


Abstract
Many endogenous compounds and xenobiotics are organic cations that rely on polyspecific organic cation transporters (OCTs) to traverse cell membranes. We recently cloned a novel human plasma membrane monoamine transporter (PMAT) that belongs to the equilibrative nucleoside transporter (ENT) family. We have reported previously that, unlike other ENTs, PMAT (also known as ENT4) is a Na+-independent and membrane potential-sensitive transporter that transports monoamine neurotransmitters and the neurotoxin 1-methyl-4-phenylpyridinium (MPP+). These compounds are the known substrates for OCTs, which raises the possibility that PMAT functions as a polyspecific transporter like the OCTs. In the present study, we analyzed the interaction of PMAT with a series of structurally diverse organic cations using MDCK cells stably expressing human PMAT. Our study showed that PMAT interacts with many organic cations that have heterogeneous chemical structures. PMAT transports classic OCT substrates, such as tetraethylammonium, guanidine, and histamine. Prototype OCT inhibitors, including cimetidine, and type II cations (e.g., quinidine, quinine, verapamil, and rhodamine123) are also PMAT inhibitors. An analysis of molecular structures and apparent binding affinities revealed that charge and hydrophobicity are the principal determinants for transporter-substrate/inhibitor interaction. A planar aromatic mass seems to be important for high affinity interaction. trans-Stimulation and efflux studies demonstrate that PMAT is able to mediate bidirectional transport. These functional properties of PMAT are strikingly similar to those of the OCTs. We therefore conclude that PMAT can function as a polyspecific organic cation transporter, which may play a role in organic cation transport in vivo.
Many endogenous compounds and xenobiotics, including drugs and environmental toxins, carry a net positive charge at physiological pH and are collectively termed “organic cations”. To eliminate organic cations from the body, mammalian cells have evolved complex transport systems. The organic cation transporters (OCTs) from the solute carrier 22 (SLC22) family transport relatively small and hydrophilic organic cations (i.e., the type I cations) (Koepsell and Endou, 2003; Koepsell et al., 2003; Wright and Dantzler, 2004). Transport-mediated by the OCTs is electrogenic and Na+-independent. The OCTs are considered “polyspecifc” or “multispecific” because a wide array of compounds with diverse chemical structures interact with these transporters, either as substrates or as inhibitors (Dresser et al., 2001; Koepsell et al., 2003; Wright and Dantzler, 2004). Classic substrates of the OCTs include MPP+, TEA, guanidine, choline (Busch et al., 1996; Gorboulev et al., 1997; Grundemann et al., 1999; Kekuda et al., 1998; Sweet et al., 2001), the biogenic amines, such as histamine, and the monoamine neurotransmitters (Breidert et al., 1998; Busch et al., 1998; Grundemann et al., 1998; Jonker and Schinkel, 2004). Bulkier and more hydrophobic cations (i.e., the type II cations) are often potent inhibitors, but not substrates, for the OCTs (Nagel et al., 1997). So far, three OCT isoforms, OCT1, OCT2, and OCT3, which share high sequence similarities and a common 12-transmembrane domain structure, have been identified and characterized from human and other mammalian species (Dresser et al., 2001; Koepsell et al., 2003; Wright and Dantzler, 2004). OCTs 1-3 display a large overlap in substrate and inhibitor specificity and are differentially expressed in various tissues. In humans, OCT1 and OCT2 are mainly expressed in the liver and kidney, respectively, where they are believed to play a key role in systemic elimination of organic cations (Gorboulev et al., 1997; Zhang et al., 1997; Wright and Dantzler, 2004). OCT3 has a broad distribution and is found in various tissues, including the liver, heart, placenta, skeletal muscle, kidney, and brain (Grundemann et al., 1998; Wu et al., 2000). OCT3 is also alternatively called “extraneuronal monoamine transporter” because its functional properties match those of the corticosterone-sensitive catecholamine transport system originally characterized in non-neuronal cells, such as cardiac myocytes and smooth muscle cells (Iversen, 1965; Grundemann et al., 1998; Wu et al., 1998; Eisenhofer, 2001).
We recently cloned a novel membrane transporter, PMAT, that belongs to the equilibrative nucleoside transporter (ENT) family (SLC29) (Engel et al., 2004; Kong et al., 2004). PMAT is a protein of 530 amino acid residues with 11 predicted transmembrane domains. In humans, PMAT mRNA is most strongly expressed in the brain and skeletal muscle, but transcripts are also found in the liver, kidney, and heart (Engel et al., 2004). By gene ontology, PMAT is the fourth isoform of the ENT family, and it was therefore alternatively designated ENT4 (Acimovic and Coe, 2002; Baldwin et al., 2003). However, unlike other ENT isoforms, namely ENT1-3, which exclusively transport nucleosides and nucleoside analogs (Kong et al., 2004; Baldwin et al., 2005), PMAT does not significantly interact with nucleosides and structurally related analogs (Engel et al., 2004). We recently showed that PMAT is a low-affinity, high-capacity plasma membrane transporter for monoamine neurotransmitters such as dopamine and serotonin. We also proposed the use of the functional name plasma membrane monoamine transporter (PMAT) in preference to ENT4 (Engel et al., 2004).
