


Abstract
Many cytochrome P450 isoforms are known to be drug-inducible. The anticonvulsant phenytoin has been reported to be an inducer of human CYP2B6, CYP3A4, and murine CYP2C29. However, the molecular mechanism mediating phenytoin induction remains unclear. Herein, we used in vivo and in vitro gene reporter assays of the Cyp2c29 promoter to delineate the phenytoin-response activity to a phenytoin-responsive module located at -1371 kb upstream of the Cyp2c29 translation start site. The phenytoin-responsive module, consisting of two motifs of two imperfect direct repeat hexamers spaced by four nucleotides and a putative CCAAT/enhancer-binding protein-binding site, mediated luciferase reporter induction by phenytoin in mouse livers in vivo and was activated by CAR in HepG2 cells. Hepatic CYP2C29 mRNA was induced by phenytoin in wild-type but not in CAR-null mice, indicating that constitutive active or androstane receptor (CAR) regulates phenytoin-induced transcription of the Cyp2c29 gene. Furthermore, the constitutive levels of CYP2C29 mRNA were reduced ∼77-fold in CAR-null mice compared with those in the wild-type mice, suggesting that CAR may also regulate the constitutive expression of the Cyp2c29 gene either directly or indirectly.
P450s are a superfamily of heme-containing monooxygenases that metabolize a wide range of endogenous and exogenous compounds. Regulation of P450 expression is a major source of variability in the metabolism of xenobiotics and endogenous compounds. Induction of P450 expression can lead to alterations in drug efficacy and create the potential for drug-drug interactions (Honkakoski and Negishi, 2000; Sueyoshi and Negishi, 2001). Drug induction of P450s is often regulated by so-called xenobiotic sensing nuclear receptors such as CAR, pregnane X receptor (PXR), and peroxisome proliferator-activated receptor. Although phenytoin is reported to induce the Cyp2c29 gene (Meyer et al., 2001a,b), the phenytoin response element and the nuclear receptor that regulate the element remain unidentified.
Phenytoin and PB are known to be inducers of several P450 isoforms. PB is the prototypical inducer for a structurally diverse group of chemicals that increase the expression of several isoforms in the CYP2B and CYP2C subfamilies (Honkakoski et al., 1998a; Sueyoshi et al., 1999; Sueyoshi and Negishi, 2001). PB induction of Cyp2b10 is regulated by CAR (Honkakoski et al., 1998b; Honkakoski and Negishi, 2000; Sueyoshi and Negishi, 2001). In mouse hepatocytes or in mouse liver in vivo, CAR is predominately sequestered in the cytoplasm and translocates into the nucleus after treatment with PB (Honkakoski and Negishi, 2000; Sueyoshi and Negishi, 2001; Kobayashi et al., 2003; Yoshinari et al., 2003). In the nucleus, CAR forms heterodimers with the retinoid X receptor (RXR) and binds to enhancer elements such as the phenobarbital-responsive enhancer module (PBREM) in the mouse Cyp2b10 and human CYP2B6 genes (Honkakoski et al., 1998b; Sueyoshi et al., 1999; Honkakoski and Negishi, 2000; Sueyoshi and Negishi, 2001). The nuclear receptor binding motifs within the PBREM are organized into two imperfect direct repeat hexamers spaced by four nucleotides (DR-4) (Honkakoski et al., 1998a, 1998b). Related DR-4 binding motifs, capable of binding CAR, are also found in other genes such as human CYP3A4 and CYP2C9 (Honkakoski and Negishi, 2000; Sueyoshi and Negishi, 2001; Ferguson et al., 2002; Gerbal-Chaloin et al., 2002; Goodwin et al., 2002).
Unlike CAR, PXR requires ligand binding for activation (Kliewer et al., 2002). Prototypical ligands of PXR increase the expression of CYP3A4 (Honkakoski and Negishi, 2000; Sueyoshi and Negishi, 2001; Kast et al., 2002; Luo et al., 2002). Once activated, PXR forms heterodimers with RXR and binds enhancer elements such as the xenobiotic-responsive enhancer module (XREM) within the regulatory region of the human CYP3A4 gene (Honkakoski and Negishi, 2000; Sueyoshi and Negishi, 2001; Liddle and Goodwin, 2002). The XREM is composed of two nuclear receptor-binding motifs spaced by 29 nucleotides, two imperfect direct-repeat hexamers spaced by three nucleotides, and an imperfect everted hexamer repeat spaced by six nucleotides. Although the XREM is considered to be the archetypical PXR binding site, the DNA binding preferences of PXR and CAR are known to be quite similar (Honkakoski and Negishi, 2000; Makinen et al., 2002).
The epileptic drug phenytoin was recently found to induce CYP3A4 and CYP2B6 mRNA in primary human hepatocytes (Luo et al., 2002; Raucy, 2003; Wang et al., 2003). Raucy (2003) showed that phenytoin produced a marked increase in CYP3A4 mRNA, equivalent to that seen with rifampicin. Using a CYP3A4 XREM luciferase promoter construct in HepG2 cells transfected with human PXR, promoter activation by phenytoin was surprisingly much weaker than that produced by rifampicin (Luo et al., 2002; Raucy, 2003).
A recent study suggested that phenytoin and PB induce CYP2C29, the major murine CYP2C, protein in liver microsomes (Meyer et al., 2001a,b). However, this preliminary conclusion was based on Western blot data with a polyclonal antibody to CYP2C29. Knowing the highly homologous nature of murine CYP2C29 to the four other published members of this subfamily (84-85% similar to CYP2C38 and CYP2C39), it is likely that this polyclonal antibody would recognize several CYP2C isoforms (Luo et al., 1998). Because of the recent interest in phenytoin as an inducer of P450 enzymes and the lack of conclusive evidence indicating whether this induction is mediated by CAR or PXR, the murine model provides an excellent system to investigate the molecular mechanism of this induction event. The goals of the present study were 1) to determine whether transcription of the Cyp2c29 gene is inducible by phenytoin and PB, 2) to identify putative nuclear receptor-responsive elements within the upstream promoter region, 3) to examine putative element function using wild-type and mutant Cyp2c29 luciferase promoter constructs, and 4) to determine whether CAR mediates induction of murine Cyp2c29 by phenytoin and PB using CAR-null mice.
