2c genes and pseudogenes were given formal names by the Committee on Standardized P450 Nomenclature (see http://drnelson.utmem.edu/CytochromeP450.html)

Organization of the mouse CYP2C cluster on chromosome 19. Exonic sequences of known mouse, rat, and human CYP2C subfamily members were used to search the mouse genomic sequences in the Celera Discovery System. Fifteen Cyp2c genes (solid arrows) and four Cyp2c pseudogenes (hatched arrows) were identified. All of them are located in one contig in a 1.5-Mb cluster on chromosome 19 (shown proximal to distal), except for Cyp2c44, which is located 3.8 Mb downstream. The distance between the top three lines is enlarged to show the direction of transcription of the individual P450 genes (indicated by arrows).
Cloning of the CYP2C50, CYP2C54, and CYP2C55 cDNAs. Total RNA was prepared from female C57BL/6 mouse liver, heart, and colon using an RNeasy Mini Kit according to the manufacturer's instructions (QIAGEN, Valencia, CA). Based on the cDNA sequences derived from the Celera Discovery System analysis, primer pairs (Table 1) were designed to amplify the coding regions of the CYP2C50, CYP2C54, and CYP2C55 cDNAs. RT-PCR was done with a ProSTAR Ultra HF RT-PCR System (Stratagene, La Jolla, CA). Briefly, first strand cDNA was synthesized from 500 ng of total RNA using StrataScript reverse transcriptase with oligo(dT) priming. PCR amplifications were performed with 1.0 μl of cDNA template, 0.4 μM concentrations of each primer, 0.2 μM dNTPs and 2.5 U of PfuTurbo DNA polymerase (Stratagene) in a total volume of 50 μl. PCR conditions were as follows: 95°C for 1 min; 40 cycles of 95°C for 30 s, 50°C for 30 s, and 68°C for 5 min; final extension step at 68°C for 10 min. PCR products were analyzed on 1.2% agarose gels containing ethidium bromide and cloned into pCR II vectors using a TA cloning kit from Invitrogen (Carlsbad, CA). Positive clones were identified by blue-white selection and restriction enzyme digestion. Plasmids were prepared from positive clones using a Plasmid Mini-prep kit (QIAGEN) and completely sequenced using an ABI Prism BigDye DNA sequencing kit (Applied Biosystems, Foster City, CA).
Designation of primers for cloning of CYP2C cDNAs
Based on the cDNA sequences derived from the Celera Discovery System analysis, primer pairs were designed to amplify the coding regions of the three CYP2C cDNAs. The initiation codon ATG is in bold. Positions are relative to the ATG as +1.
Expression and Partial Purification of Mouse CYP2Cs. All three mouse CYP2C cDNAs were expressed in E. coli using methods described previously (Barnes, 1996). To enhance expression levels, the open reading frames were subcloned into the pCW bacterial expression vector after modification of their N termini by replacing the initial eight amino acid residues with the corresponding ones of the modified bovine P450 17α-hydroxylase (Barnes et al., 1991). As shown in Table 2, the forward primers were 57 to 61 nucleotides in length and contained eight nucleotides at the 5′-end preceding an NdeI restriction site, the modified N-terminal sequence, and an additional 21 to 25 nucleotides at the 3′-end, which were specific for the individual CYP2C cDNA. Modification was accomplished by PCR amplification of the open reading frames using PfuTurbo DNA polymerase. The PCR fragments with the NdeI site at the 5′-end and either a HindIII site (CYP2C50) or a SalI site (CYP2C54 and CYP2C55) at the 3′-end were subcloned into the corresponding sites of the pCW vector and sequenced to verify the absence of mutation and to confirm orientation. About 50 ng of expression plasmids were introduced into DH5α competent cells. A single colony was picked and cultured overnight in Luria-Bertani broth containing 100 μg/ml ampicillin. The overnight culture was diluted 10-fold in Terrific Broth and cultured for 48 to 72 h at room temperature in the presence of ampicillin (200 μg/ml), isopropyl β-d-thiogalactoside (0.5 mM) and δ-aminolevulinic acid (0.5 mM). Cytochrome P450 expression was monitored at 24-, 48-, and 72-h intervals with a DW-2/OLIS spectrophotometer as described by Omura and Sato (1964). The cultured cells were pelleted, suspended in suspension buffer (20 mM KPO4, pH 7.25, 100 mM KCl, 1 mM EDTA, 1 mM dithiothreitol), and sonicated (20 s at 40 sec intervals, repeated 20 times) in the presence of 1 mM phenylmethylsulfonyl fluoride. Cell membranes were obtained by centrifugation at 40,000 rpm for 1 h and then homogenized with a Teflon homogenizer in buffer A (10 mM KPO4, pH 7.4, 0.1 mM EDTA, 20% glycerol, and 0.1% cholate). The cytochrome P450 was solubilized from cell membranes by adding Nonidet P-40 and shaking at 4°C overnight. The supernatant obtained by centrifugation at 40,000 rpm for 1 h was applied to a hypatite C column (Clarkson Chromatography Products, South Williamsport, PA), which was pre-washed with buffer B (10 mM KPO4, pH 7.4, and 20% glycerol). The cytochrome P450 was washed with buffer A and finally eluted with elution buffer (0.5 M KPO4, pH 7.4, 0.1 mM EDTA, 20% glycerol, and 0.2% cholate). Fractions containing a high concentration of P450 were pooled and dialyzed against dialysis buffer (0.1 M KPO4, pH 7.4, 0.1 mM EDTA, and 20% glycerol) for 48 h at 4°C.
Modification of mouse CYP2C cDNAs for expression in E. coli
Underlined bases show the NdeI restriction site and modified N-terminal sequences are shown in bold.
Incubation of Recombinant CYP2Cs with AA and Linoleic Acid. Partially purified recombinant CYP2C proteins (final concentration, 0.25 μM P450) were preincubated with POR (POR/P450 molar ratio, 3:1) and dilauroylphosphatidylcholine (final concentration, 50 μg/ml) for 20 min at room temperature. This ratio of POR/P450 was found to be optimal in initial experiments. After preincubation, the enzyme mixtures were added to a buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM KCl, 10 mM MgCl2, 8 mM sodium isocitrate, and 0.5 IU/ml isocitrate dehydrogenase, and the entire mixture was then added to tubes containing [1-14C]arachidonic acid (55 μCi/μmol; final concentration, 100 μM). After temperature equilibration at 37°C, reactions were initiated by the addition of NADPH (final concentration, 1 mM) and incubated with constant stirring at 37°C for 15 min. The reaction products were extracted into diethyl ether and analyzed by reversed-phase HPLC as described previously (Zeldin et al., 1997; Ma et al., 1999). All products were identified by comparing their HPLC properties with those of authentic EET and HETE standards. To determine regiochemical distribution, fractions were collected from the reversed-phase HPLC eluent and then rechromatographed on a normal-phase HPLC system to resolve individual EET and HETE regioisomers, as described previously (Zeldin et al., 1997; Ma et al., 1999). For chiral analysis, the EETs were collected from HPLC eluents, derivatized to the corresponding EET-pentafluorobenzyl or EET-methyl esters, purified by normal phase HPLC, resolved into the corresponding enantiomers by chiral-phase HPLC, and quantified by liquid scintillation as described previously (Capdevila et al., 1991). In some experiments, [1-14C]linoleic acid (55 μCi/μmol; final concentration, 100 μM) was substituted for [1-14C]arachidonic acid, and the products were identified by coelution with authentic epoxyoctadecenoic acid (EOA) and hydroxyoctadecadienoic acid (HODE) standards on reversed-phase HPLC.