Because of its abundant expression in the brain, PMAT is thought to supplement the role of high-affinity neurotransmitter transporters in regulating brain monoamine levels under certain conditions (Engel et al., 2004). Other than the brain, PMAT is also expressed in excretory organs such as the liver and the kidney, where its role is not known. It is noteworthy that although PMAT is not genetically related to the OCTs, it exhibits significant functional similarities to the OCTs. Both PMAT and OCTs transport MPP+ and the monoamine neurotransmitters (dopamine, serotonin, and norepinephrine). Transport mediated by PMAT and OCTs is both membrane potential- and Na+-independent (Sweet and Pritchard, 1999; Koepsell et al., 2003; Engel et al., 2004; Wright and Dantzler, 2004). However, whether PMAT is able to function as a polyspecific organic cation transporter that interacts with structurally diverse compounds remains unknown. Furthermore, the molecular features important for compounds to interact with the PMAT protein are unknown. This information is not only important for revealing the physiological and pharmacological roles of PMAT but is also mechanistically interesting, given that PMAT is evolved from a family of nutrient transporters (i.e., the ENTs) that have a narrow substrate specificity and only transport compounds (i.e., nucleosides and their analogs) with related structures (Baldwin et al., 2003, 2005; Kong et al., 2004). In the present study, we investigate the interaction of PMAT with a series of structurally diverse organic cations including prototype OCT substrates and inhibitors, type II organic cations, biogenic amines, and other endogenous compounds. Our data suggest that PMAT recognizes various type I and II cations with heterogeneous structures. Our studies also revealed general structural features important for molecular interaction with PMAT.
Materials and Methods
Materials. [3H]MPP+ (80 Ci/mmol), [3H]tyramine hydrochloride (50 Ci/mmol), [14C]tryptamine bisuccinate (0.1 mCi/mmol), [3H]verapamil hydrochloride (85 Ci/mmol), [14C]tetraethylammonium bromide (55.6 mCi/mmol), [14C]choline chloride (55 mCi/mmol), and [14C]guanidine hydrochloride (55 mCi/mmol) were purchased from American Radiolabeled Chemicals (St. Louis, MO). [3H]Histamine dihydrochloride (12.4 Ci/mmol) was acquired from PerkinElmer Life and Analytical Sciences (Boston, MA). All other chemicals were obtained from Sigma (St. Louis, MO) and were of analytical grade.
Cloning of PMAT and Expression in MDCK Cells. PMAT cDNA was obtained and expressed in MDCK cells as described previously (Engel et al., 2004). In brief, PMAT cDNA, isolated from human kidney, was subcloned into the HindIII and XbaI restriction sites of the pcDNA3 vector (Invitrogen, Carlsbad, CA) and transfected into MDCK cells by liposome-mediated transfection (Lipofectamine; Invitrogen, Carlsbad, CA). A stably transfected cell line was obtained by G418 selection. PMAT- and vector-transfected MDCK cells were cultured in minimal essential medium containing 10% fetal bovine serum and 200 μg of G418/ml medium.
Uptake Experiments. Cells were plated in 24-well plates and allowed to grow at 37°C for 3 days. Growth medium was aspirated, and each well was rinsed with Krebs-Ringer-Henseleit (KRH) buffer (5.6 mM glucose, 125 mM NaCl, 4.8 mM KCl, 1.2 mM KH2PO4, 1.2 mM CaCl2, 1.2 mM MgSO4, and 25 mM HEPES, pH 7.4) and then preincubated in KRH buffer for 15 min at 37°C. Transport assays were performed at 37°C by incubating cells in KRH buffer containing a 3H- or 14C-labeled ligand. After incubation, uptake was terminated by aspirating the reaction mixture and washing the cells three times with ice-cold KRH buffer. Cells were then solubilized with 0.5 ml of 1 N NaOH. After 2 h, the solubilized cells were neutralized with 0.5 ml of 1 N HCl. This solution (0.5 ml) was quantified by liquid scintillation counting and 25 μl was used for the protein assay.
Inhibition Studies. For inhibition and Ki studies, cells were preincubated in KRH buffer for 15 min at 37°C containing various concentrations of unlabeled compound. Cells were then incubated at 37°C for 1 min in KRH buffer containing [3H]MPP+ and various concentrations of an unlabeled compound. Cells were then rinsed three times with ice-cold KRH buffer and samples were assayed as described above.
trans-Stimulation Studies. In trans-stimulation experiments, cells were preincubated for 20 min with either KRH buffer (control) or KRH buffer containing an unlabeled compound (50 μM for rhodamine123 and 1 mM for all other compounds). Cells were then rinsed three times with ice-cold KRH buffer before incubation at 37°C for 1 min in KRH buffer containing 1 μM [3H]MPP+. Cells were then rinsed three times with ice-cold KRH buffer, and samples were assayed as described above.
Efflux Studies. For efflux studies, cells were preloaded with KRH buffer containing 1 μM [3H]MPP+ at 37°C for 2 h. The buffer was then removed, and the cells were washed three times with ice-cold KRH buffer. Efflux was initiated by adding 1 ml of KRH buffer (37°C) with or without a PMAT inhibitor (decynium-22). After various incubation times, 0.5 ml of the buffer was transferred to scintillation vials for determination of effluxed radioactivity. At the end of each incubation time, efflux was terminated by aspirating the reaction mixture and washing the cells three times with ice-cold KRH buffer. Cells were then solubilized, and MPP+ remaining in cells was determined by liquid scintillation counting. The total amount of preloaded MPP+ in each well was determined by calculating the sum of MPP+ effluxed into buffer and MPP+ remained within cells.
Protein Assay. Protein content was determined in each uptake well. Twenty-five microliters out of 1 ml of solubilized cells were measured using a BCA protein assay kit (Pierce, Rockford, IL). The amount of protein was calculated from the standard curve generated by use of the known amounts of bovine serum albumin, and the uptake in each well was normalized to its protein content.