Materials and Methods
Materials and Reagents. PB (sodium salt), phenytoin (5,5-diphenylhydantoin sodium salt), and 5α-androstenol were purchased from Sigma-Aldrich (St. Louis, MO). DMSO and other common reagents not listed were also purchased from Sigma-Aldrich or standard sources. 1,4-bis[2-(3,5-Dichloropyridyloxy)]benzene (TCPOBOP) was a kind gift from Dr. James Sideway (AstraZeneca Pharmaceuticals LP, Wilmington, DE). Cell culture media, fetal bovine serum, and trypsin/EDTA were purchased from Invitrogen (Carlsbad, CA). Penicillin and streptomycin were purchased from Sigma-Aldrich. Oligonucleotides were purchased from Genosys, Inc. (The Woodlands, TX) at 50 μM scale and desalted. HepG2 cells were purchased from American Type Culture Collection. (Manassas, VA)
Isolation of Total RNA and Quantitative RT-PCR Analysis. Total RNA was extracted using an ABI 6100 Nucleic Acid PrepStation. All chemicals for the ABI 6100 were purchased from Applied Biosystems (Foster City, CA). Total RNA was isolated and stored at -80°C individually. Before reverse transcription, a portion of each total RNA isolate was pooled within each experimental group. Quantitative RT-PCR analysis was performed in two steps by an initial reaction with murine leukemia virus MuLV reverse transcriptase (Applied Biosystems, Foster City, CA), followed by PCR using 2× SYBR Green master mix. Reverse transcription was performed with 120 ng of total RNA combined with a buffer containing 0.4 μl (8 units) of RNase inhibitor (Applied Biosystems), 1× PCR buffer II, 5.5 mM MgCl2, 0.5 mM each dATP, dCTP, dTTP, and dGTP, 2.5 μM random hexamers, and 0.5 μl (25 units) of murine leukemia virus reverse transcriptase in a final volume of 20 μl. Reverse transcription reactions were incubated using a PCR System 9700 Thermocycler (Applied Biosystems) using the following cycling parameters: 25°C for 10 min, 42°C for 60 min, 95°C for 5 min (inactivation), and 4°C hold. Quantitative PCR was performed on an ABI Prism 7900HT Sequence Detection System (Applied Biosystems). PCR reaction consisted of 2 μl of cDNA template, 1× SYBR Green buffer master mix, and 5 pmol of each primer in a final volume of 20 μl. Sequences of specific primers for CYP2B10, CYP2C29, and β-actin are shown in Table 1. Quantitative PCR parameters for cycling were as follows: 50°C for 2 min hold, 95°C for 10 min hold, denaturation at 94°C for 30 s for 42 cycles, annealing at 60°C (CYP2B10 and β-actin primers) or at 55°C (CYP2C29 primers) for 30 s for 42 cycles, and 72°C extension for 30 s for 42 cycles. PCR products were determined to be single products for each gene-specific primer set using 3% agarose gels stained with ethidium bromide and dissociation curve analyses followed by dye terminator DNA sequencing (Applied Biosystems). Standard curves (log of template dilution versus cycle threshold value) for each gene specific primer set were used to determine relative mRNA content for each target gene.
Oligonucleotides
Plasmids and Cloning of Cyp2c29 Promoter Region. pGL3-Basic vector was purchased from Promega (Madison, WI). Author M.N. provided nuclear receptor expression plasmids pCR3.0-mCAR and pSG5-hPXR. PCR was performed to isolate the Cyp2c29 -1.5 kb upstream promoter region using mouse genomic DNA as the template (Promega). Primers 52F and 53R (Table 1) were used to amplify the Cyp2c29 -1.5 kb promoter region. Primer 52F introduced a XhoI restriction site 5′ and primer 53R introduced an NcoI site 3′. The isolated amplicon was subcloned into pCRTopoTA vector (Invitrogen). After sequence verification with genomic sequence data, the XhoI and NcoI sites were used to remove the Cyp2c29 -1.5 kb promoter region and insert it into the pGL3-Basic vector. Mutant Cyp2c29 1.5 kb promoter constructs were produced by site-directed mutagenesis using the QuikChange system (Stratagene, La Jolla, CA). Mutants were sequenced to ensure that only the intended nucleotides were changed. Subsequently, the 1.5-kb promoter region was removed and inserted into fresh pGL3Basic vector at XhoI and NcoI sites. Mutant oligonucleotides used in mutagenesis reactions are shown in Table 2 (mutated nucleotides are underlined). Dye terminator DNA sequencing (Applied Biosystems) was used to verify all sequences.
EMSA and mutagenesis oligonucleotides Mutated nucleotides are underlined.
Culture and Transfection of HepG2 Cells. HepG2 cells were cultured in Eagle's minimal essential medium with 30 mg/l penicillin and 50 mg/l streptomycin and supplemented with 10% fetal bovine serum. Transfections were performed with Effectene transfection reagent (QIAGEN, Valencia, CA) using the manufacturer's recommended procedures. Cells were plated in 24-well plates and transfected for 12 to 18 h before drug treatment.
Transcriptional Activation Assays. HepG2 cells were maintained in Eagle's minimal essential medium with 10% fetal bovine serum. Cells were passaged at a density of approximately 100,000 cells/well into 24 well plates. Transfections typically included 100 ng of receptor, 100 ng of reporter, and 1 ng of pRL-tk transfection control. Twelve to 18 h after transfection, cells were washed per manufacturer's recommendations, treated with drugs or vehicle and incubated for 24 h. Cells were subsequently lysed with 100 μl of passive lysis buffer (Promega) for 30 min at room temp with gentle rocking, and dual luciferase assays (Promega) were then performed on cell lysates per the manufacturer's procedures.