Northern Blotting and RT-PCR Analysis. Northern blots containing mRNAs from 16 different tissues of C57BL/6 mice were purchased from BD Biosciences Clontech (Palo Alto, CA). Blots were hybridized with either CYP2C cDNA inserts, CYP2C sequence-specific oligonucleotide probes (Table 3), or β-actin cDNA. cDNA probes were labeled with [α-32P]dCTP using a random primed labeling method (High Prime; Roche Applied Science, Indianapolis, IN). Oligonucleotide probes were end-labeled with [γ-32P]ATP using T4 polynucleotide kinase. Hybridizations were conducted for 1 h at 68°C for cDNA probes and at 37°C for oligonucleotide probes in ExpressHyb hybridization solution (BD Biosciences Clontech). After hybridizations, blots were washed four times for 40 min at room temperature and four times for 40 min at 68°C for cDNA probes and at 37°C for oligonucleotide probes. Blots were stripped by washing twice (total 30 min) with heated sterile water containing 0.5% SDS before reprobing. Northern blot results were independently confirmed by PCR amplification from reverse-transcribed mouse RNAs. Total RNA was prepared from various C57BL/6 mouse tissues using an RNeasy Mini Kit (QIAGEN). PCR primers (Table 3) were designed to specifically amplify individual CYP2C cDNAs under appropriate conditions. Forty cycles were performed with annealing at 54°C for 30 s for CYP2C50, and 35 cycles were performed with annealing at 56°C for 30 s for CYP2C54 and CYP2C55. Mouse β-actin cDNA was amplified to control for RNA quality and quantity with the forward primer 5′-GACAGGATGCAGAAGGAGATCAC-3′ (corresponding to nucleotides 1011 to 1033 of the mouse β-actin cDNA) and the reverse primer 5′-GCTGATCCACATCTGCTGGAA-3′ (corresponding to nucleotides 1133 to 1154 of the mouse β-actin cDNA). PCR products were analyzed on 1.5% agarose gels using ethidium bromide staining.
Sequences of gene-specific primers for RT-PCR
Unique nucleotides are underlined.
Protein Immunoblotting. Microsomal fractions were prepared from adult C57BL/6 mouse tissues by differential centrifugation at 4°C as described previously (Zeldin et al., 1997; Ma et al., 1999). Peptides specific to the deduced amino acid sequences of CYP2C50, CYP2C54, and CYP2C55 were designed based on the sequence alignments with the other known CYP2 family and CYP2C subfamily members (Fig. 2). The peptides were synthesized, HPLC-purified, and coupled to keyhole limpet hemocyanin via a C-terminal cysteine to enhance antigenicity by Research Genetics (Huntsville, AL). Polyclonal antibodies against these peptides (anti-CYP2C50pep1, anti-CYP2C54pep1, and anti-CYP2C55pep1) were raised in New Zealand white rabbits at Covance Research Products, Inc. (Denver, PA). Purified recombinant CYP2C29, CYP2C37, CYP2C38, CYP2C39, CYP2C40, and CYP2C44 were prepared as described previously (Luo et al., 1998). Microsomes from Sf9 cells infected with recombinant CYP2J5 baculovirus were prepared as described previously (Ma et al., 1999). Tissue microsomal fractions (30 μg/lane) or recombinant P450s (0.5 pmol of P450/lane) were electrophoresed in 12% Tris glycine gels from Novex (San Diego, CA), and the resolved proteins were transferred onto nitrocellulose membranes. Membranes were immunoblotted using the various rabbit anti-mouse CYP2C antibodies (1:1000 dilution), goat anti-rabbit IgG (1:10,000 dilution) conjugated to horseradish peroxidase (Amersham Biosciences), and the Supersignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL). Protein determinations were performed using reagents from Bio-Rad (Hercules, CA).

Deduced amino acid sequences for CYP2C50, CYP2C54, and CYP2C55. The putative heme-binding region and the putative recognition site for cAMP-dependent kinase are underlined. The six putative CYP2C substrate recognition sites are boxed. The locations of the peptides that were used to prepare the anti-CYP2C50pep1, anti-CYP2C54pep1, and anti-CYP2C55pep1 antibodies are highlighted in gray. Asterisks indicate identical amino acids; dots indicate conservatively replaced amino acids.
Immunohistochemistry. Immunohistochemical staining of CYP2C55 in mouse intestinal tissues was performed using methods similar to those described previously(Zeldin et al., 1997). The anti-CYP2C55pep1 IgG was used at a dilution of 1:500. Rabbit preimmune IgG was used as the negative control. Antibody-antigen detection was performed with the ABC kit (Vector Laboratories, Burlingame, CA). The end product was visualized by exposure to 3,3′-diaminobenzidine (DakoCytomation California Inc., Carpinteria, CA). To validate immunohistochemical specificity, the anti-CYP2C55pep1 IgG was incubated overnight at 4°C with excess peptide (0.05 mg/ml CYP2C55pep1), and the antibody-protein mixture was then used in place of primary antibody for immunohistochemistry. The resultant staining was compared with that observed without the blocking peptide.
