


Abstract
We investigated the impact of promoter polymorphisms on transcription of the human CYP2B6 gene. In total, 98 DNA samples from white persons from a previously characterized liver bank were sequenced throughout 2.3 kilobases of upstream sequence and haplotype structures were determined using additional coding sequence information. HepG2 cells and primary rat and human hepatocytes were transfected with luciferase reporter gene constructs driven by 2033 base pairs (bp) of the most frequent promoter variants. The novel haplotype *22 (–1848C→ A, –801G→ T, –750T→ C, and –82T→ C) showed 3- to 9-fold enhanced transcriptional activity in all transfected cells. Constructs containing single mutations surprisingly revealed –82T→ C, predicted to disrupt a putative TATA box, to be alone responsible for this effect. In silico analysis and electrophoretic mobility shift assay demonstrated conversion of the putative TATA box into a functional CCAAT/enhancer-binding protein binding site. Analysis of transcriptional start sites showed the mutant promoter to be transcribed from a start site located approximately 30 bp downstream of the wild-type start site, consistent with the use of a noncanonical TATA box at –55 bp. Median CYP2B6 mRNA expression and bupropion hydroxylase activity as a selective marker of CYP2B6 catalytic activity were approximately 2-fold higher in livers genotyped –82TC as in those genotyped –82TT (20.4 versus 9.8 arbitrary units, p = 0.007, and 201.8 versus 106.7 pmol/mg/min, p = 0.042, respectively). This promoter polymorphism thus contributes to CYP2B6 functional variability and represents a novel mechanism by which mutations can enhance transcription. Furthermore, a detailed interspecies comparison of CYP2B promoters and transcriptional start sites provided novel insights into evolutionary relationships.
The human cytochrome P450 gene superfamily currently consists of 57 functional genes and 58 pseudogenes (Nelson et al., 2004). The members of families CYP1, CYP2, and CYP3 are localized in the endoplasmic reticulum of both liver and extrahepatic tissues where they catalyze a variety of biotransformations of numerous endogenous and exogenous substrates, including many drugs currently in use. The family member CYP2B6 was first described in 1989 (Yamano et al., 1989) as the human ortholog to the phenobarbital-inducible CYP2B genes in rodents. Initially underestimated, the number of drugs recognized as CYP2B6 substrates has been constantly increasing, and several clinically important substances are now known to be preferred substrates of this enzyme. These include the anticancer prodrug cyclophosphamide (Roy et al., 1999); the narcotic propofol (Court et al., 2001); the antidepressant bupropion, which is now the most commonly used probe drug for CYP2B6 (Faucette et al., 2000); the antimalarial drug artemisinin (Svensson and Ashton, 1999); and the reverse transcriptase inhibitor efavirenz (Ward et al., 2003).
In vitro studies revealed a large degree of interindividual variability in hepatic CYP2B6 expression at the mRNA, protein, and catalytic activity level (Code et al., 1997; Lang et al., 2001). Therefore, population studies demonstrated a broad interindividual variability of in vivo pharmacokinetic parameters of several CYP2B6 drug substrates, including cyclophosphamide (Yule et al., 1996), bupropion (Kirchheiner et al., 2003) and efavirenz (Csajka et al., 2003), which could be the cause for nonresponse or toxicity. Studies conducted by several laboratories in recent years have provided evidence that part of the human CYP2B6 variability is caused by its drug-inducible regulation via proximal and distal response elements termed phenobarbital-responsive enhancer module at –1.7 kb (Goodwin et al., 2001) and xenobiotic-responsive enhancer module at –8.5 kb (Wang et al., 2003). Recent work from a number of laboratories, including our own, focused on the genetic variations as an additional source for interindividual variability in expression (Lang et al., 2001, 2004; Lamba et al., 2003; Hesse et al., 2004). CYP2B6 was found to be highly polymorphic within exons and introns as well as in its promoter region. Some of the common nonsynonymous polymorphisms [e.g., those of alleles CYP2B6*5 (R487C) and CYP2B6*6 (Q172H, K262R)], are associated with decreased liver protein expression (Lang et al., 2001; Hesse et al., 2004) or changes in function (Ariyoshi et al., 2001). A recent clinical study with human immunodeficiency patients revealed significantly elevated efavirenz plasma levels in homozygous CYP2B6*6/*6 carriers (Tsuchiya et al., 2004), suggesting that CYP2B6 polymorphisms can have a significant impact on the in vivo pharmacokinetics of certain drugs. In addition, several more rare nonsynonymous SNPs were found to result in absent or nonfunctional proteins (Lang et al., 2004). In contrast, the numerous polymorphisms within the CYP2B6 promoter region that seem to be linked to the coding SNPs in a complex manner (Lamba et al., 2003; Hesse et al., 2004) have not been investigated mechanistically with respect to their potential impact on transcription of the CYP2B6 gene. In addition, because only few studies have so far been carried out to investigate its constitutive regulation, no systematic promoter analysis has been carried out, and the transcriptional start site has not been determined.
The aim of this study was to identify the common promoter haplotypes of the CYP2B6 gene that are present among white persons and to investigate their potential impact on transcription. For this purpose, we generated reporter gene constructs and transfected them into different cell types (hepatoma cells and liver-derived primary hepatocytes). The TATA box polymorphism –82T→ C was found to enhance transcriptional activity in vitro and to be associated with increased hepatic expression in vivo, which prompted us to further investigate the mechanism in detail. The results illustrated a novel and unique mechanism by which a natural SNP leads to enhanced gene transcription. Furthermore, this study contributes to a better understanding of the evolution and constitutive regulation of CYP2B6 gene expression in the liver.
Materials and Methods
DNA and Liver Samples. The human liver samples used for this study were obtained from nontumorous tissue from white persons who underwent liver surgery for various reasons at the Department of General, Visceral, and Transplantation Surgery (Humboldt University, Berlin) (Wolbold et al., 2003). The preparation of genomic DNA from corresponding blood samples and of liver microsomes had been described previously (Lang et al., 2001). Patients with known severe liver diseases (viral hepatitis and cirrhosis) or alcohol abuse as well as two individuals known to be induced by treatment with carbamazepine (Ketter et al., 1995) were excluded from this investigation. The study has been carried out in accordance with the Declaration of Helsinki and approved by the ethics committee of the Medical Faculty of the Charité (Humboldt University), and written informed consent was obtained from each patient.
