


Abstract
The drug transporter P-glycoprotein (ABCB1) plays an important role in drug distribution and elimination, and when overexpressed it may confer multidrug resistance (MDR). P-glycoprotein is localized in the plasma membrane, especially within rafts and caveolae, characterized as detergent-resistant membranes (DRMs). This study investigated the effect of cholesterol depletion and repletion as well as saturation on subcellular localization and function of P-glycoprotein to determine the effect of DRM localization on P-glycoprotein–mediated drug efflux. In L-MDR1 overexpressing human P-glycoprotein, cholesterol depletion removed P-glycoprotein from the raft membranes into non-DRM fractions, whereas repletion fully reconstituted raft localization. P-glycoprotein function was assessed by realtime monitoring with confocal laser scanning microscopy using BODIPY-verapamil as substrate. Cholesterol depletion reduced P-glycoprotein function in L-MDR1 cells resulting in intracellular substrate accumulation (159% ± 43, p < 0.001; control = 100%). Cholesterol repletion reduced intracellular substrate fluorescence (120% ± 36, p < 0.001) and restored the transporter activity. Addition of surplus cholesterol (saturation) even enhanced drug efflux in L-MDR1 cells, leading to reduced intracellular accumulation of BODIPY-verapamil (69% ± 10, p < 0.001). Transport of BODIPY-verapamil in cells not expressing human P-glycoprotein (LLC-PK1) was not susceptible to cholesterol alterations. These results demonstrate that cholesterol alterations influence P-glycoprotein localization and function, which might contribute to the large interindividual variability of P-glycoprotein activity known from in vivo studies.
P-glycoprotein (ABCB1) is a drug transporter and a member of the ATP-binding cassette transporter superfamily, which confers efflux of its substrates from the cytosol into the extracellular fluid, thereby reducing intracellular substrate concentrations (Schinkel and Jonker, 2003). Expressed in many tissues like liver and kidneys and at the intestinal and the blood-brain barrier, P-glycoprotein may alter the distribution and elimination of its substrates (Schinkel et al., 1994; Kusuhara et al., 1998; van Asperen et al., 1998; Chiou et al., 2000). In case of overexpression, P-glycoprotein may confer multidrug resistance (MDR), leading to failure of drug therapies for cancer (Gottesman et al., 2002) and AIDS (Lee et al., 1998).
In addition to its function as a drug efflux pump, P-glycoprotein acts as a flippase of phospholipids (e.g., phosphatidylserine), which moves within the bilayer of the plasma membrane and therefore contributes to the asymmetric lipid distribution in the outer and inner leaflet of the phospholipid bilayer (Romsicki and Sharom, 2001; Pohl et al., 2002; van Meer, 2002).
The interactions between P-glycoprotein and the plasma membrane are complex and do not only affect the transporter's role as a flippase. The plasma membrane harbors P-glycoprotein, and its hydrophobic part provides the environment in which the drug transporter may interact with its substrates and exports them in an ATP-dependent process across the membrane (Romsicki and Sharom, 1999; Gottesman et al., 2002). P-glycoprotein is located in membrane microdomains, which are detergent-resistant (detergent-resistant membranes, DRMs), and are up-regulated in MDR (Lavie et al., 1998; Liscovitch and Lavie, 2000). Moreover, it has been suggested that microdomains themselves might be important for the development of MDR in tumor cells (Liscovitch and Lavie, 2000).
DRMs are constituted of lipid rafts and caveolae, which are laterally separated into cholesterol- and sphingolipid-rich domains of the plasma membrane (Simons and Toomre, 2000; Razani et al., 2002; van Meer, 2002). These microdomains are characterized physicochemically by a relative rigidity and reduced fluidity compared with the surrounding plasma membrane, which is in part caused by their cholesterol content (London et al., 2000). On the basis of their lipid composition, lipid rafts and caveolae may “trap” proteins, which harbor special lipid moieties or have a special amino acid sequence in the transmembrane segments of the protein and thereby confer a higher affinity to membrane microdomains than to the surrounding plasma membrane (Sprong et al., 2001; Fastenberg et al., 2003). Lipid rafts are highly dynamic and may rapidly assemble and disassemble, leading to a dynamic segregation of proteins (Simons and Toomre, 2000; Fastenberg et al., 2003).
Raft localization is shown to modulate a variety of proteins, such as receptor activities and therefore signal transduction (Gimpl et al., 1997; Pike and Casey, 2002). Cholesterol alterations, which affect raft structure, also affect receptor function (Gimpl et al., 1997) as well as the ATPase activity of P-glycoprotein (Rothnie et al., 2001; Garrigues et al., 2002). Ghetie and coworkers (2004) have shown recently that rafts comprise the compartment in which P-glycoprotein exists in a more active state than when localized outside the rafts.
Membrane fluidization has been shown to impair P-glycoprotein function when assessed with daunomycin and vinblastine accumulation in rat canalicular membrane vesicles (Sinicrope et al., 1992) or with an ATPase assay in vesicles of P-glycoprotein overexpressing Chinese hamster ovary cells (Regev et al., 1999). Moreover, it has been demonstrated that cholesterol depletion of the plasma membrane inhibits transport of the P-glycoprotein substrate rhodamine123 in Caco-2 cells (Arima et al., 2001) and acute depletion of cholesterol impacts P-glycoprotein mediated drug transport in a substrate- and cell-type–specific manner (Luker et al., 2000).