Gel Mobility Shift Assays. Electrophoretic mobility shift assays were performed by methods analogous to the procedure described by Ferguson et al. (2002). In brief, human RXR, mouse CAR, and human PXR were synthesized in vitro using the TnT quick-coupled in vitro transcription/translation system (Promega) following the manufacturer's protocol. Oligonucleotides (Table 2) were labeled with [γ-32P]dCTP, and the probe was purified by Microspin G-25 columns (Amersham Biosciences, Piscataway, NJ). A volume of labeled probe (∼35,000 cpm per reaction) was applied to each binding reaction in 10 mM HEPES, pH 7.6, 0.5 mM dithiothreitol, 15% glycerol, 0.05% Nonidet P-40, 50 mM NaCl, 2 μg of poly(dI-dC), and 1 μl of in vitro transcribed/translated proteins in a final volume of 10 μl. The reactions were incubated at room temperature for 20 min after addition of probe, then loaded on a 5% acrylamide gel in Tris/acetate/EDTA buffer, dried, and exposed to film for 6 to 18 h at -70°C.
Animals. Male C57BL/6NCrlBR (C57BL/6) and C3H/HeNCrlBR (C3H) mice were purchased from Charles River Laboratory (Wilmington, MA). A CAR-null mouse (Ueda et al., 2002) was first cross-bred with C3H to generate CAR heterozygous offspring. Later, CAR heterozygous offspring were repeatedly backcrossed with C3H mice until the genetic background became more than 95% C3H. The obtained heterozygous mice were bred to produce the wild-type and CAR-null C3H mice. Mice were fed with a standard solid diet and tap water ad libitum for 5 days. Animals received corn oil (vehicle), PB (80 mg/kg), or phenytoin (80 mg/kg) via oral gavage at a volume of 10 ml/kg for 4 days. Animals were sacrificed on the fifth day, and the livers were removed for total RNA isolation or luciferase activity quantitation. The NIEHS committee for the humane care and treatment of research animals approved all animal procedures.
Tail Vein Injections. Mice were injected using the TransIT in vivo gene delivery system (Mirus Corp., Madison, WI). Injections consisted of 6 μg of the Cyp2c29 promoter reporter along with 4 μg of pRL-tk transfection control reporter and Mirus proprietary polymers. Injections were performed following the manufacturer's suggested procedure on the fourth day of drug treatment. Animals were sacrificed on the next day, and the livers were removed for quantitation of luciferase activity.
Western Blot Analysis. Mice were treated intraperitoneally with vehicle (DMSO, 10 ml/kg), PB (80 mg/kg), or phenytoin (80 mg/kg). Livers were removed 3 h after treatment and nuclear extracts were prepared as described previously (Sueyoshi et al., 1995). Immunoblotting of the nuclear extract was performed as described previously (Honkakoski et al., 1998b) using a polyclonal antibody for mCAR.
Results
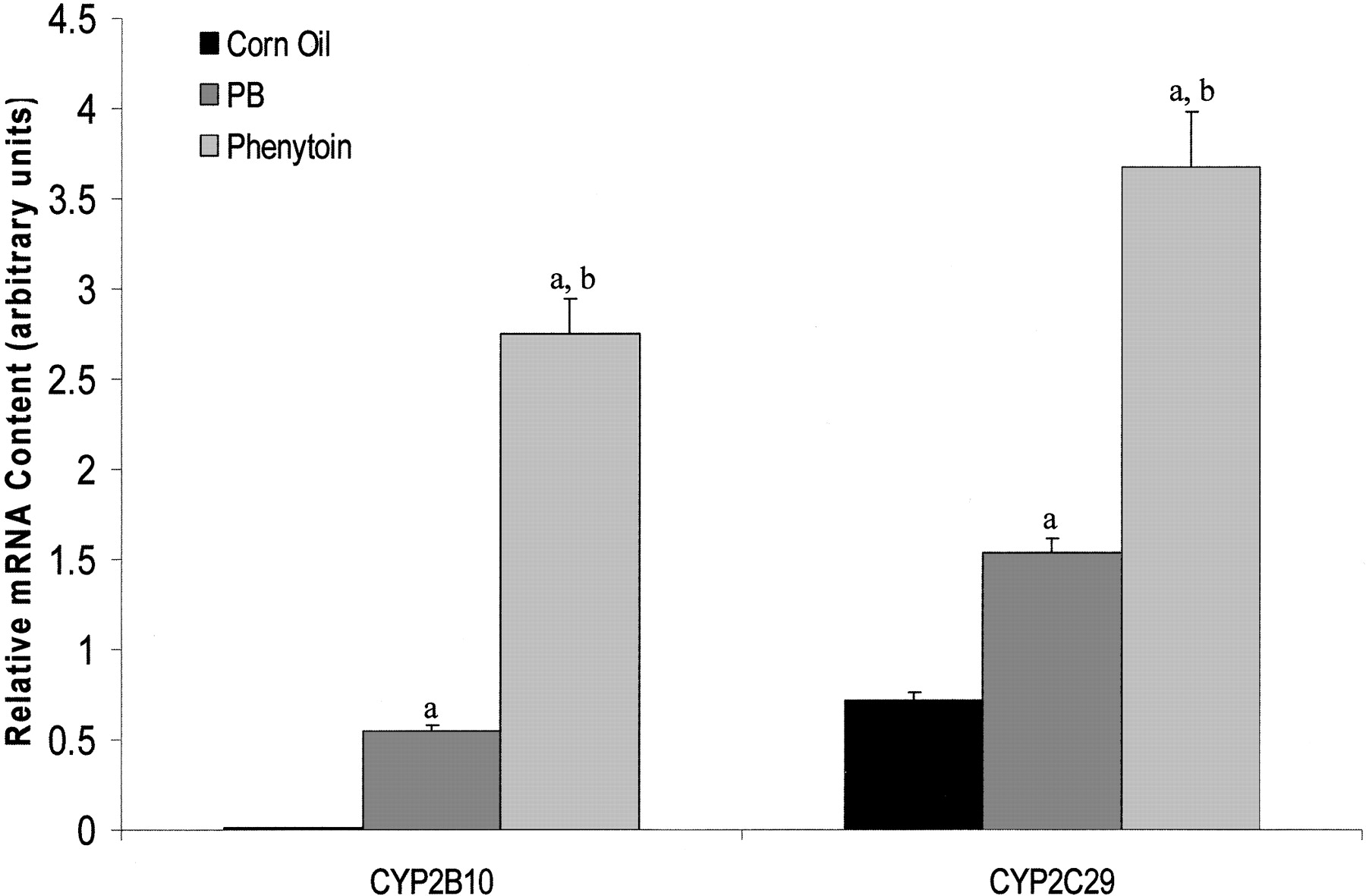
Drug Response of CYP2C29 mRNA. In initial experiments, quantitative RT-PCR was used to determine the induction response of CYP2C29 mRNA. Phenytoin and PB increased CYP2C29 mRNA by ∼5- and ∼2.5-fold, respectively, in C57BL/6 mice (Fig. 1). CYP2B10 mRNA, a positive control, was increased ∼25-fold by PB. An ∼122-fold induction of CYP2B10 mRNA was observed in response to phenytoin. Thus, CYP2C29 and CYP2B10 mRNAs were induced by phenytoin to a greater extent than PB at the concentrations tested.