CYP2B6 Expression and Activity in Human Liver Samples. Total RNA was prepared from liver tissue using the RNeasy midi kit (QIAGEN GmbH, Hilden, Germany). Quantification of β-actin and CYP2B6 mRNA was performed by specific TaqMan real-time reverse transcription-PCR assays on an ABI Prism 7500 system (Applied Biosystems, Foster City, CA). For β-actin, TaqMan predeveloped assay reagents were used. For CYP2B6, the intron-spanning primers TQ-CYP2B6FOR/REV (Table 1) and the 5-carboxyfluorescein-labeled CYP2B6-MGB probe were used at concentrations of 400 nM each, and specificity against CYP2B7P1 was confirmed using DNA plasmids of both genes as template (data not shown). PCR was performed using 2× universal PCR master mix (Applied Biosystems) in a final volume of 25 μl and the following cycling conditions: 50°C for 2 min, 95°C for 10 min followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. CYP2B6 mRNA expression was normalized to β-actin expression, and the lowest value was arbitrarily set to 1. CYP2B6 protein was quantified by Western blotting with a monoclonal antibody from BD Gentest (Woburn, MA) according to standard procedures using SDS-polyacrylamide gel electrophoresis (10% polyacrylamide gels) with 25 μg of liver microsomal protein per lane. Recombinant CYP2B6 (BD Gentest) was coanalyzed on each gel to generate a standard curve using the manufacturer's information on specific protein content (Lang et al., 2001), and CYP2B6 apoprotein content was expressed as picomoles of CYP2B6 per milligram of microsomal protein. Bupropion hydroxylation activity was measured in human liver microsomes (50 μg) in a final volume of 250 μl in 0.1 M sodium phosphate buffer, pH 7.4, using 50 μM bupropion as substrate. After equilibrating the reaction mixture at 37°C for 3 min, enzyme reactions were started by adding 25 μl of 10-fold concentrated NADPH-regenerating system (final concentrations 5 mM MgCl2, 4 mM glucose 6-phosphate, 0.5 mM NADP+, and 4 U/ml glucose 6-phosphate dehydrogenase). After 30 min at 37°C, the reactions were stopped by adding 50 μl of 1 N HCl. After addition of 100 pmol of the internal standard d3-hydroxy-bupropion, the samples were centrifuged at 16,000g for 5 min. The supernatant was directly injected into a high-performance liquid chromatography system, and the metabolite hydroxy-bupropion was detected by high-performance liquid chromatography-electrospray ionization-mass spectrometry as described previously (Richter et al., 2004).
Oligonucleotides used in this study
Sequencing of the CYP2B6 Promoter Region and Construction of Reporter Plasmids. For direct sequencing, a 2.3-kb fragment of the CYP2B6 promoter region was amplified from genomic DNA of 98 white persons using the Expand High-Fidelity PCR system (Roche Diagnostics, Basel, Switzerland). Forward primer 2B6(–2253)F (Table 1) and reverse primer 2B6(+16)R were designed to contain mismatches (in lowercase) to create an MluI or a BglII site, respectively. The products were then sequenced using different primers summarized in Table 1. Bases were numbered according to the recommendations of the Nomenclature Working Group (Antonarakis and the Nomenclature Working Group, 1998), denoting the base 5′ to the translation start as –1. The reference sequence (also termed wild type) for the CYP2B6 promoter was extracted from GenBank accession number 24497632.
At first, we attempted to prepare reporter gene constructs containing the entire amplified upstream sequence described above. For this purpose, the 2.3-kb fragment was prepared from two genotyped white persons selected to carry one wild-type and three variant alleles representing the most frequent mutations, cloned into the pCR4-TOPO vector (Invitrogen, Carlsbad, CA), and sequenced. The MluI/BglII fragment was then subcloned into the reporter gene vector pGL3-Basic (Promega, Madison, WI). However, after transformation and plasmid preparation, the desired products were not obtained, despite several attempts, indicating that they may be unstable or toxic for Escherichia coli. Therefore, a shorter ScaI/BglII fragment was cloned into pGL3-Basic sequentially digested with SmaI and BglII, yielding the pGL3-2B6(–2033) series. Four different plasmids corresponding to the four different alleles of the two individuals were obtained. Single mutations were introduced by in vitro mutagenesis into the wild-type construct using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) and mutagenesis primers that are described in Table 1.
Genotyping –2320T→C and Exonic Mutations. Because the previously described polymorphism –2320T→C (Lamba et al., 2003) was not included in the sequenced fragment, a PCR-restriction fragment length polymorphism assay was developed to genotype for this mutation. Using the primers 2B6(–3010)F and 2B6(–1894)R (Table 1), we amplified a 1.1-kb fragment of the CYP2B6 promoter. After purification of the PCR product, this fragment was digested with EcoRV at 37°C for 2 h. In the presence of the mutation, the EcoRV site was abolished, whereas in absence of the mutation, the reaction yielded two products of 0.7 and 0.4 kb. The DNA fragments were visualized by ethidium bromide staining on a 2% agarose gel. The exonic mutations c.64C→T (exon 1), c.516G→T (exon 4), c.777C→A, c.785A→G (both exon 5), and c.1459C→T (exon 9) were genotyped using PCR-restriction fragment length polymorphism assays described previously (Lang et al., 2001).
Reconstruction of Haplotypes and Allele Designation. Genotype data for 10 promoter- and four exonic SNPs from 96 white persons were used to infer haplotypes using the program PHASE version 2.0.2 (Stephens et al., 2001). The exonic SNP 777C→A was omitted from the analysis because it was not observed in this population. Runs were conducted five times to ensure model stability. Phase calls with a probability of >95% were considered unambiguous. Identified haplotypes were compared with alleles described by the Human Cytochrome P450 Allele Nomenclature Committee (http://www.imm.ki.se/CYPalleles/). Novel alleles were provisionally designated in concordance with the published inclusion criteria.
Cell Culture and Transient Transfection. HepG2 cells were cultured in minimum essential medium supplemented with 10% fetal calf serum, 1% penicillin/streptomycin, and 1% l-glutamine (Invitrogen, Carlsbad, CA). The day before transfection, cells were seeded in 24-well plates with a density of approximately 150,000 cells per well and 0.5 ml of medium. After transfection, medium was changed daily. Primary rat hepatocytes were isolated from adult male Wistar rats (180–220 g) by collagenase perfusion (Seglen, 1976) and prepared for transfection as described previously (Hirsch-Ernst et al., 2001). Primary human hepatocytes were isolated as described previously (Dorko et al., 1994), plated in collagen-coated six-well plates with a density of 750,000 cells per well, and grown in 2 ml of William's E medium supplemented with 10% dialyzed fetal calf serum (Invitrogen), 1% penicillin/streptomycin, 32 U/l insulin, 1.4 × 10–6 M hydrocortisone, and 15 mM HEPES buffer.