Our study investigated the effect of cholesterol modification on the subcellular localization of P-glycoprotein in rafts and caveolae and on P-glycoprotein activity using its specific substrate BODIPY-verapamil in a cell model overexpressing human P-glycoprotein (L-MDR1). We were able to characterize the time response of the effects of cholesterol depletion, repletion, and saturation on P-glycoprotein activity using real-time confocal laser scanning microscopy and to demonstrate a fast and saturable response of P-glycoprotein function upon modulation of cellular cholesterol concentrations.
Materials and Methods
Materials. Medium, medium supplements, and Hanks' balanced salt solution were purchased from Invitrogen (Karlsruhe, Germany), fetal calf serum was from Biochrom AG (Berlin, Germany), and rhodamine123 and vincristine were from Calbiochem (Bad Soden, Germany). The ECL Plus kit was from Amersham Biosciences (Freiburg, Germany). Aprotinin, leupeptin, cholesterol (5-cholesten-3β-ol), methyl-β-cyclodextrin (MβCD), Tween 20, Nonidet P-40, Triton X-100, and the monoclonal P-glycoprotein antibody (clone F4) were purchased from Sigma-Aldrich (Taufkirchen, Germany), and the monoclonal caveolin-1 antibody (clone 2297) was obtained from BD Biosciences (Heidelberg, Germany). NaCl and SDS were from AppliChem (Darmstadt, Germany), Na3VO4 was from Acros Organics (Geel, Belgium), and Na4P2O7 as well as chloroform were from Merck Biosciences (Darmstadt, Germany). The cellulose nitrate membrane for Western blotting was purchased from Schleicher & Schüll (Dassel, Germany). Millipore filters (0.22 μM) were purchased from Millipore Corporation (Bedford, MA), and MES, Tris-HCl, EDTA, and EGTA were from BIOMOL Research Laboratories (Hamburg, Germany). Rotiphorese gel (30% acrylamide and 0.8% bisacrylamide), methanol, and phenylmethylsulfonyl fluoride were purchased from Roth (Karlsruhe, Germany), the DC Protein Assay was from Bio-Rad Laboratories GmbH (Munich, Germany), and the Amplex Red Cholesterol Assay Kit was from Molecular Probes (MoBiTec, Göttingen, Germany). LY335979 was obtained from Eli Lilly & Company (Bad Homburg, Germany), and SDZ-PSC833 was from Novartis (Basel, Switzerland).
MβCD and MβCD Cholesterol Inclusion Complexes. The incubation media for cholesterol depletion (MβCD in HHBSS, 10 mM for the sucrose density gradients and 5 mM for the assay with the confocal laser scanning microscope) and cholesterol inclusion complexes (cholesterol/MβCD) were prepared as described previously (Troost et al., 2004). In brief, 200 μl of cholesterol in CHCl3/CH3OH (1:2 v/v) were added to a final concentration of 2 mM in 10 ml of HHBSS containing 5 mM MβCD under stirring at 80°C. The solution was homogenized by sonication (60 kHz, 3 × 20 s) yielding cholesterol-saturated MβCD (cholesterol inclusion complexes), which was shown to deliver cholesterol into plasma membranes (Klein et al., 1995). The solution was sterile-filtered through a 0.22 μM Millipore filter, maintained at 37°C, and used immediately after preparation.
LLC-PK1 and L-MDR1 Cells. As model for multidrug-resistant cells, we used L-MDR1 cells, a porcine kidney epithelial cell line overexpressing the human MDR1 gene (Schinkel et al., 1996) and the corresponding parental cell line LLC-PK1 (available from American Type Culture Collection, Manassas, VA) with only low levels of porcine pgp1A (Weiss et al., 2003a) as a control. The L-MDR1 cell line was kindly provided by Dr. A. H. Schinkel (The Netherlands Cancer Institute, Amsterdam, the Netherlands). The cells were cultured as described previously (Weiss et al., 2003a). One day before the assay, both cell lines were fed with vincristine-free culture medium.
Confocal Laser Scanning Microscopy. Intracellular accumulation of the P-glycoprotein substrate BODIPY-verapamil (Lelong et al., 1991) during cholesterol depletion, repletion, and saturation was analyzed with a DM IRE 2 TCS SP2 confocal laser scanning microscope from Leica (Bensheim, Germany). For excitation, a 488-nm argon laser line was used, and a 500- to 550-nm band-pass filter was used to detect emission. The suitability of this assay to quantify P-glycoprotein activity has been shown previously (Weiss et al., 2003b).