Drug response of hepatic CYP2C29 and CYP2B10 mRNA in C57BL/6 mice. Mice were treated with phenytoin (80 mg/kg), PB (80 mg/kg), or corn oil (10 ml/kg) via oral gavage for 4 days. Mice were sacrificed 24 h after the fourth dose and livers were removed for total RNA isolation. Quantitative RT-PCR was performed to determine the CYP2C29 and CYP2B10 mRNA content. Target gene values are normalized to β-Actin values. CYP2B10 mRNA was increased ∼122- and ∼34-fold by phenytoin and PB, respectively. Likewise, CYP2C29 mRNA content increased ∼5.2-fold with phenytoin and ∼2.2-fold with PB. RT-PCR products were analyzed by gel electrophoresis, dissociation curve analyses, and autosequencing to verify gene product specificity. Values expressed above represent relative amounts of gene specific mRNA ± S.E. a, Significantly greater than corn oil treated mice, p < 0.05. b, significantly greater than PB treated mice, p < 0.05.
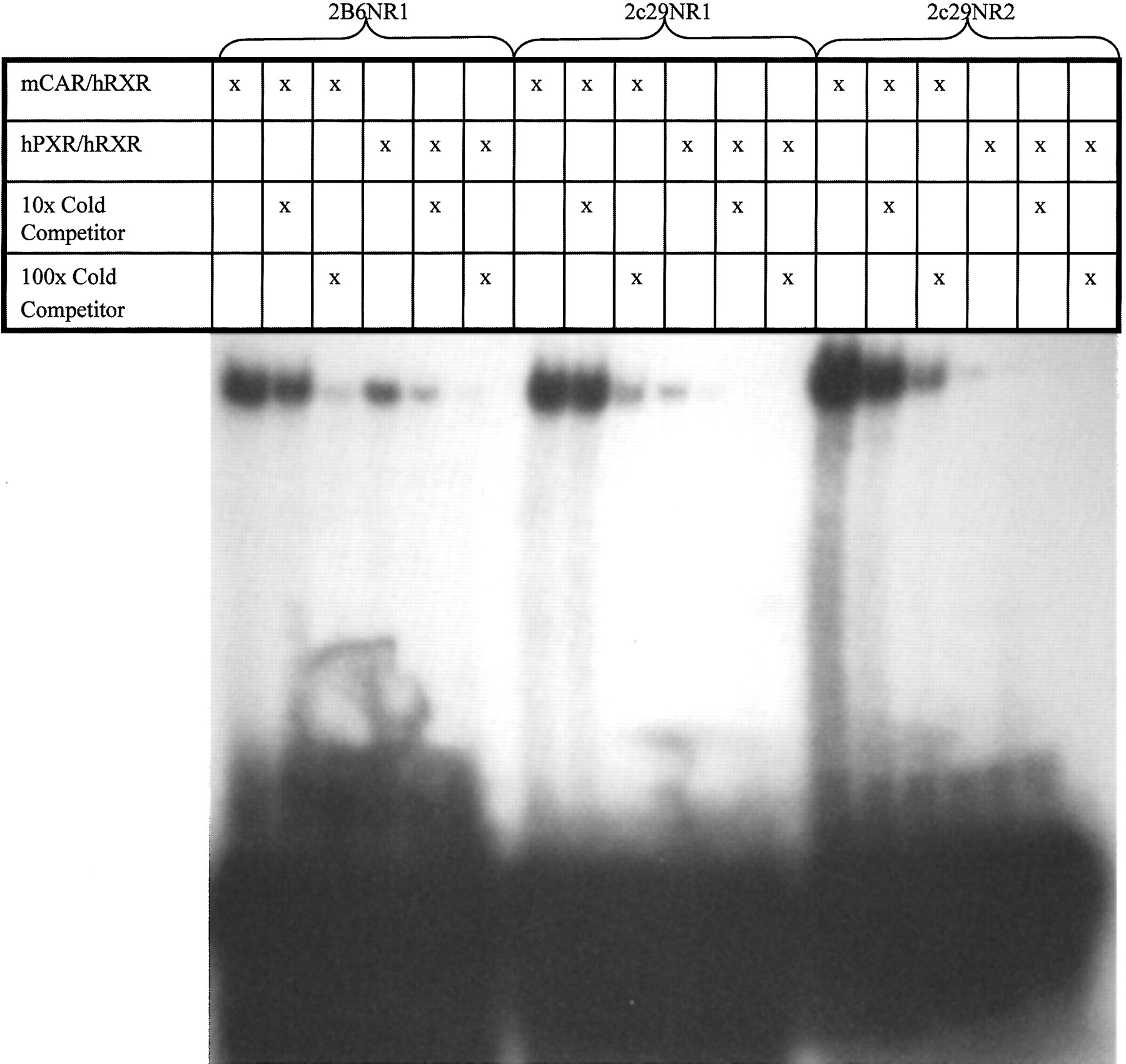
Element Identification and Binding Analysis. A software-based analysis (SeqLab, GCG; Accelrys, San Diego, CA) of 10 kb of the 5′-flanking region of Cyp2c29 revealed two DR-4 imperfect repeats of AGGTCA located at -1371 bp upstream of the translation start site which we herein designate as a phenytoin-responsive module (PHREM) (Fig. 2). These motifs resemble known CAR-binding sites within the enhancer modules of the Cyp2b10 and CYP2B6 genes (Honkakoski et al., 1998a; Sueyoshi et al., 1999; Sueyoshi and Negishi, 2001). In addition, a putative c/EBP site was discovered separating the DR-4 motifs of Cyp2c29 in contrast to the NF-1 site found in Cyp2b10 (Fig. 2) (Honkakoski et al., 1998b). Using electrophoretic mobility shift assays, both putative DR-4 motifs were examined for their ability to bind nuclear receptors CAR and PXR. Both NR1 and NR2 sites bound to mCAR in the presence of RXR, which was competed out with 100× unlabeled oligonucleotides (Fig. 3). Human PXR bound weakly to both DR-4 sites of the Cyp2c29 gene relative to the Cyp2B6 NR1 control.