Results
Organization of the Mouse Cyp2c Locus on Chromosome 19. By BLAST searching the mouse genomic database in the Celera Discovery System with exonic sequences of known mouse, rat, and human CYP2Cs, we mapped all five of the known mouse Cyp2c genes (including Cyp2c29, Cyp2c37, Cyp2c38, Cyp2c39, and Cyp2c40) to a 5.5-Mb region on chromosome 19 (contig GA_x6K02T2RE5P: 29000001.34500000) (Fig. 1). We also found 10 potentially new Cyp2c genes (designated Cyp2c44, Cyp2c50, Cyp2c54, Cyp2c55, Cyp2c65, Cyp2c66, Cyp2c67, Cyp2c68, Cyp2c69, and Cyp2c70) and four new Cyp2c pseudogenes (designated Cyp2c52-ps, Cyp2c53-ps, Cyp2c71-ps, and Cyp2c72-ps) in this region. Each of the pseudogenes has missing exons or critical amino acids. For example, several nucleotides encoding amino acids PYTD in K-helix are absent in Cyp2c52-ps, Cyp2c53-ps does not contain an open reading frame, Cyp2c71-ps is missing exons 2, 3, 4, 6, and 7, and Cyp2c72-ps contains only exons 1, 2, and 3. Cyp2c52-ps, Cyp2c55, Cyp2c65, Cyp2c66, Cyp2c53p, Cyp2c29, Cyp2c72-ps, Cyp2c39, Cyp2c71-ps, Cyp2c37, and Cyp2c50 are situated in a positive orientation (5′ to 3′), and Cyp2c38, Cyp2c67, Cyp2c68, Cyp2c40, Cyp2c69, Cyp2c54, Cyp2c70, and Cyp2c44 are in a negative orientation (3′ to 5′) on chromosome 19 (Fig. 1). Each of the Cyp2c genes is located within a 1.5-Mb cluster except for Cyp2c44, which is least homologous with other Cyp2c subfamily members (56-61% identical) and is located ∼3.8 Mb downstream. Approximately 57 genes unrelated to the P450 superfamily are located between Cyp2c70 and Cyp2c44. Examples include pyrroline 5-carboxylate synthetase, ectonucleoside triphosphate diphosphohydrolase, and the ATP-binding cassette subfamily C transporter MRP2. Highly homologous mouse Cyp2c genes tend to be located adjacent to one another on the chromosome. Examples include Cyp2c37, Cyp2c50, and Cyp2c54 (95-96% identical), Cyp2c40, Cyp2c67, Cyp2c68, and Cyp2c69 (97-98% identical), Cyp2c38 and Cyp2c39 (93% homologous), and Cyp2c65 and Cyp2c66 (95% identical).
cDNA Cloning of CYP2C50, CYP2C54, and CYP2C55. The CYP2C50, CYP2C54, and CYP2C55 cDNAs were cloned by PCR from reverse-transcribed mouse heart, liver, and colon RNAs, respectively. The cDNA sequences have been deposited into GenBank with the following accession numbers: CYP2C50 (AY206873), CYP2C54 (AY206874), and CYP2C55 (AY206875). These sequences are 99 to 100% identical to the predicted cDNA sequences derived from the Celera mouse genomic database. Each cDNA contains an open reading frame that encodes a 490-amino acid polypeptide that includes a putative heme binding peptide and an invariant cysteine at position 435 (Fig. 2). Each polypeptide also contains structural features common to other CYP2 family members, including an N-terminal hydrophobic peptide, a proline cluster between residues 30 and 37, and six putative substrate recognition sites (Gotoh, 1992). In addition, CYP2C50 and CYP2C55 contain a putative recognition site for cAMP-dependent kinase (Arg-Arg-Phe-Ser) at positions 124 to 127, which is believed to be a cytochrome b5 binding site (Muller et al., 1985; Jansson et al., 1987). The three new CYP2C enzymes are 65 to 94% identical at the amino acid level with the five known CYP2C enzymes (Table 4).
Deduced amino acid sequence identity of the three new mouse CYP2C enzymes with each other and with known mouse CYP2Cs
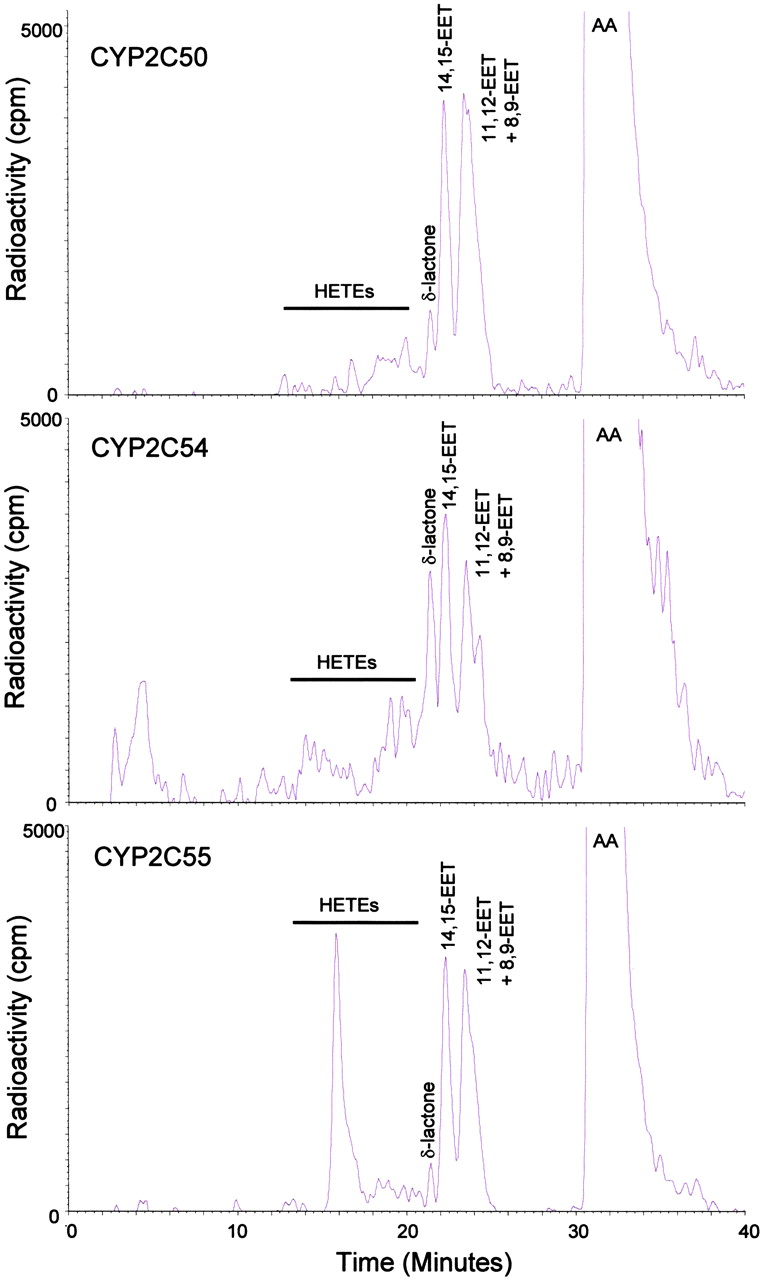
Heterologous Expression and Enzymatic Characterization of Recombinant CYP2Cs. The three mouse CYP2C cDNAs were heterologously expressed in E. coli using the pCW vector with slight modification at the N terminus to optimize expression. Expression varied from 128 to 346 nmol of P450/liter of cultured cells. All three recombinant CYP2Cs gave typical b-type cytochrome P450 reduced CO-difference spectrum with Soret maxima at 450 nm (data not shown). When the partially purified cytochromes were reconstituted with POR and incubated with radiolabeled AA in the presence of NADPH and an NADPH-regenerating system, all three enzymes were active in the metabolism of AA, albeit with different catalytic efficiencies and profiles (Fig. 3). CYP2C50 and CYP2C54 were primarily AA epoxygenases in that EETs accounted for approximately 83 to 87% of the total reaction products. In contrast, CYP2C55 was both an AA epoxygenase and AA hydroxylase, producing mixtures of EETs (57% of total) and HETEs (43% of total). Catalytic turnover numbers for combined EET and HETE formation by CYP2C50, CYP2C54, and CYP2C55 were 0.7, 1.0, and 1.2 nmol of product/nmol of P450/min at 37°C, respectively.