Confluent monolayers of LLC-PK1 and L-MDR1 cultured on collagen-coated coverslips were preincubated with 1 μM BODIPY-verapamil with or without P-glycoprotein inhibitor (10 μM LY335979 or SDZ-PSC833) in HHBSS for 15 min to reach a steady-state situation in the cells with this P-glycoprotein substrate and the inhibitors (Table 1). Afterward, the coverslip was transferred into a closed miniperfusion chamber (H. Saur, Reutlingen, Germany). Thereafter, preincubation solution was exchanged by the solution for cholesterol depletion (HHBSS containing MβCD) enclosing 1 μM BODIPY-verapamil with or without P-glycoprotein inhibitor (10 μM LY335979 or SDZ-PSC833), and time series in xyt-modus over a period of 15 min were recorded with one picture per minute. Afterward, the depletion solution (MβCD in HHBSS) was substituted by the cholesterol-repletion solution (cholesterol/MβCD in HHBSS) containing 1 μM BODIPY-verapamil with or without P-glycoprotein inhibitor, and another time series over 15 min was recorded as described.
Cholesterol modification procedure Cholesterol alterations were carried out as described under Materials and Methods.
Cholesterol-saturation experiments were carried out accordingly. After preincubation, cholesterol/MβCD inclusion complexes with or without P-glycoprotein inhibitor were added to the cells within the closed miniperfusion chamber, and time series in xy-plane over a period of 15 min were recorded with one picture per minute.
Fluorescence was quantified with the profile function in each series, which measures a region of interest with an area of 30,000 μm2 (approximately 20–30 cells) drawn automatically in the image. The software (Leica Software version 2.5.1104, quantify modus) calculates the statistical mean average. The experiments were performed at least in triplicate on different days.
Sucrose Density Gradients. Isolation of DRMs was carried out as described previously (Song et al., 1996; Lavie et al., 1998). In brief, two dishes (150 mm diameter) of cells with 80% confluence were lysed in homogenization buffer (25 mM MES, pH 6.5, 150 mM NaCl, 1% Triton X-100, 1 mM Na3VO4, 1 mM Na4P2O7, 1 mM phenylmethylsulfonyl fluoride, and aprotinin and leupeptin at a final concentration of 10 μg/ml each) after or without prior cholesterol alteration treatment according to Table 1. The total cell lysate was adjusted to a volume of 2 ml containing 40% sucrose. This volume was placed at the bottom of an ultracentrifuge tube, and a discontinuous gradient was formed by adding 4 ml of 30% and 4 ml of 5% sucrose solution. The sample was centrifuged at 190,000g for 18 h at 4°C in an SW41 rotor (Beckman Coulter, Krefeld, Germany). The gradient was fractionized into 12 fractions of 0.9 ml volume, whereby the last fraction (pellet) was resuspended in MES/NaCl buffer in a volume of 0.9 ml. The protein content of the samples was determined according to Lowry et al. (1951), using the commercially available kit DC Protein Assay from Bio-Rad.
Detection of P-Glycoprotein by Immunoblotting. Expression of P-glycoprotein in L-MDR1 was verified by Western blot analysis according to methods used by Lavie and coworkers (1998). Fractions of the sucrose density gradients were further separated by SDS-polyacrylamide gel electrophoresis. Equal amounts of proteins per corresponding fractions were transferred to nitrocellulose membranes and blocked by incubation for 1 h with 5% skim milk (w/v) in phosphate-buffered saline containing 0.1% Tween 20. Immunoblot analysis was carried out with a monoclonal antibody raised against P-glycoprotein (clone F4) or caveolin-1 (clone 2297) used in a dilution of 1:1000 in the blocking buffer. The blots were then washed extensively and incubated with horseradish peroxidase-linked secondary antibody. Bands were visualized by enhanced chemiluminescence using the ECL plus kit (Amersham Biosciences).
Fluorimetric Cholesterol Determination. For cholesterol determination, L-MDR1 and LLC-PK1 cells were seeded in six-well microtiter plates and cultivated for 2 days until reaching 80% confluence. After cholesterol alterations (15 min; 5 mM MβCD for depletion cholesterol, 5 mM MβCD for repletion and saturation), cells were washed once with phosphate-buffered saline. The total cholesterol of the cells was measured as cholesterol per well determined after lysis of the cells using a commercial kit derived from an oxidase/peroxidase assay (Amplex Red Cholesterol Assay Kit; Molecular Probes).
Statistical Analysis. Data are presented as means ± S.D. and were analyzed using GraphPad Prism version 4.0 (GraphPad Software Inc., San Diego, CA). Statistical significance was assessed using analysis of variance with Bonferroni's multiple comparison test for post hoc pairwise comparison of the results. A p value <0.05 was considered significant.