Module topology comparison. A schematic comparing the topology of the PHREM in the upstream sequence of Cyp2c29 compared with the PBREMs identified in the upstream sequences of murine Cyp2b10 and human CYP2B6. Sequences of each DR-4 element are shown in bold for all three genes, arrows denote the orientation of elements, and NF-1 and c/EBP sites are underlined.

Analysis of NR1 and NR2 binding to CAR and PXR by electrophoretic mobility shift assay. Radiolabeled NR1 and NR2 (35,000 cpm 32P) oligonucleotides were incubated with in vitro transcribed and translated mCAR/RXR or hPXR/RXR proteins. In parallel experiments, incubation was performed in the presence of 10- and 100-fold molar excess of unlabeled NR1 or NR2 oligonucleotides.
In Vitro PHREM Analysis. To evaluate the functional significance of each CAR-binding element in the regulation of the Cyp2c29 gene, we constructed various luciferase reporter plasmids containing 5′ flanking sequence (-1.5 kb) of Cyp2c29 and transfected them into HepG2 cells. The DR-4 sites, NR1 and NR2, were mutated individually and in combination to evaluate their contribution to the function of the module (Fig. 4A). The Cyp2c29 wild-type luciferase construct was activated ∼75-fold by mCAR in the absence of exogenous ligand. Mutation of the NR1 or NR2 binding elements significantly reduced mCAR activation in a nonadditive manner to ∼23% and ∼20% of the wild-type response, respectively (Fig. 4B). The Cyp2c29 double mutant, NR1/2mut, further reduced mCAR activation to ∼12% of the wild-type response. Mutations within the putative c/EBP binding site reduced mCAR activation by ∼50%.

Functional analysis of the proximal DR-4 elements in the 5′-flanking region of Cyp2c29. A, wild-type and mutant PHREM sequences in -1.5 kb luciferase reporter constructs. Mutated nucleotides are underlined. B, transfection experiments of the Cyp2c29 promoter constructs to evaluate the effects of mCAR and its ligands on gene reporter activity. Luciferase reporters containing wild-type and mutant PHREM of the Cyp2c29 gene were transfected in HepG2 cells with mCAR expression vector (pCR3.0-mCAR) and pRL-TK for internal transfection control. Modulation of the constitutive activity of mCAR was accomplished by treatment with androstenol (10 μM) or TCPOBOP (250 nM)/androstenol (10 μM) for 24 h. Reporter activation is expressed as a ratio of normalized luciferase activity in the presence of mCAR to the activity in the absence of mCAR ± S.E. a, cotransfection of mCAR significantly increased activity of the Cyp2c29 Wt promoter in cells treated with DMSO or TCPOBOP, p < 0.05. b, activity of the Cyp2c29 mutant constructs was significantly lower than that of Cyp2c29 Wt constructs in cells cotransfected with mCAR and treated with DMSO, or TCPOBOP, p < 0.05. c, androstenol significantly repressed activity of the Cyp2c29 Wt promoter construct in cells cotransfected with mCAR, p < 0.05. d, TCPOBOP derepressed activity of the Cyp2c29 Wt promoter in cells cotransfected with mCAR and treated with androstenol, p < 0.05. e, activity of Cyp2c29 mutant constructs was significantly lower than that of Cyp2c29 Wt in cells cotransfected with mCAR and treated with androstenol and TCPOBOP, p < 0.05.
Known ligands of mCAR, androstenol, and TCPOBOP (Forman et al., 1998; Tzameli et al., 2000), were used to further investigate the ability of mCAR to regulate Cyp2c29 transcription. Addition of 10 μM androstenol repressed transcriptional activation of the wild-type Cyp2c29 reporter. This response was derepressed by the addition of 250 nM TCPOBOP (Fig. 4B). NR1 mutant and NR2 mutant transcriptional activation was marginally repressed by androstenol, but was not consistently derepressed by the addition of TCPOBOP. The mutations of both elements resulted in the complete loss of mCAR ligand responsiveness. Although mutation of the c/EBP site decreased constitutive activation by mCAR, repression by androstenol and derepression by TCPOBOP were similar to that of the wild-type response (Fig. 4B). In contrast to TCPOBOP, neither phenytoin (100 μM) nor PB (1 mM) was able to reverse androstenol repression of mCAR constitutive activity on the wild-type luciferase construct (Fig. 5). Nor did phenytoin or phenobarbital have an effect on promoter activity in the presence or absence of mCAR. The NR2 mutation had a slightly greater effect on mCAR activation than the NR1 or NR1/NR2 mutations, consistent with results in Fig. 4.

Phenytoin and PB are unable to reverse androstenol repression of mCAR constitutive activity in HepG2 transient transfections of Cyp2c29 promoter constructs. Luciferase reporters containing wild-type and mutant PHREM of the Cyp2c29 gene were transfected in HepG2 cells with mCAR expression vector (pCR3.0-mCAR) and pRL-TK for internal transfection control. Reporter activation is expressed as a ratio of normalized luciferase activity in the presence of mCAR to the activity in the absence of mCAR ± S.E. a, cotransfection of mCAR significantly increased activity of Cyp2c29 Wt promoter in cells treated with DMSO, PB (1 mM), or phenytoin (100 μM), p < 0.05. b, activity of the Cyp2c29 mutant constructs was significantly lower than that of Cyp2c29 Wt constructs in cells contransfected with mCAR in the presence of DMSO, PB, or phenytoin, p < 0.05. c, androstenol (10 μM) significantly repressed activity of the Cyp2c29 promoter constructs in cells cotransfected with mCAR, p < 0.05, regardless of the presence of PB (1 mM) or phenytoin (100 μM).