Reversed-phase HPLC chromatograms of products formed during incubations of recombinant CYP2C enzymes with radiolabeled AA. Retention times of authentic EET and HETE standards are indicated above the respective peaks. Ordinate, radioactivity (counts per minute); abscissa, time (minutes). Results are representative of eight separate experiments with each P450 enzyme.
Regiochemical analysis of the EETs revealed a preference for epoxidation at the 11,12-olefin (30-40% of total EETs) for each of the P450s (Table 5). Epoxidation at the 14,15-olefin (25-36% of total EETs), 8,9-olefin (18-25% of total EETs), and 5,6-olefin (7-20% of total EETs) occurred less often. Stereochemical analysis showed that each of the P450s produced EETs with slight stereochemical selectivity (Table 6). Thus, CYP2C50 exhibited a slight preference for biosynthesis of 14R,15S-, 11S,12R- and 8S,9R-EET, CYP2C54 exhibited a slight preference for 14S,15R-, 11S,12R- and 8S,9R-EET, and CYP2C55 exhibited a slight preference for 14S,15R-, 11R,12S-, and 8R,9S-EET (Table 6). Regiochemical analysis of HETEs revealed that CYP2C50 made several midchain and omega-terminal HETEs; 12-HETE (24% of total HETEs) was the most abundant, followed by 11-, 15-, 8-, 16-, 19-, 18-, and 5-HETE. In contrast, CYP2C54 made primarily 12-HETE (56% of total HETEs) and CYP2C55 made primarily 19-HETE (79% of total HETEs) (Table 5).
Regioselective composition of EETs and HETEs produced by recombinant CYP2C enzymes
The regiochemical distribution of EETs and HETEs formed by incubation of recombinant mouse CYP2C enzymes with [1-14C]AA was quantified by HPLC and liquid scintillation as described under Materials and Methods. Values shown are average of at least three different experiments.
Enantioselective composition of EETs produced by recombinant CYP2C enzymes
The stereochemical distribution of EETs formed by incubation of recombinant mouse CYP2C enzymes with [1-14C]AA was quantified by chiral-phase HPLC and liquid scintillation as described under Materials and Methods. Values shown are average of at least three different experiments.
All three recombinant mouse CYP2Cs were also active in the metabolism of linoleic acid to epoxides and alcohols. Thus, CYP2C50 metabolized linoleic acid primarily to EOAs (82% of total products), whereas CYP2C54 and CYP2C55 metabolized linoleic acid to mixtures of EOAs and HODEs (Fig. 4). For CYP2C54, 53% of total products were EOAs and for CYP2C55, 57% of total products were EOAs. CYP2C50, CYP2C54, and CYP2C55 metabolized linoleic acid with catalytic turnover numbers of 1.3, 1.6, and 1.6 nmol products formed/nmol of P450/min at 37°C, respectively.