Results
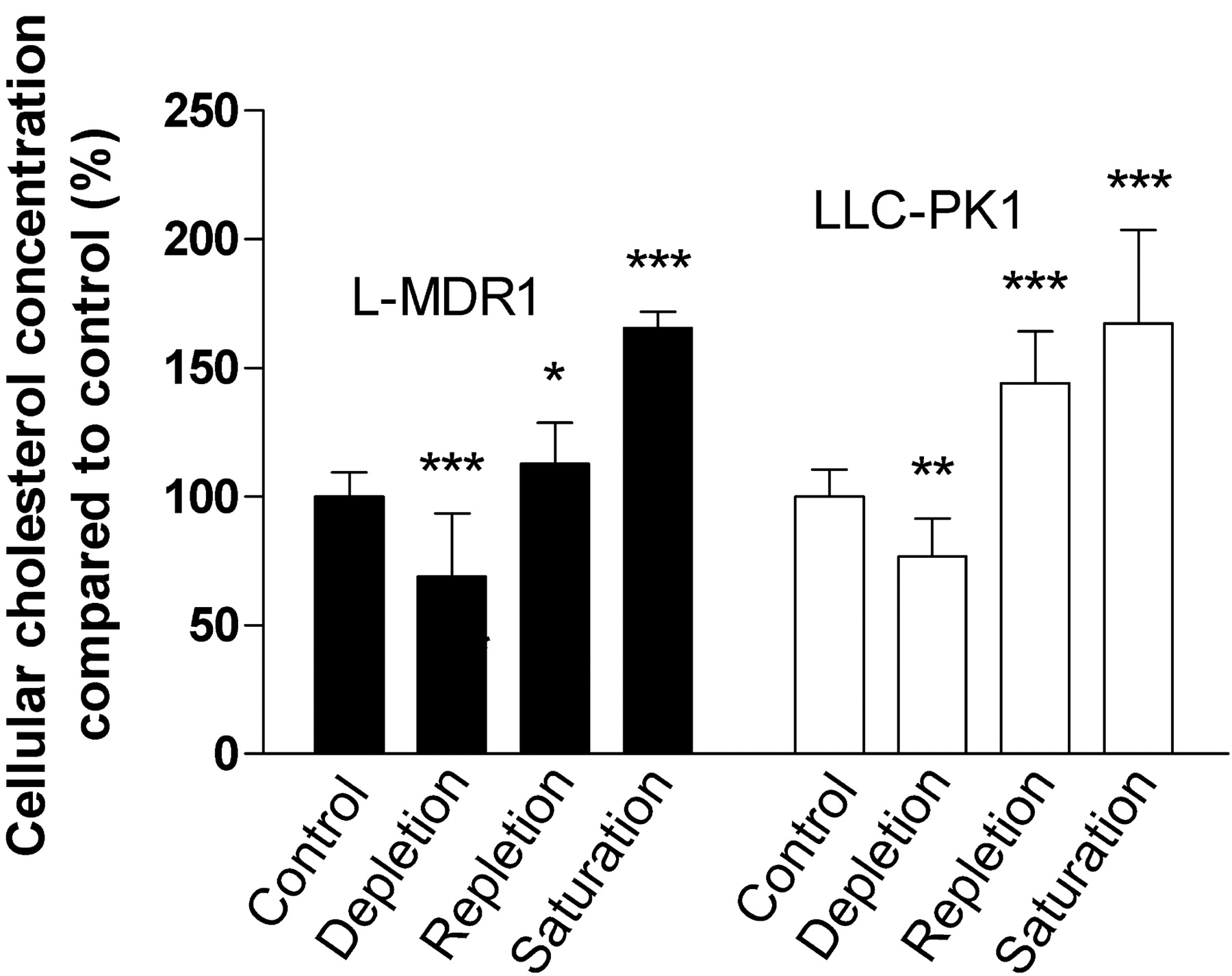
Effects of Cholesterol Alterations on the Intracellular Cholesterol Concentration of LLC-PK1 and L-MDR1 Cells. The effect of cholesterol alterations according to Table 1 on cellular cholesterol concentrations was measured in control cells (LLC-PK1) and P-glycoprotein–overexpressing L-MDR1 cells (Fig. 1). Cholesterol depletion lowered total cellular cholesterol by 31% (p < 0.001, n = 21) in L-MDR1 cells and by 23% in LLC-PK1 cells (p < 0.01, n = 21). Cholesterol repletion in L-MDR1 cells resulted in intracellular cholesterol concentration 12% higher than in the untreated control in L-MDR1 cells (p < 0.05, n = 21) and 44% higher in LLC-PK1 cells (p < 0.001, n = 21). Cholesterol saturation of L-MDR1 and LLC-PK1 cells led to an increase of intracellular cholesterol concentrations by 65% (p < 0.001, n = 12) and 67% (p < 0.001, n = 12), respectively, compared with the corresponding native cells.

Effect of cholesterol depletion, repletion, and saturation on cholesterol concentrations in L-MDR1 and LLC-PK1 cells. Values represent means ± S.D. (n = 21 for control, depletion, and repletion, and n = 12 for saturation). p Values were determined by analysis of variance with Bonferroni's multiple comparison test for post hoc pairwise comparison of the results. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Analysis of Caveolin-1 and P-Glycoprotein Expression in L-MDR1 Cells: Impact of Cholesterol Alteration on P-glycoprotein Localization to DRMs. To verify the localization of the caveolar marker protein caveolin-1 in membrane microdomains, L-MDR1 cells were fractionated. The subcellular fractions along sucrose density gradients demonstrated that a distinct proportion of caveolin-1 was segregated in the 5 to 30% sucrose interface of the gradient (fraction 5), proving the colocalization of this protein in DRMs (Fig. 2A). Upon cholesterol depletion, caveolin-1 immunoreactivity is no longer detectable in the DRM fraction 5 but reappears after repletion, whereas in detergent-soluble fractions 10 and 11, it disappears after repletion.