Transient transfections were carried out using the Effectene transfection reagent (QIAGEN GmbH) according to the manufacturer's instructions. For normalization, either the Renilla reniformis luciferase expression plasmid pRL-CMV (Promega) (3 ng/well for rat hepatocytes) or the β-galactosidase expression plasmid pCMV-β (BD Biosciences Clontech, Palo Alto, CA) (20 ng/well for HepG2 cells and 40 ng/well for human hepatocytes) was cotransfected with 400 ng/well (primary hepatocytes) or 150 ng/well (HepG2 cells) of the respective firefly luciferase reporter plasmid. Cells were harvested 48 h after transfection with passive lysis buffer (Promega), and reporter activity was measured in the cell extract using the AutoLumat Plus luminometer (Berthold Technologies, Bad Wildbad, Germany). Firefly luciferase activity was assayed with RIM+ buffer (50 μM luciferin, 2 mM ATP, 10 mM MgCl2, 27 μM CoA, and 30 mM DTT in 25 mM glycylglycine, pH 7.8); β-galactosidase activity was measured using the Galacto-Light system (Applied Biosystems). Firefly and R. reniformis luciferase activities were sequentially determined in the same samples of primary rat hepatocyte lysates with the dual luciferase assay kit (Promega).
Primer Extension Analysis. Primer 2B6cDNA(41)R (Table 1) was labeled using the primer extension system-avian myeloblastosis virus reverse transcriptase (Promega) and [γ-32P]ATP (Hartmann Analytic, Braunschweig, Germany) according to the manufacturer's instructions. One hundred femtomoles of the labeled primer was hybridized to 100 μg of total liver RNA at 58°C for 20 min in 10 μlof the provided avian myeloblastosis virus primer extension buffer and cooled down at room temperature for 10 min. Reverse transcription was carried out at 42°C for 30 min according to the manufacturer's instructions. After RNase treatment and subsequent phenol-chloroform extraction followed by an ethanol precipitation, reaction products were dissolved in 2 μl of loading dye, electrophoresed in a 6% Long Ranger sequencing gel (Cambrex, East Rutherford, NJ), and visualized using Fuji imaging plates MS 2325 and a BAS-1800II plate reader (Fuji, Kanagawa, Japan). A 35S-labeled sequencing ladder, generated with the SeqiTherm EXCEL II DNA sequencing kit (Epicenter, Madison, WI) using a plasmid containing the corresponding genomic fragment as template and the primer 2B6cDNA(41)R, was electrophoresed in parallel.
Mapping of Transcriptional Start Sites with 5′-RLM-RACE. Total RNA (1–2 μg) of two human livers was isolated as described previously (Wolbold et al., 2003) and used to map the transcriptional start site of CYP2B6 with the GeneRacer kit (Invitrogen). 5′ cDNA ends were amplified with Taq polymerase (QIAGEN GmbH) in a total volume of 50 μl and the following touchdown cycling conditions: 95°C for 5 min; five cycles of 95°C for 30 s and 72°C for 90 s; five cycles of 95°C for 30 s and 70°C for 90 s; 20 cycles of 95°C for 30 s, 68°C for 30 s, and 72°C for 60 s; and finally 72°C for 7 min. Primer 2B6cDNA(521)R was used as reverse and the GeneRacer 5′ primer as forward primer. The sequence of 2B6cDNA(521)R represents bases 521 to 546 of the CYP2B6 mRNA and is identical to the corresponding region in the CYP2B7P1 pseudogene, thus allowing to simultaneously analyze for the presence of CYP2B7P1 transcripts.
Total RNA of transfected HepG2 cells was isolated with the RNA mini kit (QIAGEN GmbH) according to the manufacturer's instructions, and 5 μg was used to map the transcription start of the luciferase mRNA transcripts. To enhance transcription, cells had been cotransfected with the expression plasmid MSV/EBPβ for murine CCAAT/enhancer-binding protein (C/EBP)β (kindly provided by Dr. Oliver Burk, Dr. Margarete Fischer-Bosch Institute of Clinical Pharmacology, Stuttgart, Germany). The cDNA ends were amplified with nested PCR using the primers luci(350)R and luci(220)R (Table 1) in combination with the GeneRacer 5′ primer or NestedPrimer in a total volume of 50 μl. Cycling conditions for initial PCR were 95°C for 5 min; five cycles of 95°C for 30 s and 72°C for 60 s; five cycles of 95°C for 30 s and 70°C for 60 s; 20 cycles of 95°C for 30 s, 68°C for 30 s and 72°C for 60 s; and finally 72°C for 7 min. For nested PCR, initial denaturation at 95°C for 5 min was followed by 25 cycles of 95°C for 30 s, 65°C for 30 s and 72°C for 60 s, and finally 72°C for 7 min.
Electrophoretic Mobility Shift Assay. Double-stranded oligonucleotides (10 pmol) with four-nucleotide extensions at both ends were labeled with Klenow fragment under the following conditions: 50 mM NaCl; 50 mM Tris, pH 7.5; 10 mM MgCl2; 200 μM dATP, dGTP, and dTTP; 100 μM[α-32P]dCTP; and 2 U of Klenow fragment. Reactions were incubated at 37°C for 1 h. Salt and unincorporated nucleotides were removed by spin-column chromatography using ProbeQuant G-50 microcolumns (Amersham Biosciences Inc., Piscataway, NJ). The purified labeled probes were diluted to contain 50,000 cpm/μl. C/EBPβ and hepatic nuclear factor (HNF)1α were produced by in vitro transcription/translation using the Quick TnT kit (Promega). The DNA templates were generated by subcloning the open reading frame of murine C/EBPβ from the MSV/EBPβ plasmid into the pcDNA 3.1+ vector (Invitrogen) with EcoRI and XhoI yielding the pcDNA3-C/EBPβ plasmid and subcloning the EcoRI fragment of pBJ5-HNF1α (kindly provided by Prof. Dr. Ronald N. Hines, Medical College of Wisconsin, Milwaukee, WI), into the pcDNA 3.1+ vector yielding the pcDNA3-HNF1α plasmid. TATA box-binding protein (TBP) was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The in vitro-translated C/EBPβ or HNF1α (2 μl) was incubated at RT for 15 min with 2 μg of poly(dI-dC) and 5 pmol of unlabeled competitor, when included, before adding 2 μl of labeled DNA and incubating for another 20 min at RT. Binding reactions contained 10 mM HEPES, pH 7.8, 60 mM KCl, 0.2% NP-40, 6% glycerol, and 2 mM DTT. The binding reactions were loaded onto 5% polyacrylamide gels (0.5× Tris borate-EDTA). The gel was subjected to electrophoresis at 200 V for 60 min before sample loading. Electrophoresis was conducted for 1 to 2 h at RT. When TBP was used in gel retardation assays, 50 ng of protein was incubated with or without 5 pmol of unlabeled competitor at 30°C for 15 min before addition of 2 μl of the labeled DNA probe and further 30-min incubation at 30°C. Binding reactions contained 20 mM Tris, pH 8.0, 10 mM MgCl2, 2 mM DTT, 80 mM KCl, 2% Ficoll, and 0.1% NP-40. The binding reactions were loaded onto 4% polyacrylamide gels (0.5× Tris borate-EDTA); gel and running buffer contained 4 mM MgCl2 and 0.01% NP-40. Running conditions were the same as described above. After electrophoresis, gels were dried and radioactive probe visualized using imaging plates MS 2325 and a BAS-1800II plate reader (Fuji).