Reversed-phase HPLC chromatograms of products formed during incubations of recombinant CYP2C enzymes with radiolabeled linoleic acid. Retention times of authentic EOA and HODE standards are indicated above the respective peaks. Ordinate, radioactivity (counts per minute); abscissa, time (minutes). Results are representative of eight separate experiments with each P450 enzyme.
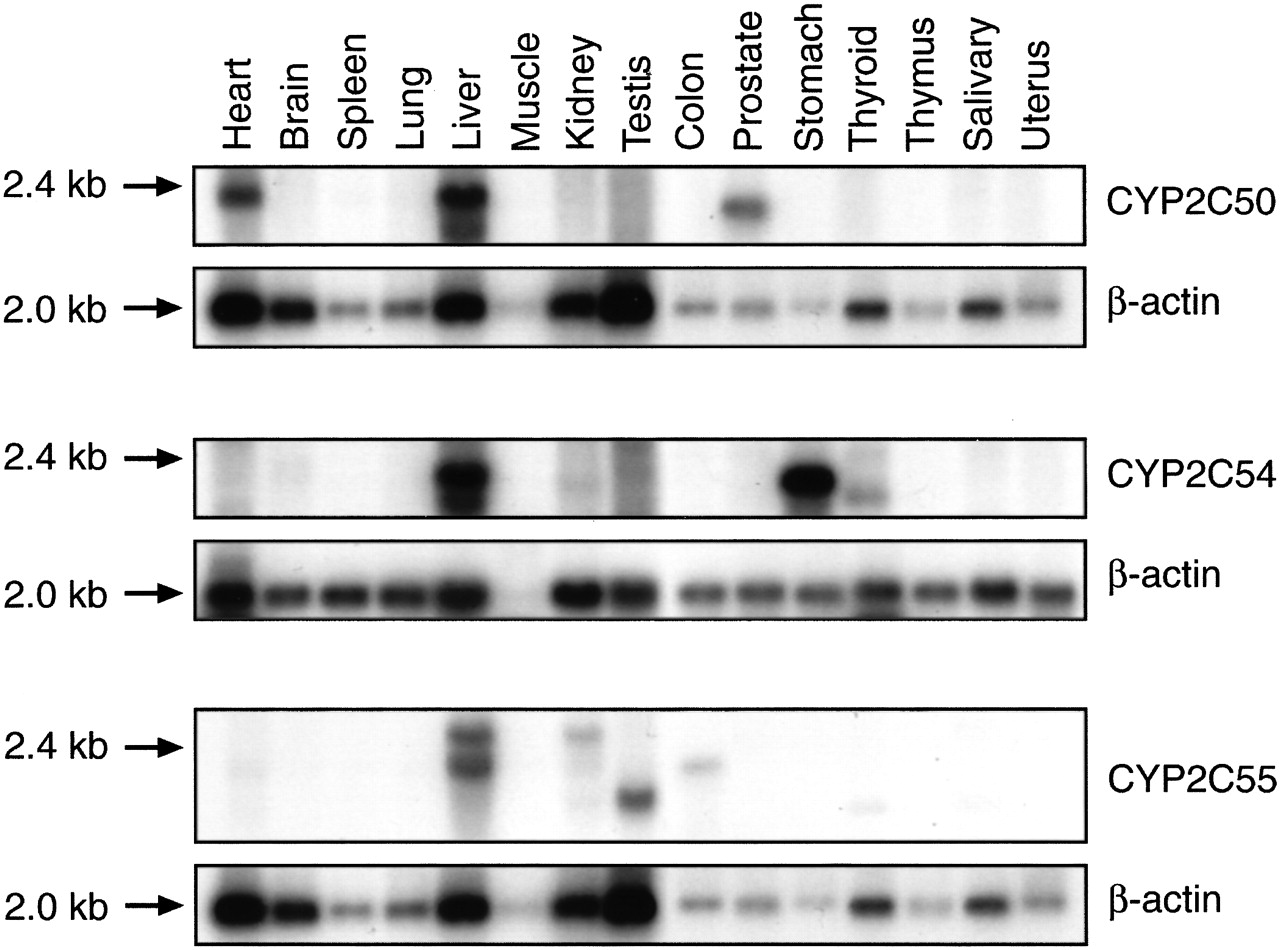
Tissue Distribution of New CYP2C mRNAs. To ascertain the relative organ abundance of the new CYP2C mRNAs, we performed Northern blotting using sequence-specific CYP2C oligonucleotide probes under high-stringency conditions and RT-PCR using CYP2C sequence-specific primer pairs. A prominent 1.8- to 2.0-kb transcript was detected in both liver and heart RNA on Northern blots probed with the CYP2C50-specific oligonucleotide (Fig. 5). A slightly smaller, less abundant transcript (1.5 kb) was also observed in prostate RNA. Consistent with these results, RT-PCR detected a prominent band of the predicted size in both liver and heart RNA using CYP2C50 sequence-specific primer pairs (Fig. 6). However, no band was detected in prostate RNA, suggesting that the identity of the smaller 1.5-kb transcript on Northern blots was either a CYP2C50 splice variant that did not contain the amplified exons or a related gene that shared nucleic acid sequence homology with CYP2C50. Northern blots with the CYP2C54-specific probe detected a prominent 1.8-kb band in liver and stomach RNA, and a less abundant band of the same size in kidney RNA (Fig. 5). A slightly smaller 1.5-kb band was also detected in thyroid RNA. RT-PCR analysis with CYP2C54 sequence-specific primer pairs confirmed expression of CYP2C54 transcripts in liver, stomach, and kidney and also detected a band of the predicted size in adrenal RNA (Fig. 6). Northern blots with the CYP2C55-specific probe detected a prominent 1.8- to 2.0-kb band in liver RNA and a less abundant band of the same size in colon and kidney RNA (Fig. 5). A slightly larger transcript (2.4 kb) was detected in liver and kidney RNA, and slightly smaller transcript (1.5 kb) was detected in testes RNA. RT-PCR analysis demonstrated that CYP2C55 transcripts were present in all mouse tissues examined (Fig. 6). Fragments of the predicted size were amplified in livers from both male and female mice using specific primers for CYP2C50, CYP2C54, and CYP2C55, and in kidneys from both male and female mice using specific primers for CYP2C54 and CYP2C55, suggesting that none of these P450s was expressed in a gender-specific fashion, as has been reported previously for some rat CYP2C enzymes (Dannan et al., 1986).
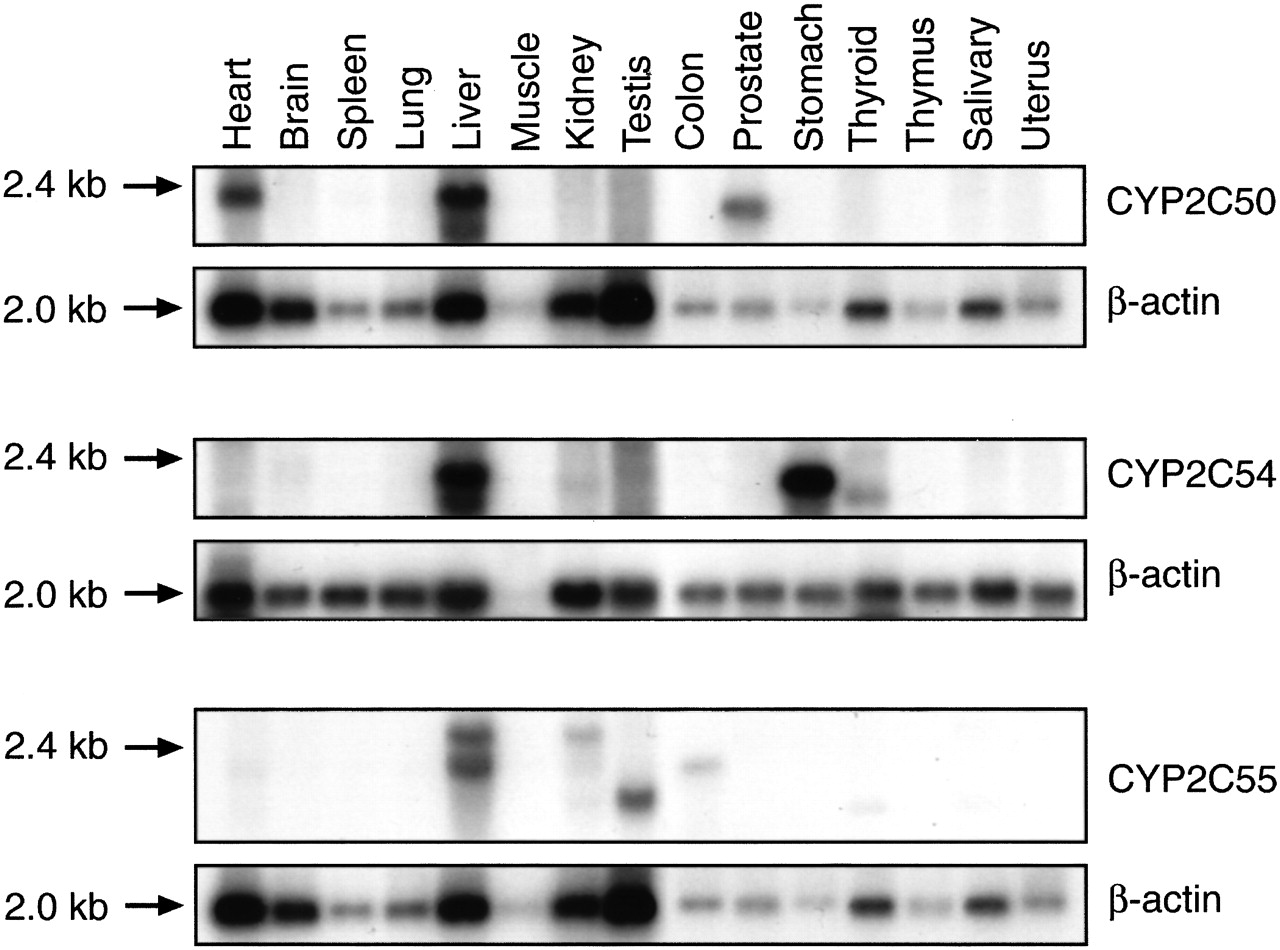
Tissue distribution of CYP2C mRNAs by Northern blotting. Mouse multiple tissue Northern blots containing 2 μg of tissue mRNA per lane were hybridized with P450 sequence-specific oligonucleotides or β-actin cDNA as described under Materials and Methods. Results are representative of three separate experiments.