Log P Value Calculation. The octanol/water partition coefficients (Log P) were obtained from the SciFinder Scholar Program (2004 edition; American Chemical Society, Washington, DC), where the Log P values are calculated using the Advanced Chemistry Development (ACD/Labs) Software Solaris ver. 4.67. The calculated Log P values for TEA and MPP+, which are not available from the SciFinder Scholar Program, were obtained from Bednarczyk et al. (2003).
Data Analysis. All experiments were performed in triplicate and repeated two to four times. Results from a representative experiment were shown. Data were expressed as mean ± S.D. Statistical significance was determined by Student's t test, and kinetic parameters were determined by nonlinear least-squares regression fitting as described previously (Wang et al., 1997). For Michaelis-Menten studies, data were fit to the equation V = Vmax[S]/(Km + [S]) using Kaleidagraph Version 3.6 (Synergy Software, Reading, PA), where V is the transport rate and [S] is the substrate concentration. The IC50 was determined by fitting the data to the equation V = Vo/[1 + (I/IC50)nH], where V is the rate of uptake of MPP+ in the presence of the inhibitor, Vo is the rate of uptake of MPP+ in the absence of inhibitor, I is the inhibitor concentration, nH is the Hill coefficient, and IC50 is the half-maximal inhibitory concentration. Assuming a competitive mechanism of inhibition, the inhibition constant (Ki) was determined by the equation Ki = IC50/(1 + C/Km), where C represents the concentration of MPP+ and Km represents the apparent affinity of MPP+ uptake. The PMAT-specific uptake was calculated by subtracting the transport activity in vector-transfected cells.
Results
cis-Inhibition Studies. To identify compounds potentially interacting with PMAT, 25 structurally diverse compounds were examined for their ability to cis-inhibit MPP+ (1 μM) uptake in MDCK cells stably transfected with the human PMAT cDNA. Except for rhodamine123, which was used at 50 μM because of poor solubility, compounds were first tested at 500 μM (Fig. 1a). All type II organic cations (quinine, quinidine, rhodamine123, and verapamil), which are potent OCT inhibitors (Zhang et al., 1998; Martel et al., 2001), greatly inhibited PMAT-mediated MPP+ uptake. The biogenic amines dopamine, serotonin, tyramine, and tryptamine significantly inhibited PMAT-mediated MPP+ uptake with the inhibitory effect of tryptamine > serotonin ≥ dopamine > tyramine. Histamine did not inhibit PMAT-mediated MPP+ uptake at 500 μM at l min. Clonidine and amantadine, two clinically used drugs that are known to interact with OCT1 and OCT2 (Zhang et al., 1997; Busch et al., 1998), strongly inhibited PMAT activity. Cimetidine and procainamide, which are classic OCT inhibitors (Urakami et al., 1998; Dresser et al., 2001; Koepsell et al., 2003), significantly decreased PMAT-mediated MPP+ uptake. Nicotine and pargyline (a monoamine oxidase inhibitor) strongly inhibited PMAT. The classic OCT substrates TEA, guanidine, choline, and N-methyl-nicotinamide (NMN) exhibited no significant inhibitory effect at 500 μM. Paraquat, a divalent cation that is structurally similar to MPP+, did not interfere with PMAT-mediated MPP+ uptake. The polyamines agmatine and thiamine exhibited no inhibitory effect. Creatinine and l-DOPA (a zwitterion) also did not inhibit the uptake of PMAT. Compounds that did not show a significant effect at 500 μM were tested further at a higher concentration (2 mM) (Fig. 1b). At this concentration, small but significant (p < 0.01) inhibitory effects were observed for choline, guanidine, histamine, and NMN. A marginal inhibitory effect was observed for TEA, although it was not statistically significant. Paraquat, creatinine, l-DOPA, agmatine, and thiamine did not exhibit any inhibitory effects toward PMAT at 2 mM.