Nuclear Translocation of CAR. To determine whether phenytoin could induce nuclear translocation of CAR in a manner similar to that observed after PB exposure, immunoblots were performed on hepatic nuclear extracts isolated from mice treated with corn oil, phenytoin (80 mg/kg), or PB (80 mg/kg) using an antibody to mCAR. Immunoblot analysis showed a marked increase in nuclear CAR content in phenytoin- and PB-treated mice, indicating that both phenytoin and PB initiate nuclear translocation of CAR (Fig. 6).
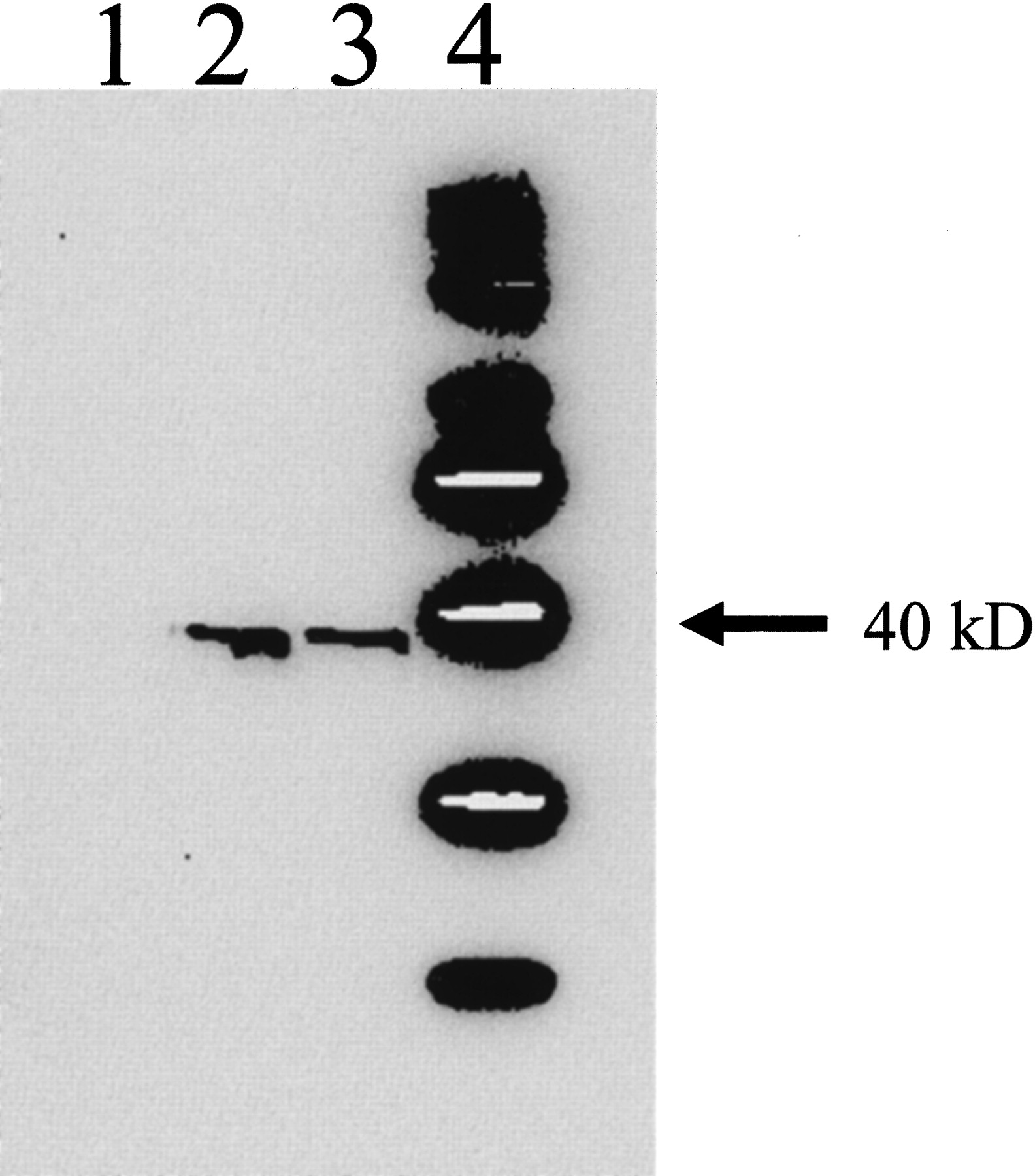
Nuclear translocation of mCAR by phenytoin and PB. Hepatic nuclear extracts were isolated from mice 3 h after treatment with phenytoin (80 mg/kg), PB (80 mg/kg), or corn oil (10 ml/kg) i.p. and subjected to Western blot analysis using an antibody to mCAR. Lane 1, corn oil; lane 2, PB; lane 3, phenytoin.