Western blot analysis of discontinuous sucrose gradients for caveolin-1 (A) and P-glycoprotein (B) in L-MDR1 cells. Gradients for cholesterol depletion and repletion were prepared in parallel and equal amounts of proteins are represented in the fractions. Fractions 4 to 6 are considered to be DRM. Blots depict the results of one of three experiments.
The effect of cholesterol depletion and repletion on P-glycoprotein localization in L-MDR1 cells is shown in Fig. 2B. The localization of P-glycoprotein in the gradient was very similar to the pattern observed with caveolin-1 with distinct localization in DRMs, predominantly in fraction 5 of the sucrose density gradient. Furthermore, significant proportions of P-glycoprotein are localized in the detergent-soluble fractions 9 to 11. Upon cholesterol depletion, P-glycoprotein localization in caveolar fractions is no longer detectable, suggesting that P-glycoprotein shifted into fractions 9 and 10 (containing the detergent-soluble part of the gradient). When cholesterol-depleted L-MDR1 cells were restored by cholesterol/MβCD inclusion complexes, P-glycoprotein was redistributed into fraction 5, whereas the immunoreactivity in fraction 10 and 11 declined but did not disappear like that of caveolin-1. Furthermore, cholesterol repletion led to an additional band of lower molecular weight and unknown identity originating from fraction 10 and 11 (see control and repletion gradients) to the 5 to 30% sucrose interface (DRMs). Moreover, cholesterol led to an increased formation of DRMs, visible as an increased amount of cloudy particles at the 5 to 30% interface after ultracentrifugation. Perhaps, because of this augmentation, faint bands of P-glycoprotein could be detected in fractions 4 and 6 of cholesterol-repletion gradients in contrast to the control. In contrast, neither caveolin-1 nor P-glycoprotein could be detected in LLC-PK1 cells in sucrose density gradients (data not shown).
Functional Analysis of BODIPY-Verapamil Transport in LLC-PK1 and L-MDR1 Cells: Effects of Cholesterol Alterations. Thereafter, the effect of cholesterol alterations on P-glycoprotein activity was assessed. P-glycoprotein function was determined by the efflux of the substrate BODIPY-verapamil, whose intracellular concentration was detected by real-time confocal laser scanning microscopy (Fig. 3, A–C). The basal fluorescence after incubation with BODIPY-verapamil of the P-glycoprotein overexpressing cell line L-MDR1 was approximately 50% of the fluorescence in the control cell line (LLC-PK1), demonstrating the different P-glycoprotein levels. Whereas in LLC-PK1 cells the addition of MβCD (cholesterol depletion) to the cells during the scanning procedure did not affect the intracellular fluorescence after 15 min (Fig. 3A) (100 versus 102% ± 4, n = 3 experiments), in the P-glycoprotein–overexpressing L-MDR1 cells, intracellular fluorescence increased by 59% (Fig. 3A) (100 versus 159% ± 43, n = 14 experiments; p < 0.001).

Functional analysis of P-glycoprotein in LLC-PK1 and L-MDR1 cells during cholesterol depletion (A), repletion (B), and saturation (C) with BODIPY-verapamil (1 μM) as P-glycoprotein substrate. Cholesterol repletion (B) was carried out after prior depletion (A) and fluorescence was determined in the same cells. Cells were cholesterol saturated (C) without prior depletion during an incubation period of 15 min. Individual intracellular fluorescence at the beginning of the incubation was set to 100%. Data are expressed as means ± S.D. For depletion and repletion experiments, n = 14 (L-MDR1) and n = 3 (LLC-PK1), and for saturation, n = 5 (L-MDR1) and n = 3 (LLC-PK1). p Values were determined by analysis of variance with Bonferroni's multiple comparison test for post hoc pairwise comparison of the results. For calculation of the p values for the repletion experiment intracellular fluorescence at the beginning of the repletion (t = 15 min) was used as baseline; *, p < 0.05; ***, p < 0.001.
The observed reduction of transport function concurs with the dislocation of P-glycoprotein from DRMs to detergent-soluble membranes (Fig. 2B). Restoration of cellular cholesterol by the addition of cholesterol/MβCD inclusion complexes to the prior depleted cells for an additional 15 min (15–30 min in the graph did not change the intracellular BODIPY-verapamil fluorescence in LLC-PK1 cells (Fig. 3B) (99 ± 2, t = 30 min, n = 5 experiments) but restored P-glycoprotein function in L-MDR1 cells by significantly decreasing the measured BODIPY-verapamil fluorescence in the cells to 120% of control (Fig. 3B) (120 ± 36; t = 30 min, n = 14 experiments, p < 0.001 compared with 15-min results).