Statistical Analyses. Because some data sets were not normally distributed, nonparametric methods (Mann-Whitney U test) were generally used to compare phenotypic data. Multiple comparisons were performed by one-way analysis of variance and post hoc testing with Dunnett's test. For all calculations the GraphPad InStat version 3.00 (GraphPad Software Inc., San Diego, CA) was used. The 95% CIs of proportions for mutation frequencies were calculated using the Wilson procedure with continuity correction. All statistical tests were performed two-tailed and statistical significance was defined as p < 0.05.
Results
Frequency of Promoter Polymorphisms and Haplotype Structures. To generate a basis for investigating the role of promoter polymorphisms in CYP2B6 expression, we sequenced 2.3 kb of upstream sequence in 98 DNA samples from white liver donors previously studied for exonic polymorphisms and for CYP2B6 expression (Lang et al., 2001, 2004). As shown in Table 2, the most frequent SNPs in the promoter region were –750T→C (allele frequency 57%), –2320T→C (30%), and –1456T→C (25%). The polymorphisms –1778A→G and –1186C→G both occurred with a frequency of 9%, whereas the SNPs –1848C→A, –82T→C, and the newly described SNP –801G→T were all found with a frequency of 3%. These frequencies are in good agreement with previously published data (Lamba et al., 2003; Hesse et al., 2004).
Polymorphisms in the CYP2B6 promoter and their frequencies in a white population
SNP position is relative to translation initiation.
Eleven unambiguous haplotypes were inferred by analyzing genotype data for the nine promoter and four exonic SNPs shown in Table 3. Four novel alleles could be defined, including allele *22, which contained the four linked promoter polymorphisms –1848C→A, –801G→T, –750T→C, and –82T→C and occurred at a frequency of 3%. One sample carrying this haplotype (L#110) was sequenced throughout all exons to exclude the presence of further unknown mutations. An allele similar to *4A but carrying in addition the common promoter SNPs –750T→C and –1456T→C was designated as *4D. An allele composed of the exon 9 SNP c.1459C→T and the mutation –591A→G was defined as *5C, whereas a further allele differing from *6A with regard to the presence of the –750T→C SNP was termed *6C. The most common alleles in our white population were *1A (frequency 25%), *6B (22%), and *1H (20%). Ambiguous haplotype estimates (phase probability <0.95) occurred only in four individuals (L#18, 62, 68, and 105). One of these (L#105) was the only carrier of the SNP –1578C→A.
Alleles of CYP2B6
Bases are numbered according to the recommendations of the Nomenclature Working Group. Alleles are designated according to the CYP Allele Nomenclature Committee (http://www.imm.ki.se/CYPalleles/). The names of novel alleles described in this study are printed in bold.
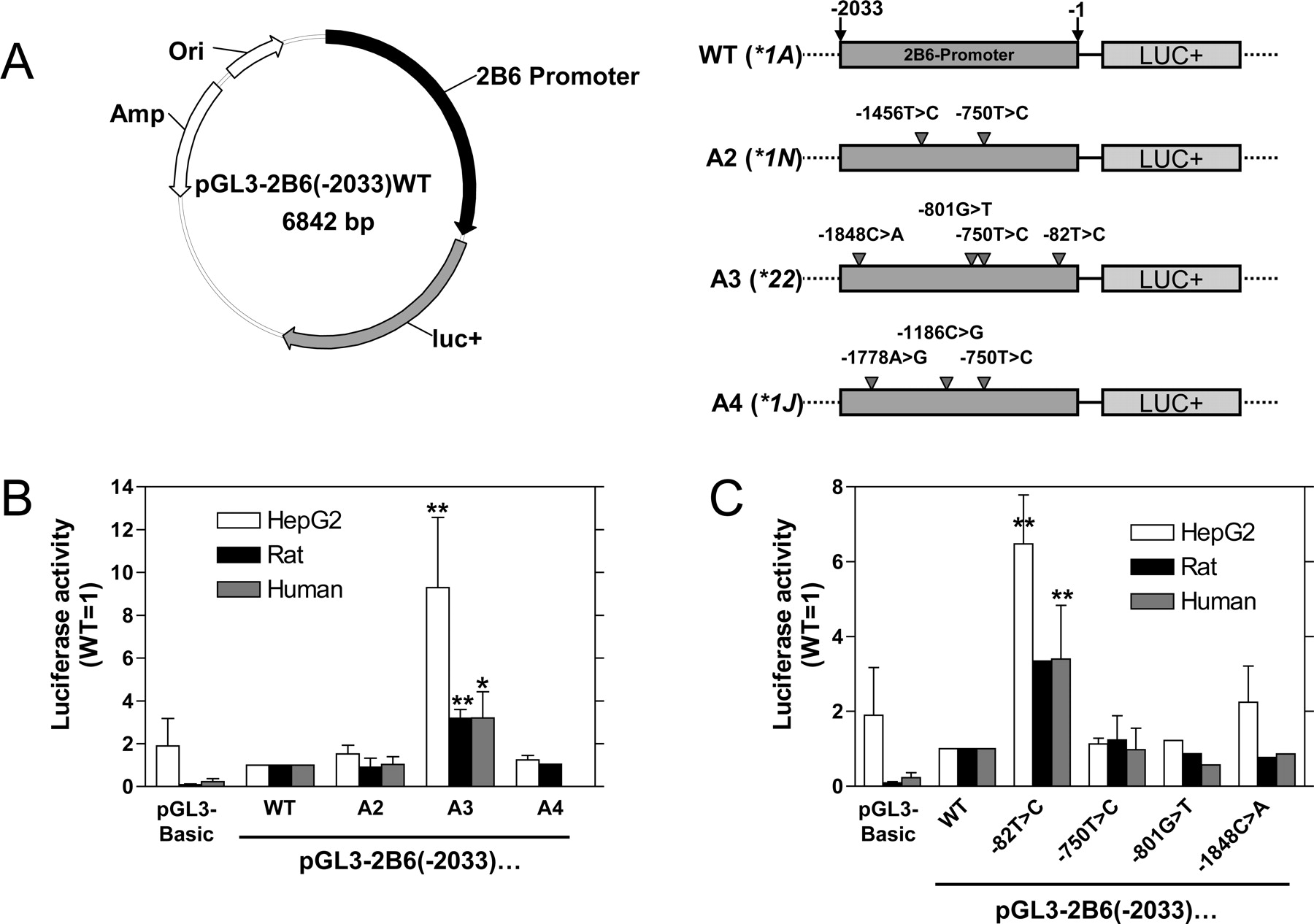
Promoter Activity in Different Cell Types. HepG2 cells were transfected with different reporter gene constructs of the pGL3-2B6(–2033) series that contain the firefly luciferase gene driven by the CYP2B6 promoter (Fig. 1A). In this hepatoma cell line, the wild-type construct did not show transcriptional activity distinguishable from the pGL3-Basic vector (Fig. 1B). In contrast, the construct pGL3-2B6(–2033)A3 consistently showed a transcriptional activity 9-fold higher than wild type, whereas the A2 and A4 constructs did not exhibit any detectable promoter activity. We then transfected HepG2 cells with constructs carrying the individual SNPs contained in A3 (Fig. 1C). The plasmid pGL3-2B6(–2033)-82T→C showed increased activity of the same magnitude as the A3 construct, whereas the other three SNPs did not alter promoter activity.