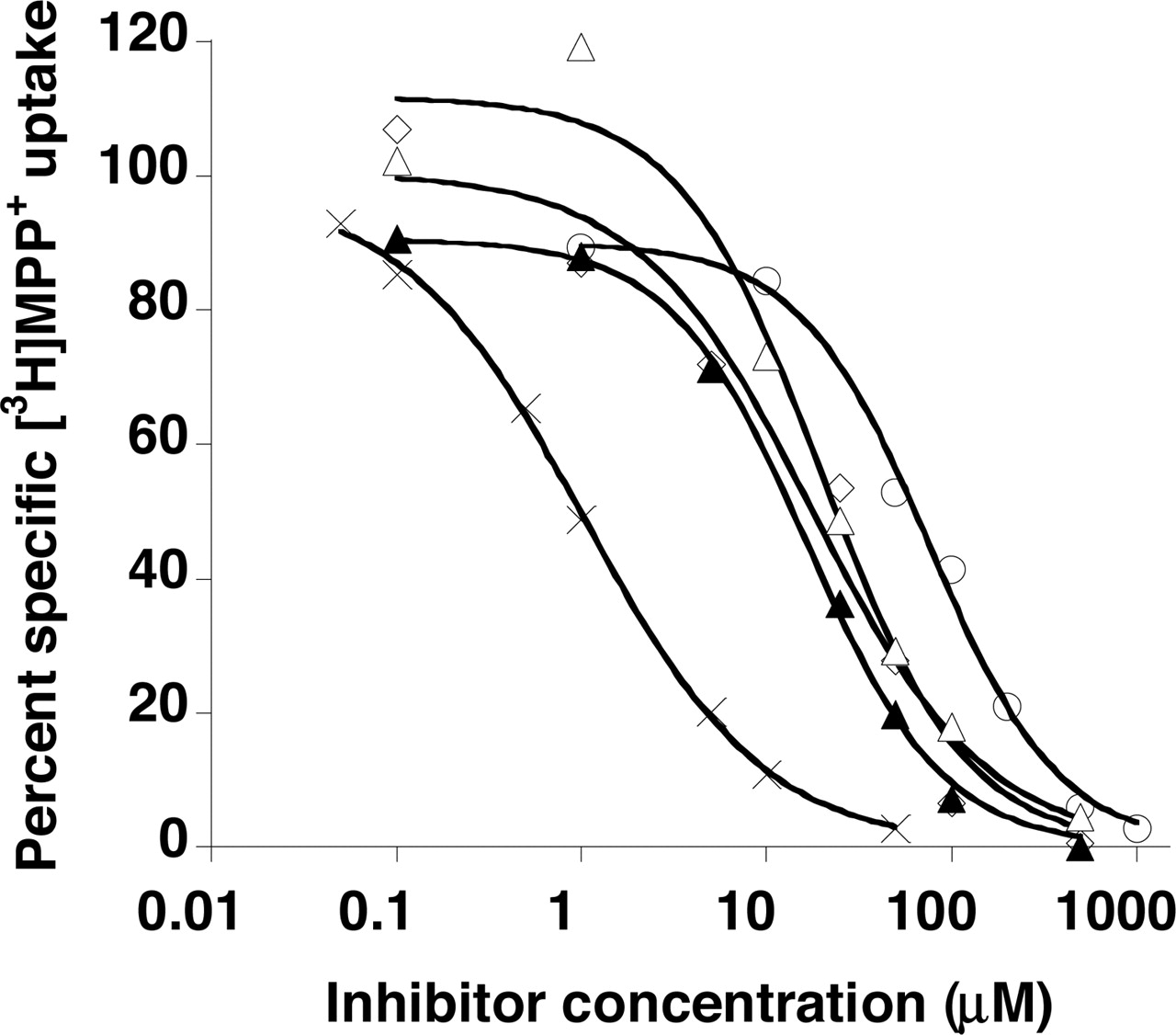
Compounds that exhibited most potent inhibitory effects (more than 80% inhibition) were tested further, and their inhibition potencies (Ki) were determined (Fig. 2). The calculated Ki values are as follows in increasing (i.e., decreasing in affinities) order: 1.02 ± 0.12 μM for rhodamine123, 18.6 ± 3.1 μM for verapamil, 25.3 ± 4.8 μM for quinidine, 26.9 ± 4.6 μM for quinine, and 62.9 ± 13.5 μM for tryptamine.

Effect of various compounds on [3H]MPP+ uptake by PMAT-expressing MDCK cells. Rhodamine123 was present at 50 μM during preincubation and incubation periods. For all other compounds, a concentration of 0.5 mM (a) and 2 mM (b) was used. Vector-transfected cells (open bars) and PMAT-transfected cells (solid bars) were incubated with 1 μM [3H]MPP+ for 1 min at 37°C in the absence or presence of an inhibitor. The values were expressed as percentages of [3H]MPP+ uptake by PMAT-expressing cells in the absence of an inhibitor (control). Each bar represents the mean ± S.D. (n = 3). Rho123, rhodamine123. *, significantly different from the control, p < 0.01.
trans-Stimulation Studies. To examine whether a compound might be a PMAT substrate, trans-stimulation, a method that has been used to test whether two molecules share a common transport pathway, was initially employed (Busch et al., 1998; Zhang et al., 1999; Wright and Dantzler, 2004). As shown in Fig. 3, after preloading PMAT-expressing MDCK cells with MPP+, dopamine, serotonin, tyramine, or histamine, [3H]MPP+ uptake was significantly enhanced (p < 0.01). Because MPP+, dopamine, and serotonin are substrates of PMAT (Engel et al., 2004), the data indicate that tyramine and histamine are likely to be transported by PMAT as well. Furthermore, the observation that known PMAT substrates trans-stimulated its uptake suggests that PMAT is capable of transporting substrates in both directions. Preincubating cells with verapamil, quinidine or quinine resulted in a substantial decrease of [3H]MPP+ uptake. Moderate to weak trans-inhibitory effects were observed when cells were preincubated with amantadine, cimetidine, clonidine, nicotine, procainamide, rhodamine123, and tryptamine. Preincubating with agmatine, choline, creatinine, l-dopa, guanidine, NMN, paraquat, pargyline, TEA, or thiamine did not result in a significant change in [3H]MPP+ uptake. Because a trans-stimulatory effect is observed only when loading of a substrate at the trans-face accelerates the in-to-out reorientation of the substrate binding site, no conclusion can be drawn regarding the substrate status for compounds that did not show a trans-stimulation effect (Busch et al., 1998; Zhang et al., 1999; Wright and Dantzler, 2004).