To evaluate the effect of cholesterol saturation without prior depletion, the effect of cholesterol/MβCD inclusion complexes on P-glycoprotein function of native LLC-PK1 and L-MDR1 cells was also assessed (cholesterol saturation) (Fig. 3C). Again, LLC-PK1 cells were not affected by cholesterol alterations over the time course of 15 min (100 versus 99% ± 1, t = 15 min, n = 3 experiments), whereas in L-MDR1 cells, intracellular fluorescence decreased, indicating accelerated efflux of BODIPY-verapamil by P-glycoprotein (100 versus 69% ± 10, t = 15 min, n = 5 experiments; p < 0.001).
To confirm that the observed effects of cholesterol alterations on intracellular substrate accumulation are mediated by P-glycoprotein, intracellular BODIPY-verapamil fluorescence was investigated in the presence of the specific P-glycoprotein inhibitors SDZ-PSC833 and LY335979. Both inhibitors significantly increased BODIPY-verapamil fluorescence in L-MDR1 but not in LLC-PK1 cells (Fig. 4), demonstrating efficient P-glycoprotein inhibition. The addition of either inhibitor in a final concentration of 10 μM abolished the modulatory effect of cholesterol on intracellular fluorescence. Again, the initial fluorescence in the cells was set to 100%, and changes of fluorescence were compared with this value during the addition of SDZPSC833 (Fig. 5, A–C) or LY335979 (Fig. 5, D–F). LLC-PK1 cells revealed no changes upon cholesterol alterations in the presence of SDZ-PSC833 in depleted cells (Fig. 5A) (n = 3 experiments), in repleted cells (Fig. 5B) (n = 3 experiments), and in saturated cells (Fig. 5C) (n = 3 experiments). The inhibition of P-glycoprotein by SDZPSC833 completely blocked the effects of cholesterol changes in L-MDR1 cells in depleted cells (Fig. 5A) (n = 3 experiments), in repleted cells (Fig. 5B) (n = 3 experiments), and in saturated cells (Fig. 5C) (n = 3 experiments). SDZPSC833 offset the effects of cholesterol modulation in L-MDR1 cells, as did LY335979 (Fig. 5, D–F) (n = 3 experiments).
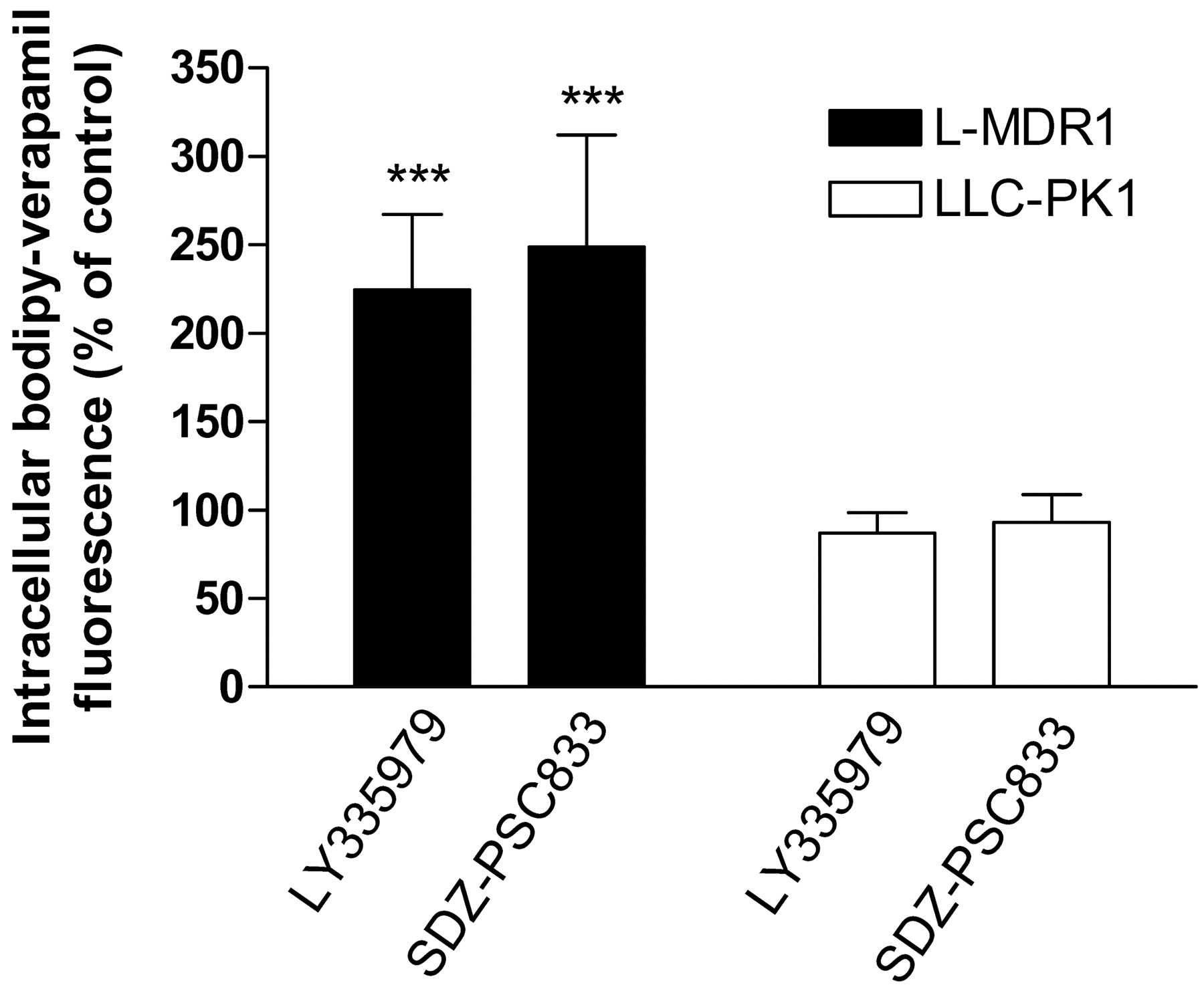
Increase in intracellular BODIPY-verapamil fluorescence in LMDR1 and LLC-PK1 cells by SDZ-PSC833 and LY335979. Data are expressed as means ± S.D. (n = 3). p Values were determined by analysis of variance with Bonferroni's multiple comparison test for post hoc pairwise comparison of the results.