Transcriptional activity of the CYP2B6 promoter. A, firefly luciferase reporter gene plasmids carrying 2033 bp of the wild-type (WT) CYP2B6 5′-flanking region or different combinations of mutations (A2, A3, and A4) representing the haplotypes *1N, *22, and *1J of the CYP2B6 promoter were constructed as described under Materials and Methods. B, HepG2 cells and primary rat and human hepatocytes were cotransfected with the various plasmids and a β-galactosidase control plasmid (HepG2, human hepatocytes) or an R. reniformis luciferase control plasmid (rat hepatocytes). β-Galactosidase-normalized or R. reniformis luciferase-normalized firefly luciferase activity is shown in relation to the wild-type construct, which was set to 1.0 for each cell type. C, plasmids carrying the individual mutations of the pGL3-2B6(–2033)A3 construct were generated by in vitro mutagenesis and analyzed as described under B. The means and standard deviations of at least two independent experiments are shown; *, p < 0.05; **, p < 0.01 (analysis of variance followed by Dunnett's multiple comparison test).
To confirm these findings in noncancerous cells, we transfected primary rat and human hepatocytes. In contrast to HepG2 cells, basal promoter activity of the wild-type construct was approximately 10-fold higher compared with pGL3-Basic in both species. The pGL3-2B6(–2033)A3 construct again showed a significant increase in transcriptional activity compared with the wild type, whereas the other constructs did not differ (Fig. 1B). Because of the elevated basal activity in primary hepatocytes, the increase was approximately 3-fold, somewhat lower than in HepG2 cells. Similar to the analysis in HepG2 cells, only the single mutation –82T→C conferred enhanced transcription activity on the CYP2B6 promoter (Fig. 1C). These results were reproduced in at least three independently performed transfections with different preparations of human hepatocytes.
cis-Elements in the Promoter Region. We used the MatInspector Software (Quandt et al., 1995) to analyze the polymorphic regions of the novel CYP2B6*22 allele for transcription factor binding sites. The predicted alterations included the loss of a putative HNF1 site at –750 bp and disruption of a C/EBP binding site at –801 bp (Fig. 2). It is remarkable that the mutation –82T→C was not only predicted to disrupt a consensus TATA box, but to simultaneously create a putative C/EBP binding site (Fig. 2, bottom). Because the transcriptional start site of the human CYP2B6 gene had not been determined so far, the significance of the loss of the putative TATA box was unclear. It is interesting that an additional noncanonical TATA box was detected at –55 bp (GATAAA). To analyze the ability of these promoter regions to interact with trans-acting factors, electrophoretic mobility shift assays were performed. When reticulocyte-translated C/EBP protein was incubated with a 32P-labeled oligonucleotide containing the TATA box at –82, no specific DNA-protein complex was observed (Fig. 3A, lane 2). However, in the presence of the mutation –82T→C, a shifted band of a specific DNA-protein complex was observed (lane 8), confirming that a functional C/EBP binding site had been created. In the presence of HepG2 nuclear extract, two specific complexes with different electrophoretic mobilities compared with the complex with recombinant C/EBP were formed with mutant, but not with wild-type probe. The reason for these differences is most likely related to the multiplicity of cellular C/EBP factors. We then determined the ability of the two putative TATA boxes to bind TBP. As evident from Fig. 3B, strong specific binding of TBP occurred to the GATA motif at –55 bp (lane 10), whereas the TATA motif at –82 bp exhibited markedly weaker binding (lane 2). This interaction was even weaker in the presence of the mutation –82T→C (lane 6). In addition, the TATA and CATA motifs were not able to fully compete the GATA-TBP complex, even at 50-fold excess (lanes 12 and 13). Thus, both the GATA and the TATA motifs were shown to bind TBP and may therefore be able to act as functional TATA boxes.

Putative cis-elements in the CYP2B6 promoter affected by the *22 allele. The promoter region of CYP2B6 was analyzed using the MatInspector software (Quandt et al., 1995). Transcription factors with their consensus sequences are shown for reference sequence (above) and mutated sequence (below), with polymorphic sites in bold. PAX6, Pax-6 paired domain binding site; ATF6, activating transcription factor 6; MAZ, Myc-associated zinc finger protein; NF-E2p45, nuclear factor, erythroid-derived 2; IRF2, interferon regulatory factor 2; DBP, albumin D-box binding protein.

Electrophoretic mobility shift assay. A, in vitro translated C/EBP or nuclear extract of HepG2 cells was incubated with labeled probes representing bases –94 to –70 of the CYP2B6 promoter (containing a putative TATA box) in absence or presence of the SNP –82T→ C (probes TATA and CATA). Unlabeled oligonucleotides were added as cold competitor (CC), as indicated. For unspecific competition, a 25-bp double-stranded oligonucleotide without C/EBP binding sites was used. Protein-DNA complexes of C/EBP are marked by an arrow. B, recombinant TBP was incubated with probes TATA and CATA (described above) as well as probe GATA (representing bases –64 to –40 of the CYP2B6 promoter containing a putative noncanonical TATA box). Unlabeled oligonucleotides were added as CC, as indicated. TBP-DNA complexes are marked by an arrow.