Tracer Flux Studies. Our trans-stimulation studies suggest that histamine and tyramine are likely to be transported by PMAT. To confirm these results, we performed tracer flux experiments using radiolabeled compounds (Fig. 4). At a substrate concentration of 1 μM and an incubation time of 1 min, PMAT-expressing MDCK cells showed a 4- to 5-fold increase in [3H]histamine and [3H]tyramine uptake (Fig. 4), suggesting that both compounds are truly substrates for PMAT. For compounds that trans-inhibited or did not have a trans effect on MPP+ uptake, no conclusions can be made regarding their substrate status (Busch et al., 1998; Zhang et al., 1999; Wright and Dantzler, 2004). Therefore, we tested five such compounds (TEA, choline, guanidine, tryptamine, and verapamil) using 3H- or 14C-labeled chemicals and tracer flux studies. TEA, choline, and guanidine were chosen because they are prototypic OCT substrates and have been suggested to be selectively transported by the three OCT isoforms (Grundemann et al., 1999). As shown in Fig. 4, cells expressing PMAT exhibited a 2-fold increase in TEA and guanidine uptake after 1-min incubation at 1 μM. There was no difference for choline uptake between PMAT- and vector-transfected cells. Tryptamine, a serotonin precursor that both cis- and trans-inhibited PMAT (Figs. 1 and 3), exhibited 2-fold increase in uptake by PMAT, indicating that it is a substrate for PMAT. Verapamil, a type II cation that showed strong trans- and cis-inhibitory effects on PMAT (Figs. 1 and 3), exhibited a high background uptake, and there was no difference in verapamil uptake between PMAT-expressing cells and vector-transfected control cells.
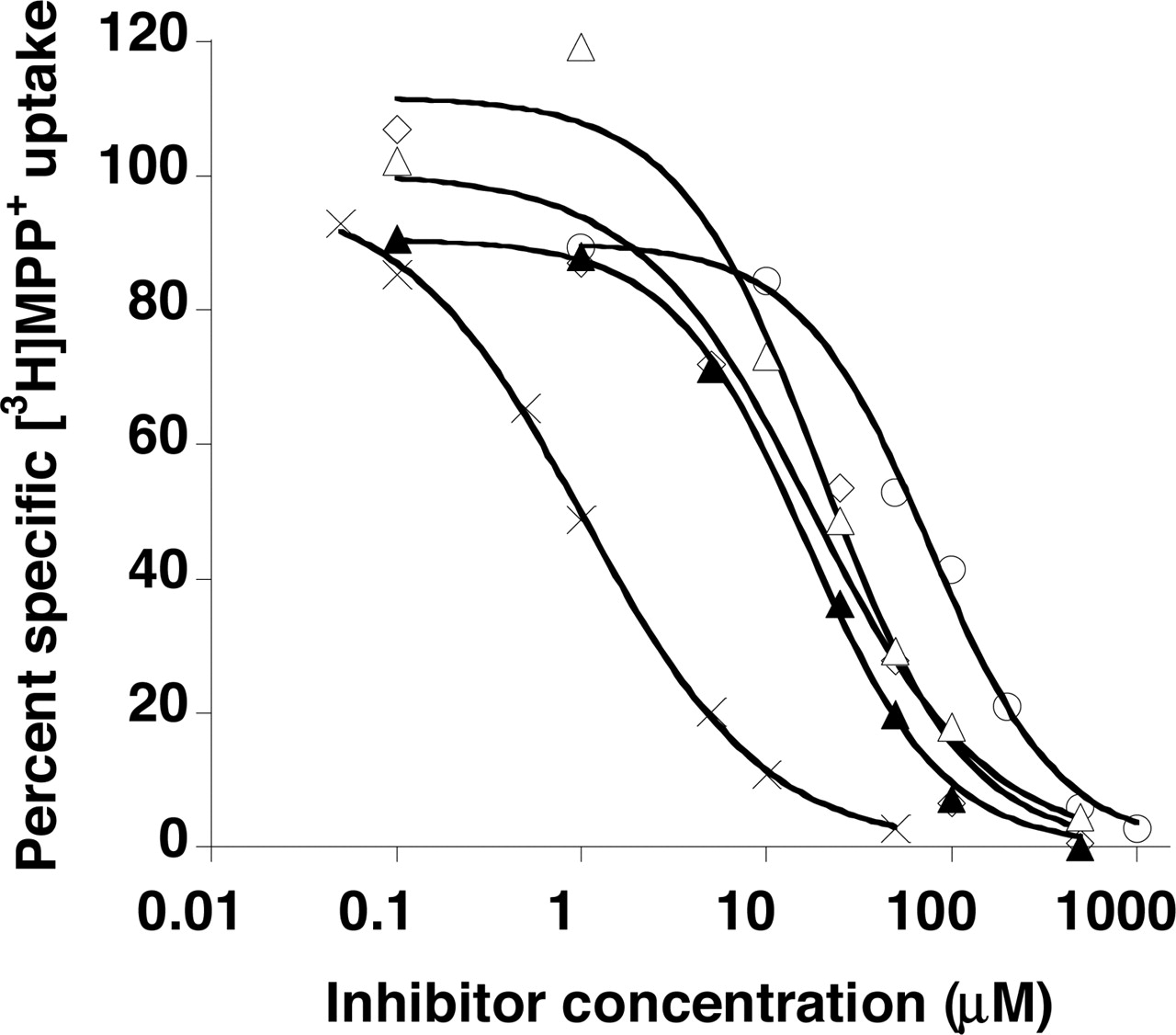
Concentration-dependent inhibition of specific MPP+ uptake by rhodamine123 (×), quinidine (⋄), quinine (▵) verapamil (▴), and tryptamine (○) in PMAT-expressing MDCK cells. Cells were incubated at 37°C with 0.1 μM [3H]MPP+ for 1 min in the presence of varying concentrations of inhibitors. The PMAT-specific uptake was calculated by subtracting the transport activity in vector-transfected cells.