Functional analysis of P-glycoprotein in LLC-PK1 and L-MDR1 cells by cholesterol depletion (A and D), repletion (B and E), and saturation (C and F) in the presence of the P-glycoprotein substrate BODIPY-verapamil (1 μM) and the P-glycoprotein inhibitors SDZ-PSC833 (10 μM; A, B, and C) and LY335979 (10 μM; D, E, and F). Depletion was carried out for 15 min (0–15 min) and repletion for further 15 min (15–30 min). Cholesterol saturation was assessed over 15 min. Intracellular fluorescence at the beginning of the incubation was set to 100% in each cell line. Data are expressed as means of n = 3 experiments ± S.D. p Values were determined by analysis of variance with Bonferroni's multiple comparison test for post hoc pairwise comparison of the results.
Discussion
Membrane microdomains of the plasma membrane are characterized by sphingolipids and cholesterol as the two major lipid constituents and are divided into lipid rafts and caveolae (Brown and London, 2000). The concentration and the ratio of these lipid constituents within microdomains differ between lipid rafts and caveolae on the one hand and the surrounding plasma membrane on the other. The lipid composition of microdomains is associated with a reduced fluidity and resistance against solubilization by some detergents such as Triton X-100 at low temperatures (London and Brown, 2000). These membrane sections are therefore termed DRMs. Altering the cholesterol concentration of the plasma membrane (Smart and Anderson, 2002) may consequently modify raft structure and function as well as the activity of P-glycoprotein, which is localized in membrane microdomains (Lavie et al., 1998; Liscovitch and Lavie, 2000). The effects of cholesterol on P-glycoprotein activity have been assessed in liposomes and membrane vesicles by means of ATPase assays (Rothnie et al., 2001; Garrigues et al., 2002), by rhodamine 123 efflux in Caco-2 cells (Arima et al., 2001; Yunomae et al., 2003), or by [99mTc]sestamibi and [3H]daunomycin in different cell lines (Luker et al., 2000). Moreover, the influence of cholesterol on azidopine binding by P-glycoprotein reconstituted into liposomes has been investigated (Saeki et al., 1992). These studies revealed that P-glycoprotein localizes to rafts and caveolae to an extent of 40% of total detectable P-glycoprotein in MDR cells (Lavie et al., 1998; Demeule et al., 2000; Luker et al., 2000) and that cholesterol may affect P-glycoprotein function and subcellular localization, depending on the cell system used.
We have shown previously that altering total cellular cholesterol modulates P-glycoprotein–mediated transport of rhodamine 123 in human peripheral blood mononuclear blood cells (Troost et al., 2004). The objective of this study was to investigate the underlying mechanisms of the cholesterol-mediated modulation of P-glycoprotein function in a P-glycoprotein overexpressing cell line (L-MDR1) and the corresponding native cell line (LLC-PK1). The study aimed to assess in real time whether and to which degree P-glycoprotein function would be affected by changing cellular cholesterol levels, leading to a shift of a proportion of P-glycoprotein from DRMs to the detergent-soluble membranes. Two approaches were chosen: 1) the effect of cellular cholesterol alterations on subcellular localization of P-glycoprotein was investigated to assess its distribution in DRMs versus detergent-soluble membranes; and 2) functional aspects of cholesterol alterations were tested for the first time using real-time monitoring of P-glycoprotein function by confocal laser scanning microscopy.
Our study demonstrates that in L-MDR1 cells, parts of P-glycoprotein are present in DRMs and supports previous work in different cell lines (Lavie et al., 1998; Demeule et al., 2000; Luker et al., 2000). Furthermore, changes of the cholesterol concentrations in P-glycoprotein overexpressing LMDR1 cells shifted P-glycoprotein from detergent-insoluble membranes to detergent-soluble parts of the membrane and vice versa. This shift of a substantial portion of P-glycoprotein in L-MDR1 cells was accompanied by marked changes in P-glycoprotein function. In L-MDR1 cells, MβCD had a clear and significant effect on P-glycoprotein transport by enhancing the intracellular fluorescent substrate concentration upon cholesterol depletion, which is equivalent to a reduced P-glycoprotein function. This effect was reversible upon cholesterol repletion, and cholesterol saturation enhanced P-glycoprotein–mediated substrate efflux.