Tissue distribution of CYP2C mRNAs by RT-PCR. Total RNAs extracted from C57BL/6 mouse liver, kidney, lung, heart, brain, spleen, colon, small intestine, stomach, testis, muscle, aorta, adrenal, prostate, and ovary (M, male; F, female; all others not specified were from males, except ovary) were analyzed by RT-PCR using sequence-specific primer pairs (Table 3) as described under Materials and Methods. Primer specificity was assessed using the mouse CYP2C cDNAs as templates. Mouse β-actin was amplified to control for RNA quality and quantity. PCR products were electrophoresed on 1.5% agarose gels and visualized with ethidium bromide staining. Results are representative of three independent experiments.
Tissue Distribution of New CYP2C Proteins. Based on multiple amino acid sequence alignments of the mouse CYP2C subfamily members, we developed polyclonal antibodies to peptides that were unique to each CYP2C enzyme (anti-CYP2C50pep1, anti-CYP2C54pep1, and anti-CYP2C55pep1). As shown in Fig. 7, both anti-CYP2C54pep1 and anti-CYP2C55pep1 were highly immunospecific for their respective recombinant proteins and did not cross-react with other known CYP2C subfamily members. In contrast, anti-CYP2C50pep1 recognized recombinant CYP2C50 and cross-reacted with recombinant CYP2C54 and, to a lesser extent, with recombinant CYP2C37. This finding is not surprising given that CYP2C37, CYP2C50, and CYP2C54 are >90% identical at the amino acid level (Table 4). Indeed, despite multiple attempts, we were unable to develop a specific antibody to CYP2C50. None of these antibodies cross-reacted with three new mouse P450 proteins (CYP2C65, CYP2C66, or CYP2C70) that we are in the process of characterizing (data not shown). Therefore, anti-CYP2C54pep1 and anti-CYP2C55pep1 seem to be highly immunospecific for their respective P450 proteins with the caveat that we have not expressed CYP2C67, CYP2C68, or CYP2C69. However, because these three P450s are 98% identical to CYP2C40 at the nucleic acid level, cross-reactivity seems highly unlikely.

Tissue distribution of CYP2C proteins by immunoblotting. Purified recombinant CYP2C29, CYP2C37, CYP2C38, CYP2C39, CYP2C40, CYP2C50, CYP2C54, and CYP2C55, or microsomes prepared from Sf9 cells infected with recombinant CYP2J5 baculovirus stock (0.5 pmol of P450/lane), or microsomes prepared from various C57BL/6 mouse tissues (M, male; F, female; adrenal from pooled male and female tissues, others not specified were from male, 30 μg of protein/lane) were electrophoresed, transferred to nitrocellulose, and immunoblotted with anti-CYP2C50pep1 (A), anti-CYP2C54pep1 (B), or anti-CYP2C55pep1 (C) as described under Materials and Methods. Results are representative of three independent experiments.
We performed immunoblotting of microsomal fractions prepared from various mouse tissues with the three peptide-based antibodies to examine the tissue distribution of the new CYP2C proteins. Immunoblotting with the anti-CYP2C54pep1 detected a ∼50-kDa band in female > male liver microsomes and a ∼51-kDa band of approximately equal intensity in liver microsomes from male and female mice (Fig. 7). The ∼50-kDa band may represent CYP2C54, because RT-PCR experiments showed a somewhat greater abundance of CYP2C54 transcripts in liver RNA from female versus male mice (Fig. 6). Anti-CYP2C54pep1 also detected a band of slightly higher molecular mass (∼53 kDa) in lung and adrenal microsomes (Fig. 7). The identity of these latter bands remains unknown, but they are not likely to be CYP2C54 because CYP2C54 transcripts were not detected in these tissues (Fig. 6). Previous RT-PCR analysis indicated that CYP2C29 is present in lung (Tsao et al., 2001); therefore, it is possible that this band could represent CYP2C29. Interestingly, we failed to detect any immunoreactive protein bands with the anti-CYP2C54pep1 antibody in stomach and kidney microsomes, despite the presence of CYP2C54 transcripts in these tissues (Figs. 6 and 7). Immunoblotting with the anti-CYP2C55pep1 produced a prominent ∼50-kDa band in colon microsomes (Fig. 7). This band was also present, albeit at lower intensity, in male and female liver and small intestine microsomes. The presence of the ∼50-kDa band in liver, colon, and small intestine are consistent with Northern blotting and RT-PCR data showing the presence of CYP2C55 transcripts in these tissues (Figs. 5 and 6). We did not detect any immunoreactive protein bands with the anti-CYP2C55pep1 antibody in kidney microsomes, despite the presence of CYP2C55 transcripts in these tissues. Immunoblotting with the anti-CYP2C50pep1 produced a complex pattern, consistent with the known cross-reactivity of this antibody with several mouse CYP2C proteins. We detected a prominent band at ∼51-kDa in heart and liver microsomes (Fig. 7), which is consistent with the abundance of CYP2C50 transcripts in these tissues (Figs. 5 and 6). We also detected the ∼51-kDa band in kidney and brain microsomes and a smaller band (∼49-kDa) in liver microsomes. The immunoreactive bands in kidney and brain are unlikely to be CYP2C50 because CYP2C50 transcripts were not detected in these tissues.