Trans-stimulation of [3H]MPP+ uptake in PMAT-expressing MDCK cells. PMAT-transfected cells (solid bars) and vector-transfected cells (empty bars) were preincubated for 20 min at 37°C in the absence (control) or the presence of various compounds at 1 mM except for rhodamine123 (50 μM). Compounds were removed from extracellular space by washing three times with ice-cold buffer before the cells were incubated for 1 min at 37°C with 1 μM [3H]MPP+. Each bar represents the mean ± S.D. (n = 3). Rho123, rhodamine123. *, significantly different from the control, p < 0.01.

Uptake of radiolabeled compounds at 1 μM. PMAT-transfected cells (solid bars) and vector-transfected cells (empty bars) were incubated for 1 min at 37°C. Each bar represents the mean ± S.D. (n = 3). *, significantly different from the vector-transfected cells, p < 0.01.
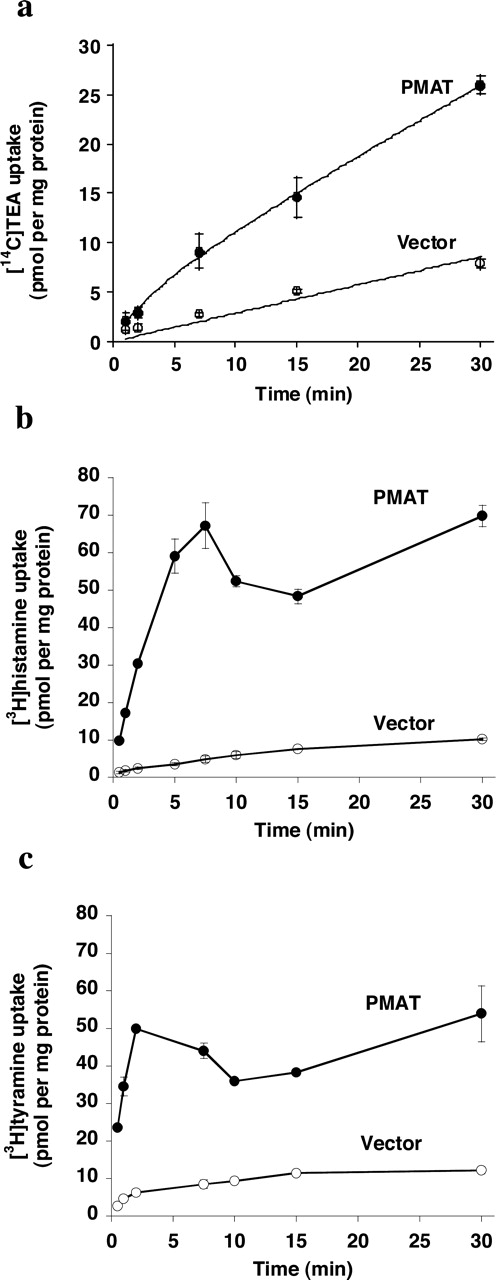
Transport Kinetics. Our tracer flux studies at 1 min incubation interval identified TEA, guanidine, histamine, tyramine, and tryptamine as new substrates for PMAT (Fig. 4). We further determined the time courses of TEA, histamine, and tyramine uptake in PMAT- and vector-transfected cells (Fig. 5). The data demonstrated that uptake of all three compounds was significantly higher in PMAT-expressing cells at all time points tested; substantial accumulation of histamine and tyramine was achieved after 5- to 10-min incubation in PMAT-expressing cells. To obtain quantitative information on substrate-transporter interaction, transport kinetics were determined from saturation studies using 1 min as the initial rate period (Fig. 6). PMAT-mediated uptake of [14C]TEA, [3H]histamine, and [3H]tyramine were saturable with apparent affinities (Km) of 6.6 ± 1.7 mM, 10.5 ± 2.6 mM, and 283 ± 23 μM, respectively. The respective maximal transport velocities (Vmax) were 5.8 ± 0.9, 99.6 ± 17.3, and 5.1 ± 0.1 nmol/min/mg of protein. The high Km values of TEA and histamine explained the weak inhibitory effect of these two compounds toward PMAT at relatively lower concentrations (Fig. 1).

Time courses of TEA (a), histamine (b), and tyramine (c) uptake by PMAT and vector-transfected (control) cells. Vector-transfected cells (○) and PMAT-transfected cells (•) were incubated at 37°C with 1 μM concentrations of each compound. Each point represents the mean ± S.D. (n = 3).

Concentration-dependent transport of TEA (a), histamine (b), and tyramine (c). PMAT-transfected cells and vector-transfected (control) cells were incubated with varying concentrations of each substrate for 1 min at 37°C. The PMAT-specific uptake (• and dotted lines) was calculated by subtracting the transport activity of control cells (⋄) from PMAT-transfected cells (○).
In Table 1, we summarize the kinetic information for all known PMAT substrates. The rank order of apparent affinities (i.e., inverse of Km) is MPP+ > serotonin > tyramine ≥ dopamine > norepinephrine > TEA > histamine > epinephrine. However, despite low affinity, histamine is transported with a high Vmax. Because our PMAT-expressing cell line is stable and we generally observed a relatively consistent MPP+ uptake activity (less than 2-fold change) over different batches of cells used (data not shown), we compared the transport efficiency (Vmax/Km) of all tested PMAT substrates assuming a constant PMAT expression level (Table 1). The data suggest that MPP+, serotonin, and dopamine are transported by PMAT at the highest efficiency. Tyramine, histamine, and norepinephrine are transported at intermediate efficiency. Epinephrine and TEA are transported at the lowest efficiency.