In contrast, in LLC-PK1 cells, a cell line with no detectable levels of P-glycoprotein and caveolin-1 in sucrose density gradients, the accumulation of BODIPY-verapamil was not affected by MβCD, although the intracellular cholesterol concentration significantly decreased, like in L-MDR1 cells. In accordance with this finding, the treatment with cholesterol/MβCD inclusion complexes for cholesterol repletion and saturation did not affect the intracellular concentration of the P-glycoprotein substrate in LLC-PK1 cells.
The comparison of LLC-PK1 and L-MDR1 cells demonstrates that changes of cholesterol concentrations in the membrane per se do not cause accumulation or extrusion of BODIPY-verapamil in the cells. Moreover, the effects in LMDR1 cells only occur if P-glycoprotein is not disabled by a specific inhibitor like LY335979. We therefore conclude that the effects of cholesterol are not caused by unspecific changes in permeability and rather suggest that the expression of P-glycoprotein is a prerequisite for changes in transport activity during cholesterol modulation. Hence, the presence of the drug transporter P-glycoprotein is necessary to accomplish the cholesterol-mediated effects. Real-time monitoring of P-glycoprotein function by laser scanning microscopy revealed that the effects of cholesterol modifications in L-MDR1 cells start immediately and reach plateau effects within a few minutes. Cholesterol added to the cells in the form of cholesterol/MβCD inclusion complexes without prior depletion enhanced the substrate efflux, suggesting that a shift of free P-glycoprotein into the DRM fraction occurred (see below). The application of the potent and efficient P-glycoprotein inhibitors SDZ-PSC833 and LY335979 blocked the modulatory effects of cholesterol completely, further confirming the involvement of P-glycoprotein in the changes of BODIPY-verapamil transport.
In contrast to these results, Pallarés-Trujillo et al. (1993) reported an influence of cholesterol on cellular permeability to the P-glycoprotein substrate vincristine not only in multidrug-resistant but also in sensitive cells, indicating an unspecific modulation of the permeation of vincristine through the plasma membrane by cholesterol. However, the experimental design as well as the investigated substrate and the cell lines were not identical, making a direct comparison impossible. Moreover, the lack of effect in LLC-PK1 cells in our study clearly indicates a specific P-glycoprotein modulatory effect.
The study confirmed that P-glycoprotein function is linked to the cholesterol content of the plasma membrane. Because the shift of P-glycoprotein into the detergent-soluble parts of the gradients is accompanied by reduced P-glycoprotein activity, the DRM compartment may play a supportive role for P-glycoprotein function, possibly mediated by the particular physicochemical properties of the microdomain membrane compartment and its influence on the binding process of the often lipophilic P-glycoprotein substrates (Romsicki and Sharom, 1999). However, it cannot be excluded that cholesterol itself may influence P-glycoprotein activity.
Our study suggests that cholesterol modifications have the potential to inhibit or increase P-glycoprotein function and that this effect evolves rapidly. Whether extracellular cholesterol also controls P-glycoprotein activity in vivo is currently unclear. It is also unknown whether continuous cholesterol changes will result in similar changes or whether counter-regulatory intracellular pathways will be activated that might modulate the effect of cholesterol. Previous studies have already demonstrated that cholesterol affects P-glycoprotein transport in human peripheral blood mononuclear cells ex vivo (Troost et al., 2004), which represent an important drug target in vivo (e.g., in AIDS therapy). The effect of cholesterol on P-glycoprotein activity might be of particular interest because P-glycoprotein plays an important role in the pharmacokinetics of a wide variety of drugs. Reduced activity of P-glycoprotein may cause toxicity of its substrates, and in case of induction or activation, multidrug resistance in cancer or human immunodeficiency virus therapy may occur. Therefore, the effects shown in this study could contribute to our understanding of drug distribution and elimination of P-glycoprotein substrates in the context of cholesterol metabolism, and individual differences in cholesterol concentrations might contribute to the large interindividual variability of P-glycoprotein activity known from in vivo studies.
Acknowledgments
L-MDR1 cells were a kind gift from Dr. A. H. Schinkel (The Netherlands Cancer Institute, Amsterdam, The Netherlands). LY335979 was generously provided by Eli Lilly & Company (Bad Homburg, Germany), and SDZ-PSC833 was provided by Novartis (Basel, Switzerland).
Footnotes
-
This work was supported by grant 01EC9902 from the German Ministry for Education and Research (Bundesministerium für Bildung und Forschung).
-
J.T. and H.L. contributed equally to this work.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.104.002329.
-
ABBREVIATIONS: MDR, multidrug resistance; DRM, detergent-resistant membrane; HHBSS, HEPES-supplemented Hanks' balanced salt solution; LY335979, zosuquidar; MβCD, methyl-β-cyclodextrin; MES, 2-(N-morpholino)ethanesulfonic acid; SDZ-PSC833, valspodar.
- Received May 4, 2004.
- Accepted August 12, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}