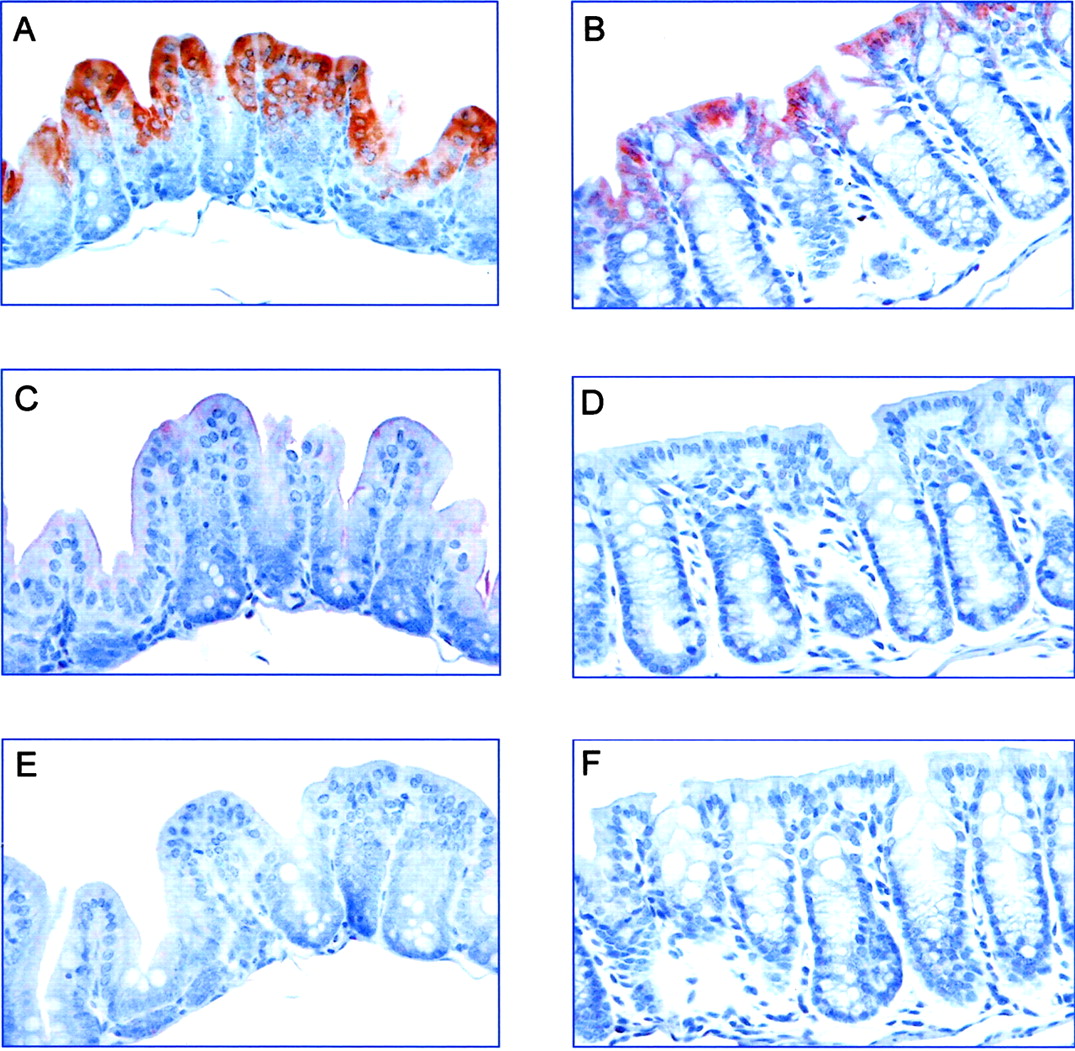
Localization of CYP2C55 Protein Expression in the Intestine. To determine the cellular localization of CYP2C55 protein expression in the intestine, we stained formalin-fixed, paraffin-embedded mouse intestinal tissues with the anti-2C55pep1. Strong staining was noted in the surface columnar epithelium in both the cecum and colon (Fig. 8, A and B). No significant staining was observed in the small intestine (data not shown). There was little or no staining with preimmune IgG (Fig. 8, C and D) or after peptide inhibition (Fig. 8, E and F), thus documenting the specificity of the immunostaining method.

Immunohistochemical staining of CYP2C55 protein in the large intestine. Sections of cecum (A, C, and E) and colon (B, D, and F) were immunostained with anti-CYP2C55pep1 (A and B), preimmune IgG (C and D), or anti-CYP2C55pep1 preincubated with blocking peptide (E and F) as described under Materials and Methods. Strong positive staining of CYP2C55 was observed in the surface columnar epithelium in the cecum and to a lesser extent in the colon. There was little or no staining with preimmune IgG and after peptide inhibition. Results are representative of three independent experiments.
Discussion
Cytochromes P450 represent a large superfamily of ubiquitous hemoproteins that metabolize a diverse array of endogenous and exogenous compounds (Guengerich, 1992; Nelson et al., 1996). The human CYP2C subfamily consists of four members (CYP2C8, CYP2C9, CYP2C18, and CYP2C19) that play a major role in the metabolism of drugs (Goldstein and de Morais, 1994; Pelkonen et al., 1998) and endogenous fatty acids (Daikh et al., 1994; Zeldin et al., 1995). The mouse CYP2C subfamily is substantially larger and more complex than its human counterpart. Five members (CYP2C29, CYP2C37, CYP2C38, CYP2C39, and CYP2C40) have previously been reported (Matsunaga et al., 1994; Luo et al., 1998; Tsao et al., 2000, 2001). Herein, we describe the cDNA cloning, enzymatic characterization, and tissue distribution of three new members of this P450 subfamily (CYP2C50, CYP2C54, and CYP2C55). We also characterize the mouse Cyp2c locus on chromosome 19, which contains an additional seven genes and four pseudogenes. Thus, in mouse, there exists a total of 15 Cyp2c genes and four Cyp2c pseudogenes.
The four human CYP2C genes are all located in a single 500-kb cluster on chromosome 10 (Gray et al., 1995; Nelson et al., 1996). Each of the mouse Cyp2c genes, except Cyp2c44, is located in a ∼1.5-Mb cluster on the syntenic region of mouse chromosome 19. Cyp2c44, also located on chromosome 19, is ∼3.8 Mb downstream of this cluster. It is interesting that CYP2C44 is least homologous to other mouse CYP2C members (50-60% identical at the amino acid level). In contrast, CYP2C subfamily members that are highly homologous (e.g., CYP2C40, CYP2C67, CYP2C68, and CYP2C69) are located next to each other on the chromosome. This pattern reflects the creation of new Cyp2c genes in the mouse by the process of tandem duplication. Indeed, the mouse-human split is estimated to have occurred approximately 75 million years ago, which is sufficient time for changes in gene cluster size and organization to take place (Waterston et al., 2002).
At the amino acid level, the three new mouse CYP2C enzymes reported herein are 67 to 92% identical. Sequence alignments reveal that the three proteins are most divergent within the six putative SRSs, consistent with the differing substrate specificities and catalytic properties of these proteins. Specifically, although each of the new CYP2Cs is active in the metabolism of AA, the regio- and stereochemical profiles differ markedly. Thus, CYP2C50 and CYP2C54 produce mainly EETs, whereas CYP2C55 produces roughly equivalent amounts of EETs and HETEs. It is of interest that the two enzymes found to be highly expressed in colon, CYP2C55 and CYP2C40 (Tsao et al. 12000), produce large amounts of HETEs. Although the regiochemistry of olefin epoxidation is comparable for CYP2C50, CYP2C54, and CYP2C55, the regiochemistry of hydroxylation is enzyme-specific. Thus, CYP2C50 produces multiple midchain and omega-terminal HETEs, CYP2C54 produces mainly 12-HETE, and CYP2C55 produces mainly 19-HETE. Moreover, stereochemical analysis of the EETs reveals subtle differences between the three P450 enzymes. For example, CYP2C50 favors epoxidation at the ri,si face of the 14,15-olefin, CYP2C55 favors epoxidation at the si,ri face, and CYP2C54 produces 14,15-EET in a nearly equal racemic mixture.