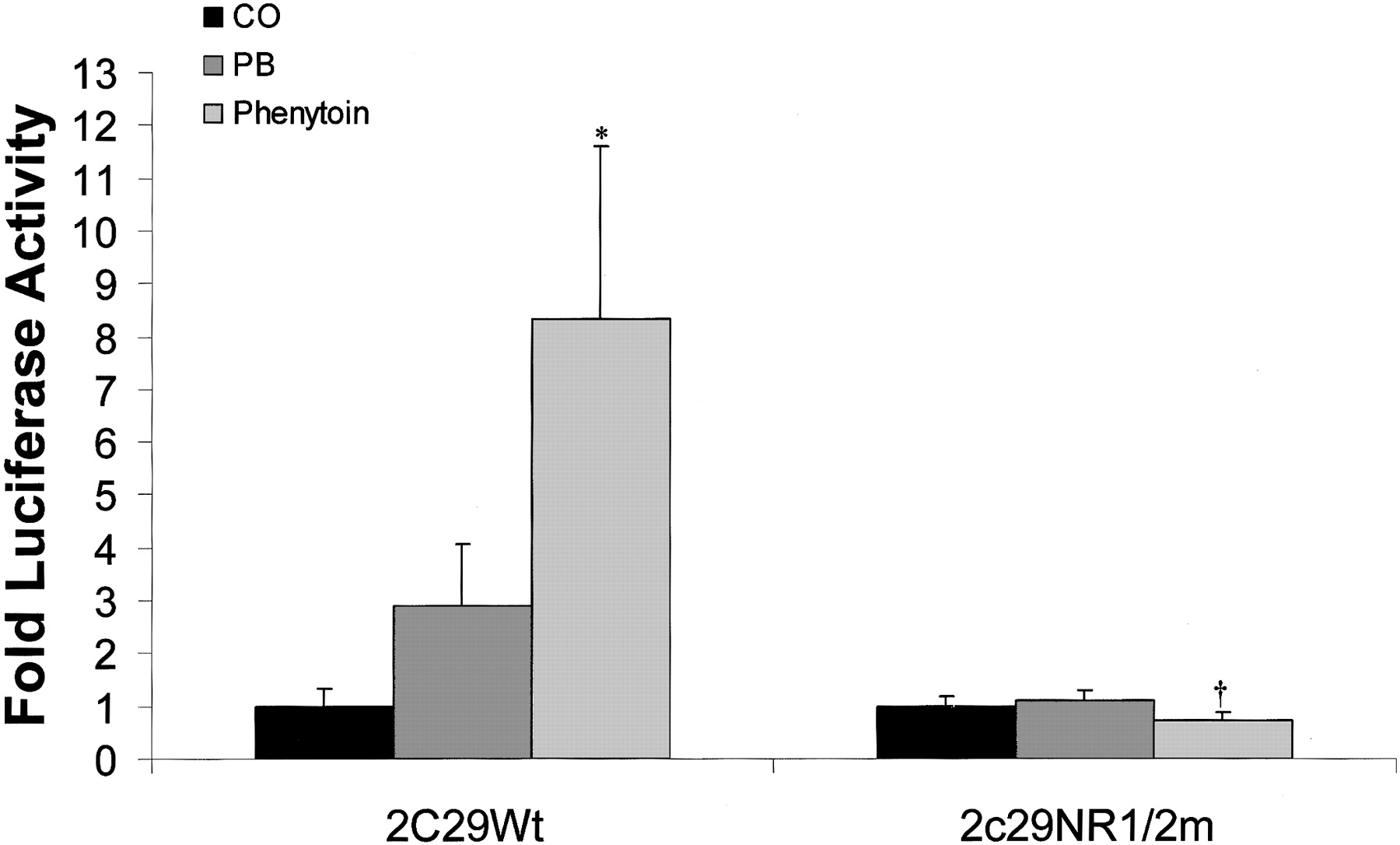
In Vivo PHREM Analysis. When Cyp2c29 -1.5 kb promoter constructs were injected into C3H mice, luciferase activity of the Cyp2c29 wild-type reporter was induced ∼8-fold by phenytoin and ∼3-fold by PB (Fig. 7). Mutation of both DR-4 sites (Fig. 4A) within the Cyp2c29 NR1/2mut promoter construct eliminated induction of luciferase activity (Fig. 7).

In vivo identification of a PHREM in the 5′-flanking region of Cyp2c29. C3H mice were treated with phenytoin (80 mg/kg), PB (80 mg/kg), or corn oil (10 ml/kg) for 4 days. On the fourth day, wild-type and NR1/2m Cyp2c29 luciferase promoter constructs and pRL-TK (transfection control plasmid) were injected into mice via tail veins using a gene delivery system from Mirus, as described under Materials and Methods. Animals were sacrificed 24 h after tail vein injection, and livers were removed for luciferase assay. Induction is expressed as a ratio of normalized luciferase activity in the presence of drug to the activity in the absence of drug ± S.E. *, significantly greater than corn oil-treated mice injected with Cyp2c29 Wt reporter, p < 0.05. †, significantly lower than phenytoin treated mice injected with Cyp2c29 Wt reporter, p < 0.01.
C3H Wild-Type versus CAR-Null. To further investigate the role of mCAR in the induction of Cyp2c29 by phenytoin and PB, wild-type and CAR-null mice were treated with either inducer. Quantitative RT-PCR indicated that CYP2C29 mRNA was elevated ∼6.2-fold in response to phenytoin and ∼2.6-fold in response to PB in wild-type mice (Fig. 8), which is consistent with our previous results. In addition, CYP2B10 mRNA was increased ∼332-fold by phenytoin and ∼60-fold by PB in wild-type mice. In CAR-null mice, CYP2B10 mRNA induction by PB was eliminated, whereas phenytoin induction was ∼99.3% lower than the response observed in wild-type mice. Induction of CYP2C29 mRNA by PB and phenytoin in CAR-null mice was decreased by ∼96 and ∼97% respectively, compared with the response in wild-type mice. The constitutive level of CYP2C29 mRNA was surprisingly dramatically reduced in the CAR-null mice (∼77-fold) (Fig. 8B), whereas constitutive expression of CYP2B10 mRNA was slightly reduced (∼6-fold).

Drug response of hepatic CYP2C29 mRNA in C3H wild-type versus CAR-null mice. A, mice were treated with phenytoin (80 mg/kg), PB (80 mg/kg), or corn oil (10 ml/kg) as described under Materials and Methods. Mice were sacrificed 24 h after the fourth dose and livers were removed for total RNA isolation. Quantitative RT-PCR was performed to determine the CYP2C29 and CYP2B10 mRNA content. Gene-specific data were normalized to β-Actin. Values expressed above represent relative amounts of gene specific mRNA ± S.E. a, significantly greater than corn oil treated wild-type mice, p < 0.01. b, significantly greater than PB treated wild-type mice, p < 0.01. c, drug-treated CAR-null mice significantly lower than respective drug treated wild-type mice, p < 0.01. B, wild-type versus CAR-null constitutive expression of Cyp2c29. CAR-null mice show a substantial reduction (∼77-fold) in Cyp2c29 constitutive expression, indicating that CAR is an important factor in the constitutive regulation of this gene. **, significantly lower than corn oil treated wild-type mice, p < 0.01.
Discussion
In the present study, we used CAR-null mice to show that phenytoin induces CYP2C29 via the nuclear receptor CAR. Furthermore, we identified a PHREM in the upstream region of the Cyp2c29 gene that mediates phenytoin induction. We also showed that CAR is involved in regulating the constitutive expression of the Cyp2c29 gene.