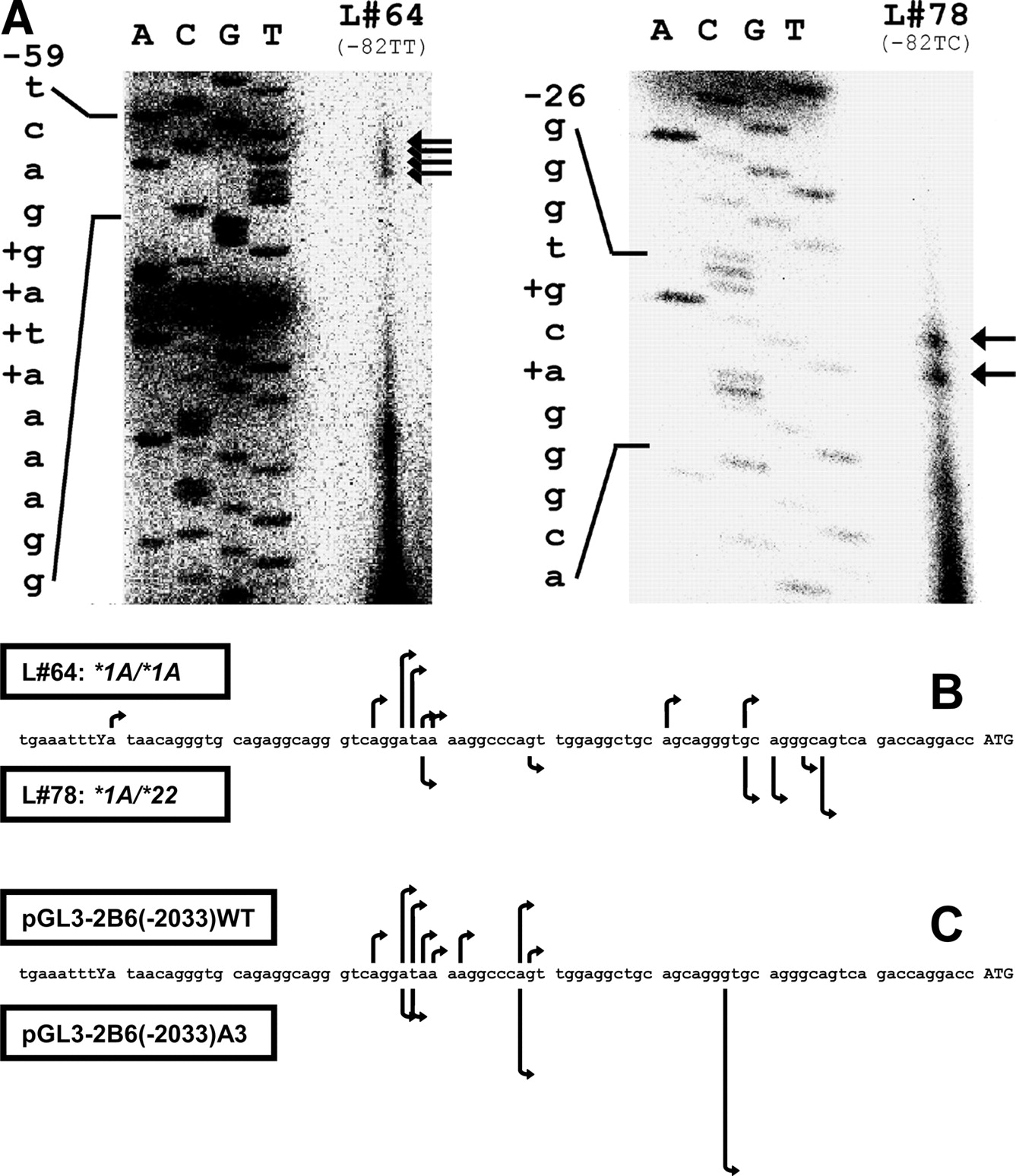
Analysis of the Transcriptional Start Site of CYP2B6. To investigate whether the two putative TATA boxes are involved in transcription, primer extension analyses were performed using total liver RNA of two persons with different genotypes at –82 bp (Fig. 4A). In the wild-type liver (L#64), we localized transcriptional start sites around –53 bp, which were not seen in the liver genotyped –82TC (L#78). In this sample, two other transcriptional start sites at –22 and –20 bp were clearly recognized. To be able to analyze use of these alternative transcriptional start site regions in a more quantitative manner, 5′-RLM-RACE was performed using the same RNA samples (Fig. 4B). Indeed, we could confirm the existence of the two different transcriptional start sites for the CYP2B6 gene around –54 and –20 bp, demonstrating excellent agreement between the two methods. For the individual with the wild-type promoter (top arrows), 13 of 18 transcripts originated from –57 to –51 bp. Additional minor transcriptional start sites were mapped to –81, –30, and –22 bp. In contrast, 11 of 14 analyzed transcripts of the *1A/*22 individual started between –22 and –15 bp (bottom arrows), whereas only two clones were derived from transcripts using the transcriptional start site at –52 bp.

Transcriptional start site of CYP2B6. A, primer extension was performed with 100 μg of total liver RNA of two individuals with different genotypes at –82 bp and a 32P-labeled primer located 41 bp downstream from the translation initiation site. The corresponding genomic fragment was sequenced using the same primer. Primer extension products are marked by arrows, and the corresponding transcriptional start sites (TSS) in the sequence are marked by +. B, TSS of CYP2B6 was determined in cDNA of two human livers with different genotype by using 5′-RLM-RACE. The length of the arrows corresponds to the number of clones representing the respective TSS. C, HepG2 cells were transfected with two reporter gene constructs reflecting the two different alleles CYP2B6*1A and CYP2B6*22. The TSS of the transcribed luciferase gene driven by 2033 bp of the CYP2B6 promoter was mapped and displayed as described above.
In addition, we also found a transcript of the recently described splice variant SV4 (Lamba et al., 2003) that contained the cryptic exon 3A, as well as a transcript of a new splice variant that used the alternate acceptor site in exon 4 without including the cryptic exon 3A. The resulting transcript contained a 32-bp deletion leading to a frameshift and a premature stop codon (data not shown). In RNA of both livers, no transcripts of the pseudogene CYP2B7P1 were found among the 34 clones analyzed.