Kinetic constants of PMAT substrates Values are given as means ± S.D. with n = 3.
Molecular Features for Interaction. To obtain a comprehensive understanding of the general structural features of a compound important for recognition by PMAT, we compiled in Table 2 the data from the present study along with information from a previous study (Engel et al., 2004). An analysis of the molecular structures (Fig. 7) of the tested compounds revealed that charge and hydrophobicity are the principal determinants for interaction with PMAT. For a molecule to be recognized by PMAT, a positive charge seems to be essential. Neutral (e.g., l-DOPA, GABA) and negatively charged molecules (e.g., p-aminohippurate) did not interact with PMAT. An exception is that corticosterone, a noncharged molecule, inhibited PMAT with low affinity. A hydrophobic mass, preferably with at least one aromatic planar structure, is needed for high-affinity interaction. Type II cations (quinidine, quinine, verapamil, rhodamine123, decynium-22, etc.) with multiple aromatic ring structures are potent PMAT inhibitors. All high-affinity substrates (e.g., MPP+, tyramine, dopamine, serotonin) possess at least one phenyl ring. On the other hand, cations without a planar hydrophobic surface (e.g., TEA, guanidine, choline) are either low-affinity substrates or did not interact with PMAT. Indeed, when hydrophobicity, expressed as calculated Log P, is plotted against Ki or Km, a trend of positive correlation between the Log P values and the apparent binding affinities (inverse of Ki or Km) was observed (Fig. 8). The correlation became statistically significant (r = 0.74) when MPP+, TEA, and rhodamine123 were treated as outliers. A balance between hydrophobicity and hydrophilicity seems to be critical in determining whether an interacting compound is a substrate or an inhibitor. Cations with intermediate hydrophobicity (e.g., serotonin, dopamine, tyramine) clustered in the middle region (Fig. 8) and were all good substrates for PMAT (Km, 114-329 μM). Cations that are more hydrophilic [low Log P (e.g., TEA, histamine, epinephrine)] clustered in the higher left corner. These compounds are low-affinity substrates (Km > 6.5 mM). Highly hydrophobic cations (e.g., quinine, quinidine, verapamil, desipramine), on the other hand, clustered on the lower right corner. These compounds are potent inhibitors but may not be transported by PMAT (e.g., verapamil in Fig. 4). It is noteworthy that MPP+, TEA, and rhodamine123 significantly deviated from the linear regression line, which suggests that molecular descriptors other than charge and hydrophobicity also play a role in transporter-substrate/inhibitor interaction.
Interaction of various compounds with PMAT Values are given as means ± S.D. with n = 3.
PMAT-Mediated Efflux of MPP+. Our previous study showed that PMAT is a Na+-independent and membrane potential-sensitive transporter that is likely to operate as an electrogenic facilitated carrier (Engel et al., 2004). Our present finding that several PMAT substrates trans-stimulated its uptake (Fig. 3) indicates that PMAT is able to function bidirectionally. To further investigate the transport mechanism of PMAT, we preloaded cells with MPP+ and measured time-dependent efflux in PMAT- and vector-transfected cells. As shown in Fig. 9a, preloaded MPP+ is rapidly released to the medium in cells expressing PMAT. MPP+ release is greatly reduced in the presence of extracellular decynium-22 (1 μM), the most potent inhibitor of PMAT tested so far (Table 2). Much less MPP+ is released from vector-transfected cells, and decynium-22 had no inhibitory effect on these cells. When MPP+ retained within the cells was measured, a corresponding rapid decline of intracellular MPP+ was seen in PMAT-expressing cells (Fig. 9b). Decynium-22 markedly slowed down the disappearance of intracellular MPP+. In contrast, efflux was much slower in vector-transfected cells and decynium-22 had no effect on MPP+ efflux rate. Under the preloading conditions, there was more MPP+ preloaded into cells expressing PMAT than control cells transfected with vector (Fig. 9b, time 0); therefore, we calculated the percentage of MPP+ remaining within cells by normalizing cellular MPP+ to total preloaded MPP+. The results clearly revealed a specific transporter-mediated efflux of MPP+ in cells expressing PMAT (Fig. 9c), which is inhibitable by the PMAT inhibitor decynium-22. Together, these data demonstrated that PMAT was able to mediate cellular efflux and thus could transport substrate in both directions.

Structures of various compounds tested for interactions with PMAT.

Relationship between calculated hydrophobicity (Log P) and the measured Km (○) or Ki (•) values of various compounds. The solid line depicts the linear regression (r = 0.74) when TEA, MPP+, and rhodamine123 are excluded.
Discussion
Many endogenous and exogenous organic cations are eliminated from the body by polyspecific membrane transporters known as the OCTs. We recently reported the cloning and characterization of a new transporter PMAT (or ENT4) that belongs to the equilibrative nucleoside transporter family. Unlike other members of the ENT family (i.e., ENT1-3), which specifically transport nucleosides and nucleoside analogs (Kong et al., 2004; Baldwin et al., 2005), PMAT is a Na+-independent and membrane potential-sensitive transporter that transports MPP+ and the monoamine neurotransmitters. Because these compounds are positively charged at physiological pH and are the known substrates for the OCTs, we hypothesized that PMAT, like the OCTs, may function as a polyspecific transporter. In this study, we tested this hypothesis by investigating the interaction of PMAT with a series of structurally diverse organic cations. We also explored the structural features of the organic molecules that are important for interaction with the PMAT protein.