Although phenytoin and PB were both able to induce both Cyp2c29 and Cyp2b10 genes, phenytoin was a stronger inducer of both P450s at the concentrations studied. We identified a novel PHREM located at -1371 bp upstream of the Cyp2c29 gene, similar to the PBREM found within both Cyp2b10 and CYP2B6 genes (Honkakoski et al., 1998a; Sueyoshi et al., 1999). Mutagenesis of both DR-4 elements of the PHREM abolished induction in phenytoin- and PB-treated mice consistent with similar studies of the PBREM removing PB induction of the Cyp2b10 gene (Honkakoski et al., 1998a,b; Sueyoshi et al., 1999). In addition, the results of the in vivo reporter assays were consistent with the magnitude of the mRNA induction, suggesting that the PHREM is the principle CAR-responsive module within the 5′-flanking region of the Cyp2c29 gene. We observed an unusual arrangement of the DR-4 elements within the PHREM; they are inverted in relation to each other. This topological arrangement of DR-4 elements is unlike those found in the PBREM motifs of the Cyp2b10 or CYP2B6 genes (Honkakoski et al., 1998a,b; Sueyoshi et al., 1999) and could be important in regulatory properties such as nuclear receptor specificity. Both elements of the PHREM were capable of binding CAR and, to a much lesser extent, PXR, which is consistent with the ability of CAR and PXR to recognize similar direct hexamer repeats but suggests marked specificity for CAR (Honkakoski and Negishi, 2000; Sueyoshi and Negishi, 2001; Liddle and Goodwin, 2002; Makinen et al., 2002).
Our studies demonstrate that the PHREM of Cyp2c29 was markedly activated by mCAR (∼75-fold) and that this activation could be modulated using mCAR-specific ligands (androstenol and TCPOBOP) (Forman et al., 1998; Tzameli et al., 2000) indicative of CAR regulated genes (Honkakoski and Negishi, 2000; Honkakoski et al., 1998b; Sueyoshi and Negishi, 2001). Mutagenesis of NR1 and NR2 abolished drug responsiveness, indicating that both DR-4 elements are necessary for mCAR-mediated induction. It is interesting that mutagenesis of either DR-4 motif reduced mCAR constitutive activation to ∼18 to 23% of the wild-type response, suggesting that for full activation, the individual DR-4 elements interact synergistically with mCAR. Mutagenesis of the putative c/EBP binding site found in the PHREM resulted in reduced mCAR constitutive activation but did not abolish drug responsiveness, indicating that the c/EBP binding site may be important for the full constitutive activation of the PHREM by mCAR. These results are somewhat analogous to the results of the NF1 mutations within the PBREM of Cyp2b10 (Honkakoski et al., 1998b).
In contrast to TCPOBOP, PB is not believed to bind mCAR (Tzameli et al., 2000; Sueyoshi and Negishi, 2001). Instead, induction is thought to occur via a presently unidentified signaling pathway that induces nuclear translocation of the constitutively active CAR. Neither phenytoin nor PB was able to reverse androstenol repression of mCAR in transiently cotransfected HepG2 cells with Cyp2c29 wild-type reporter. Thus, phenytoin is presumably not a ligand for mCAR. However, immunoblot analysis of hepatic nuclear extracts from control or PB- or phenytoin-treated mice indicated that phenytoin induces nuclear translocation of mCAR in a manner similar to PB (Fig. 6), thereby eliciting the induction response of both Cyp2c29 and Cyp2b10 genes.
Although phenytoin induction is not completely eliminated in CAR-null mice, the induction of Cyp2c29 and Cyp2b10 were reduced to ∼3 and ∼0.7% that of the wild-type response, respectively. The residual induction may possibly be explained by nuclear receptor cross-talk, because CAR and PXR recognize similar binding motifs (Honkakoski and Negishi, 2000; Sueyoshi and Negishi, 2001; Liddle and Goodwin, 2002; Makinen et al., 2002). Therefore, double-null (CAR and PXR) mice would be useful to explore this phenomenon. On the other hand, other non-CAR-mediated mechanisms may account for the residual increase in CAR-null mice.
Although CAR is currently perceived as a receptor that is involved primarily in drug induction (Sueyoshi et al., 1999; Honkakoski and Negishi, 2000; Sueyoshi and Negishi, 2001), recent evidence has suggested that CAR may play a more diverse role in gene regulation. CAR has been shown to be involved in induction, repression, and transcriptional blocking of induction and repression by PB (Ueda et al., 2002). In our study, quantitative RT-PCR in CAR-null mice revealed that constitutive expression of Cyp2c29 was substantially reduced (∼77-fold). These results clearly establish a direct or indirect role for CAR in the constitutive regulation of Cyp2c29 and are consistent with studies showing that CAR is a multifaceted transcription factor (Ueda et al., 2002).
In summary, our studies have demonstrated that phenytoin can induce both Cyp2c29 and Cyp2b10, potentially providing a useful tool for investigating P450 regulation. Using CAR-null mice, we have demonstrated that CAR is responsible for the induction of Cyp2b10 and Cyp2c29 by phenytoin, identifying a new CAR activator. CAR activation was localized to a novel PHREM located at -1371 bp upstream of the Cyp2c29 translation start site. Finally, CAR was shown to be involved in the constitutive expression of Cyp2c29, as suggested by the reduction of the basal CYP2C29 mRNA content in the CAR-null mice, providing more evidence to support the hypothesis that CAR has multiple roles in gene regulation.
Acknowledgments
We thank Kaoru Inoue for expert help in Western blot analysis. We also thank Donna Mays for information on quantitative RT-PCR.
Footnotes
-
ABBREVIATIONS: P450, cytochrome P450; PXR, pregnane X receptor; PB, phenobarbital; RXR, retinoid X receptor; PBREM, phenobarbital-responsive enhancer module; DR-4, two imperfect direct repeat hexamers spaced by four nucleotides; XREM, xenobiotic-responsive enhancer module; DMSO, dimethyl sulfoxide; TCPOBOP, 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene; PCR, polymerase chain reaction; kb, kilobase(s); PHREM, phenytoin-responsive module; c/EBP, CCAAT/enhancer-binding protein; CAR, constitutive active/androstane receptor; RT, reverse transcription; Wt, wild type.
- Received December 1, 2003.
- Accepted February 23, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}