To exclude the possibility that factors other than genotype, including gender, age, or drug exposure, were responsible for the differential use of the two transcriptional start sites, we also analyzed luciferase transcripts in transfected HepG2 cells. In cells transfected with pGL3-2B6(–2033)WT, the major transcriptional start site was mapped to a region from –57 to –49 bp, which was used by 16 of 21 analyzed clones (Fig. 4C, top arrows). The remaining five clones were derived from transcripts using a transcriptional start site at –43 bp. In contrast, when pGL3-2B6(–2033)A3 was transfected, shorter transcripts with a transcriptional start site at –24 bp dominated (14 of 24 clones; Fig. 4B, bottom arrows). Transcriptional start sites at –54 and –53 bp were found in four clones and another six transcripts originated from the site at –43 bp. This result confirmed the pivotal role of the promoter genotype in determining the transcriptional start site of CYP2B6.
Relevance of Promoter Genotype for CYP2B6 Expression in Vivo. To investigate whether the promoter genotype has functional consequences in vivo, we analyzed CYP2B6 mRNA and apoprotein expression (Fig. 5A) as well as enzyme activity in our large genotyped human liver bank. As shown in Fig. 5B, median CYP2B6 mRNA expression in livers genotyped –82TC was more than doubled compared with those genotyped –82TT (20.4 versus 9.8 arbitrary units; p = 0.007). Likewise, the median apoprotein content in livers with genotype –82TC was 66% higher compared with those genotyped –82TT (17.6 versus 10.6 pmol/mg microsomal protein), although this difference did not reach statistical significance. However, the median CYP2B6 bupropion hydroxylase activity in microsomes of livers genotyped –82TC again was almost 2-fold higher than in those with genotype –82TT (201.8 versus 106.7 pmol/mg/min; p = 0.042). Unfortunately, no –82CC homozygotes could so far be identified.

CYP2B6 mRNA and apoprotein expression and enzyme activity in human liver samples. A, a representative blot of Western immunoblot analysis which was performed as described under Materials and Methods is shown. Recombinant CYP2B6 was used to generate a standard curve for quantification. For human liver samples, the genotype at –82 bp is shown. B, CYP2B6 mRNA expression in total liver RNA and apoprotein expression as well as bupropion hydroxylase activity as selective marker for CYP2B6 catalytic activity in human liver microsomes were measured as described under Materials and Methods. Median values in samples genotyped –82TT were compared with those with genotype –82TC. Data are presented as quartiles, and the group size is given in the respective boxplot. Statistical significance is indicated (**, p < 0.01; *, p < 0.05; Mann-Whitney U test).
Discussion
Hepatic expression of CYP2B6 is highly variable and may depend on many exogenous and endogenous factors, including drug exposure, diet, sex, age, and various physiological and genetic factors. In this study, we investigated the contribution of common promoter variants to this variability by using reporter gene assays and studies in human liver samples, and we found that the mutation –82T→ C is associated with significantly increased expression and function of CYP2B6 even in heterozygous carriers. Although two previous studies have addressed the potential impact of CYP2B6 promoter variants, they used only descriptive methods to relate haplotypes to liver expression and activity, and no direct attempts were made to investigate their functional relevance (Lamba et al., 2003; Hesse et al., 2004). Our study is based on a comprehensive haplotype analysis using 2.3 kb of promoter sequence data and the genotypes for all common nonsynonymous SNPs from 96 white persons (49 men and 47 women). We identified one novel SNP (–801G→ T) as well as four novel alleles (*4D, *5C, *6C, and *22), and we confirmed the presence and frequency of the major haplotypes present among white persons. The three variant alleles *1J, *1N, and *22 were selected to be represented by reporter gene constructs because they comprised the promoter SNPs with frequencies more than 3%. When the plasmids pGL3-2B6(–2033)WT, A2, A3, and A4 were transfected into three liver cell types (HepG2, primary rat and human hepatocytes), only the A3 construct showed altered transcriptional activity (Fig. 1). Although we cannot exclude the possibility that individual SNPs of the constructs with unchanged activity (A2 and A4) may have an effect on transcription, these would be expected to be functionally compensatory in their allelic combinations. The considerably increased transcription rate of the A3-construct representing the *22 allele was entirely attributable to the TATA box disrupting SNP –82T→ C. This result seemed initially surprising, because known TATA box polymorphisms seem to decrease transcription rate, as seen, for example, in the UGT1A1 or CYP2A6 genes (Bosma et al., 1995; Pitarque et al., 2001). Using our collection of human liver samples, we could clearly demonstrate a significant impact of the mutation –82T→ C on expression and function. Median hepatic CYP2B6 mRNA expression was more than doubled in heterozygous carriers of the –82T→ C mutation compared with –82TT individuals (20.4 versus 9.8 arbitrary units; p = 0.007); accordingly, median apoprotein levels were 66% higher (17.6 versus 10.6 pmol/mg microsomal protein). The increased promoter activity even became manifest in median bupropion hydroxylase activity as a selective marker of human CYP2B6 catalytic activity (Faucette et al., 2000), which was also nearly doubled in the –82TC group (201.8 versus 106.7 pmol/mg/min; p = 0.042). These data demonstrate a close concordance between our in vitro reporter gene experiments and hepatic expression in vivo. However, a considerable variability within the TT and TC groups suggests the involvement of other factors than the –82 polymorphism. These could be either induction phenomena or additional genetic factors, because only heterozygous carriers of the mutation could be included in this study. Indeed, the lowest mRNA expression and enzyme activity in the –82TC group was found in a carrier of the *5 allele, which had been shown to result in decreased expression (Lang et al., 2001).
Our detailed promoter analysis revealed an unusual and unique scenario that seems to take place when –82T is changed into –82C (Fig. 6): The TATA box at –82 bp is predominantly used for transcription of the wild-type CYP2B6 gene, as shown by mapping the transcriptional start sites at around –54 bp using two independent methods. As indicated by electrophoretic mobility shift assay, the mutation –82T→ C seems to decrease binding of TBP and to simultaneously increase affinity for C/EBP. The enhanced binding of C/EBP would then promote usage of the alternative noncanonical TATA box at –55 bp, generating shorter transcripts initiated from transcriptional start sites around –20 bp in both transfected cells and human liver (Fig. 4). The observation that the shorter transcripts originating from the mutant allele by far dominate over the longer wild-type transcripts in liver RNA of a heterozygous individual as well as the increased mRNA expression in carriers of the –82T→ C mutation strongly supports the higher transcription rate of the *22 allele also in vivo.

Model of the mechanism of the –82T→ C polymorphism. The SNP –82T→ C converts the TATA box used by the human gene at –82 bp to a functional C/EBP binding site. Bound C/EBP supports binding of TBP to the alternative noncanonical TATA box at –55 bp, resulting in enhanced transcription from a transcriptional start site further downstream.