The catalytic properties of the newly cloned mouse CYP2Cs are different from those of the other known mouse CYP2C enzymes. Thus, CYP2C29 makes primarily 14R,15S-EET; CYP2C37 biosynthesizes mainly 11R,12S-EET and a variety of midchain HETEs; CYP2C38 makes mainly 11R,12S-EET and 12-HETE; CYP2C39 makes primarily 14S,15R-EET, 11R,12S-EET, 8R,9S-EET, and 11-HETE; and 16-HETE is the major product of CYP2C40 (Luo et al., 1998; Tsao et al., 2000, 2001). Interestingly, CYP2C37 and CYP2C50, which are 94% identical, have remarkably different metabolic profiles. CYP2C55 is unique among the mouse CYP2C enzymes in that it has both AA epoxygenase and ω-1 hydroxylase activity. Mouse CYP2J9 also metabolizes AA to EETs and 19-HETE (Qu et al., 2001).
The present study shows that each of the new mouse CYP2Cs is active in the biosynthesis of EOAs and HODEs from linoleic acid. It is noteworthy that the rates of linoleic acid metabolism are comparable with the rates of AA metabolism, suggesting that neither fatty acid substrate is preferred by these P450s. It has been previously reported that human and rat liver microsomes catalyze the epoxidation and hydroxylation of linoleic acid (Bylund et al., 1998b; Draper and Hammock, 2000). Recombinant human CYP2C8, CYP2C9, and CYP2C19 and other recombinant human P450s including CYP1A2, CYP3A4, and CYP2J2 have been shown to metabolize linoleic acid to EOAs and HODEs (Bylund et al., 1998a,b; Draper and Hammock, 2000; Moran et al., 2000). Unlike EETs and HETEs, the biological significance of the EOAs and HODEs remains largely unknown. 9,10-EOA and 12,13-EOA have been shown to be associated with death in patients with severe burns (Kosaka et al., 1994), and 9,10-EOA has been shown to be a hepatic toxin (Ozawa et al., 1986), a lung toxin that inhibits mitochondrial respiration (Sakai et al., 1995), and a potent cardiotoxin (Sugiyama et al., 1987).
We demonstrated the presence of CYP2C50 in heart at both the mRNA and protein levels. CYP2C50 metabolizes AA primarily to EETs. Importantly, high concentrations of EETs have been found in human and rat heart tissue (Wu et al., 1996, 1997). Coronary artery endothelial cells biosynthesize EETs, which hyperpolarize and relax vascular smooth muscle cells by activating calcium-sensitive potassium channels (Rosolowsky and Campbell, 1996; Campbell and Harder, 1999). In humans, CYP2C8 and CYP2C9 have been proposed to be the most likely enzymes responsible for the biosynthesis of endothelial-derived hyperpolarizing factor in the coronary microcirculation (Fisslthaler et al., 1999, 2000; Bauersachs et al., 2002). Moreover, 11,12-EET has been shown to improve recovery of heart contractile function after ischemiareperfusion injury (Wu et al., 1997). EETs can also activate ATP-sensitive potassium channels in rat cardiac myocytes and are important endogenous regulators of cardiac L-type calcium channels (Xiao et al., 1998; Lu et al., 2001, 2002). Thus, the presence of CYP2C50 in mouse heart suggests the possibility that it may play an important role in modulating cardiac function.
Members of several P450 subfamilies (including CYP1A, CYP2C, CYP2D, CYP2E, CYP2J, and CYP3A) have been detected in the human, mouse and rat gastrointestinal tract (de Waziers et al., 1990; Zeldin et al., 1997; Zhang et al., 1999; Tsao et al., 2000). In general, expression is highest in the proximal small intestine and is lowest in the colon (de Waziers et al., 1990; Zhang et al., 1999). In contrast, some P450 enzymes, such as CYP2J2 and CYP3A4, are expressed at comparable levels throughout the entire gastrointestinal tract (de Waziers et al., 1990; Zeldin et al., 1997). We previously reported that CYP2C protein expression was highest in cecum and colon using an antibody that recognizes all of the known mouse CYP2C proteins (Tsao et al., 2000). The present study shows that CYP2C55 protein is expressed at high levels in the colon and is most abundant in the surface epithelium of the cecum. Moreover, recombinant CYP2C55 is active in the metabolism of AA to EETs and 19-HETE. Mouse cecum and colon microsomes catalyze the NADPH-dependent metabolism of AA to EETs and HETEs as the major products (Tsao et al., 2000); however, the P450 enzymes that contribute to EET and HETE biosynthesis in the cecum and colon have not been well characterized. Importantly, the EETs exert potent effects on intestinal blood flow (Proctor et al., 1987) and up-regulate cyclooxygenase-2 expression in intestinal epithelial cells (Peri et al., 1998).
In summary, we identified 15 Cyp2c genes and four Cyp2c pseudogenes by searching the mouse genomic database in the Celera Discovery System. All of the mouse Cyp2c genes are located in an ∼1.5-Mb cluster on chromosome 19, except for Cyp2c44 which is located ∼3.8 Mb downstream. We report the cDNA cloning of three new mouse CYP2C enzymes (CYP2C50, CYP2C54, and CYP2C55) and demonstrate that the recombinant proteins are active in the metabolism of both AA and linoleic acid, albeit with different catalytic efficiencies and profiles. Moreover, our data show each of these new P450s is expressed in both hepatic and extrahepatic tissues. CYP2C50 is abundant in heart, where its EET products have been shown to influence vascular tone, ion transport, and postischemic contractile function. CYP2C55 is abundant in colon, where its EET products have been shown to influence vascular tone and cyclooxygenase expression. Based on this data, we conclude that these new CYP2C enzymes are probably involved in fatty acid metabolism in mouse hepatic and extrahepatic tissues.
Acknowledgments
We thank Dr. Robert Langenbach for helpful suggestions during preparation of this manuscript.
Footnotes
-
This work was supported by the National Institute of Environmental Health Sciences Division of Intramural Research.
-
ABBREVIATIONS: P450, cytochrome P450; EET, epoxyeicosatrienoic acid; HETE, hydroxyeicosatetraenoic acid; AA, arachidonic acid; POR, NADPH-cytochrome P450 oxidoreductase; RT-PCR, reverse transcriptase polymerase chain reaction; HPLC, high-performance liquid chromatography; EOA, epoxyoctadecenoic acid; HODE, hydroxyoctadecadienoic acid; Mb, megabase(s); kb, kilobase(s); RT-PCR, reverse transcription-polymerase chain reaction.
- Received November 21, 2003.
- Accepted February 17, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}