The comparison of different mammalian CYP2B promoters and transcriptional start sites further supports this model and reveals further insights into evolutionary relationships (Fig. 7). Except for Cyp2b9, all promoters analyzed contain a C at the position corresponding to –82T in human CYP2B6, and in the rat CYP2B1 promoter, this region indeed represents a functional C/EBP site (Park and Kemper, 1996). The transcriptional start sites in several rodent genes (CYP2B2, CYP2B3, Cyp2b9, Cyp2b10, and Cyp2b19) were mapped at around –25 bp (Lakso et al., 1991; Hoffmann et al., 1992; Jean et al., 1994; Honkakoski et al., 1996; Suzuki et al., 2002), indicating use of the respective noncanonical TATA boxes at –55 bp similar to the CYP2B6*22 allele. According to this analysis, the mouse Cyp2b9 gene would, however, be expected to use the TATA box at –82 bp and initiate transcription around –54 bp. That this is not the case suggests that the C/EBP site of this gene is conserved despite its –82T genotype. Apparently, –82T is compatible with C/EBP binding in the murine but not in the human sequence context, which shows an additional sequence difference at –83 bp. It is interesting that the murine –83G is compatible with the C/EBP consensus sequence, whereas the human –83T is not. It is remarkable that even in chimpanzee and in the paralogous pseudogene CYP2B7P1 the –82C is found, suggesting that the disruption of the C/EBP binding site occurred after the speciation of Homo and Pan and after the duplication of the CYP2B gene in human on the CYP2 gene cluster on chromosome 19 (Hoffman et al., 2001). This would indicate that the *22 allele (–82C) may represent the ancestral state. From an evolutionary point of view, it may be speculated that a high constitutive activity may have been disadvantageous because of the known promutagen-activating properties of CYP2B6. It remains unclear why the human GATA motif at –55 bp does not act as a functional TATA box in the absence of the mutation –82T→ C, albeit exhibiting much stronger binding to TBP than the TATA motif at –82 bp in electrophoretic mobility shift assay. Probably the flanking sequences of the GATA motif at –55 bp are not as suitable for binding other members of the Polymerase II complex as those in the TATA box at –82 bp and therefore require stabilization by C/EBP to be functional.
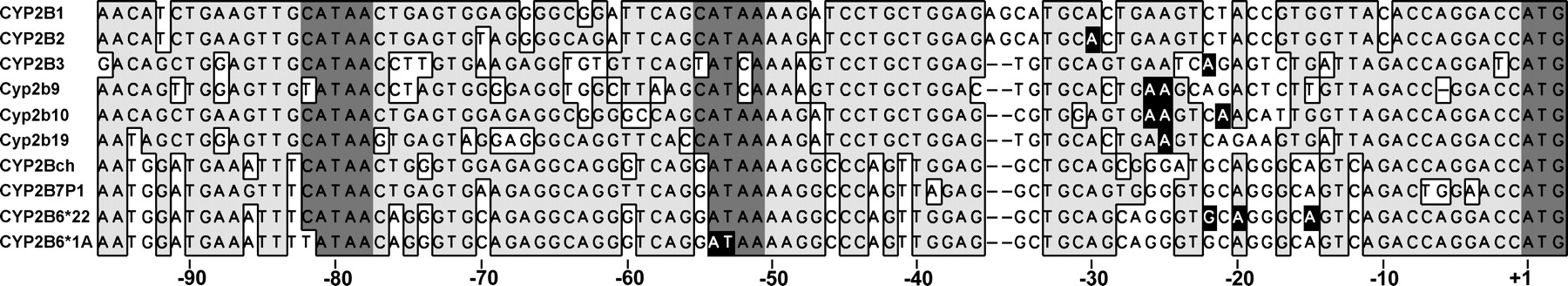
Alignment of CYP2B promoters in different species. Bases are numbered relative to the translation start ATG of CYP2B6. Available transcriptional start sites of CYP2B genes are printed white on black (Lakso et al., 1991; Hoffmann et al., 1992; Jean et al., 1994; Honkakoski et al., 1996; Suzuki et al., 2002). Putative C/EBP binding sites, TATA boxes and the translational start sites are shaded. The ortholog to human CYP2B6 in chimpanzee was designated as CYP2Bch.
In conclusion, we characterized the novel CYP2B6*22 allele as a gain-of-function allele. Promoter mutations that increase transcriptional activity have already been described, for example the UGT1A9*22 allele (Yamanaka et al., 2004) or a C→ G polymorphism in the CYP7B1 promoter (Jakobsson et al., 2004). However, to our knowledge, no TATA box mutations resulting in enhanced transcription or relocation of the transcriptional start site have been described so far. The clinical significance of this allele should be investigated as it may result in a CYP2B6 “ultrarapid metabolizer” phenotype. For CYP2D6, the well characterized and clinically relevant ultrarapid metabolizer phenotype results from a heterozygous gene duplication that increases gene dose by just 50% (Ingelman-Sundberg, 1999). Clinical implications of CYP2B6*22 may include, for example, enhanced bioactivation of cyclophosphamide, shortened duration of action of propofol, and treatment failure in human immunodeficiency virus therapy with efavirenz.
Acknowledgments
We gratefully acknowledge the excellent technical assistance of Britta Klumpp with Western blotting and protein quantification. We thank Professor Peter Neuhaus (Department of General, Visceral, and Transplantation Surgery, Humboldt University) for providing human hepatocytes and liver samples. We also thank Dr. Oliver Burk for many helpful suggestions and Dr. Katja Arnold for help with the TaqMan real-time PCR assay.
Footnotes
-
This work was supported by Deutsche Forschungsgemeinschaft (Germany) grant ZA 245/2-1 and by the Robert Bosch Foundation (Germany). Parts of this work were presented as a poster at the 15th International Symposium on Microsomes and Drug Oxidations, 2004 Jul 4–9, Mainz, Germany.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.104.008086.
-
ABBREVIATIONS: kb, kilobase(s); P450, cytochrome P450; SNP, single nucleotide polymorphism; PCR, polymerase chain reaction; RLM-RACE, RNA-ligase-mediated rapid amplification of cDNA ends; C/EBP, CCAAT/enhancer binding protein; HNF, hepatic nuclear factor; TBP, TATA box-binding protein; RT, room temperature; DTT, dithiothreitol; NP-40, Nonidet P-40; CI, confidence interval; EMSA, electrophoretic mobility shift assay.
-
↵1 Current address: Institute of Pharmacology, University of Mainz, Mainz, Germany.
- Received October 11, 2004.
- Accepted February 18, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}