Our study revealed that PMAT is able to interact with various structurally diverse organic cations including many classic OCT substrates and inhibitors (Figs. 1 and 7, Table 2). These data suggest that although PMAT is evolved from a nutrient transporter family of narrow specificity, it has gained a diversified function and qualifies for the definition of being “polyspecific”. The functional characteristics of PMAT are strikingly similar to those of the OCTs. Many classic OCT substrates, including MPP+, TEA, guanidine, and histamine, are transported by PMAT (Fig. 4). OCT inhibitors, such as cimetidine and type II cations, are also PMAT inhibitors (Fig. 1 and Table 2). An analysis of the common structural features of PMAT substrates and inhibitors revealed that the transporter requires a positive charge and a hydrophobic mass for optimal interactions (Figs. 7 and 8, Table 2). These features, i.e., charge and hydrophobicity, have also been identified as the key determinants for substrate/inhibitor binding to the OCTs (Zhang et al., 1999; Bednarczyk et al., 2003; Suhre et al., 2005). Taken together, our data suggest that the functional features of PMAT are highly similar to those of the OCTs. Because of the large overlap of substrates and inhibitors between PMAT and the OCTs, caution should be taken when interpreting organic cation transport data obtained from tissues that coexpress PMAT and the OCTs. It is also interesting to point out that the human and rat OCT1 has been reported to be able to transport certain nucleoside analogs such as deoxytubercidin and acyclovir (Chen and Nelson, 2000; Takeda et al., 2002). Therefore, some intrinsic similarities may exist between the binding sites of the OCTs and ENTs.

Efflux of MPP+ by PMAT and vector-transfected cells. After 2-h incubation in [3H]MPP+ (1 μM), efflux from PMAT-expressing cells (•, ○) and vector-expressing cells (▴,▵) into media was measured in the absence (•, ▴) or presence of 1 μM decynium-22 (○,▵). a, MPP+ released from cells into media. b, MPP+ retained within the cells. c, percentage of MPP+ left in cells after normalizing to total preloaded MPP+.
Among the three OCT isoforms, there are significant specificity and kinetic differences in substrate and inhibitor recognition (Dresser et al., 2001; Koepsell et al., 2003; Wright and Dantzler, 2004). Substantial species differences for each OCT isoform have also been documented (Dresser et al., 2000; Wright and Dantzler, 2004; Suhre et al., 2005). Because detailed kinetic information is lacking for many OCT substrates and there is considerable variability in the reported kinetic parameters (Wright and Dantzler, 2004), it is not possible to quantitatively compare PMAT with each individual OCT isoform. In general, MPP+ is a universal substrate for all three OCT isoforms and is transported by human OCTs with comparable affinities (Km = 15-47 μM) (Grundemann et al., 1999; Dresser et al., 2001; Koepsell et al., 2003). TEA is a good substrate for human OCT1 (Km = 229 μM) (Zhang et al., 1998) and OCT2 (Km = 76 μM) (Gorboulev et al., 1997), but for human OCT3, TEA is not (Grundemann et al., 1998) or is a poor substrate (Wu et al., 2000). Guanidine has been suggested to be selectively transported by OCT2 and choline selectively transported by OCT1 (Grundemann et al., 1999). Histamine is transported by human OCT2 and OCT3 with Km values of 1300 and 180 μM, respectively (Busch et al., 1998; Grundemann et al., 1999). Our data suggested that, like the OCTs, MPP+ is the best substrate identified so far for PMAT (Table 1) and is transported by PMAT with an affinity (Km = 33 μM) comparable with that of the OCTs. TEA and guanidine are recognized by PMAT as substrates but are not transported very efficiently (Fig. 4 and Table 1). Choline seems not to be transported by PMAT, although the high background transport of choline in MDCK cells may mask the activity by PMAT (Fig. 4). PMAT transports histamine efficiently despite a low affinity (Km = 10.5 mM) (Figs. 4 and 5, Table 1). Although these patterns are similar to those of OCT3 to some extent, we have previously reported substantial functional difference between PMAT and OCT3 in interacting with monoamine neurotransmitters (Engel et al., 2004). In particular, PMAT favors serotonin and dopamine over norepinephrine and epinephrine (Engel et al., 2004) whereas an opposite pattern was observed for human OCT3 (Grundemann et al., 1998). In terms of interacting with inhibitors, most inhibitors (decynium-22, quinine, quinidine, verapamil, etc.) exhibited similar or close inhibitory potencies (Ki or IC50) toward PMAT and OCTs. So far, only corticosterone can differentiate PMAT from the OCTs (Ki = 450 μM for PMAT versus IC50 < 35 μM for human OCT1-3) (Table 2) (Hayer-Zillgen et al., 2002). Taken together, these data suggest that although PMAT displays a general functional similarity to the OCTs, it processes unique functional characteristics and does not behave exactly like any of the OCT isoforms.
An analysis of the structural-activity relationship of all tested compounds indicated that a net positive charge and a hydrophobic mass are the principal determinants for substrate/inhibitor interaction with PMAT (Table 2, Figs. 7 and 8). These data suggest substrate recognition may involve a negatively charged residue(s) and hydrophobic residue(s) located in the substrate binding sites of PMAT. It is interesting that an aromatic planar structure seems to be needed for high affinity interaction with the PMAT transporter. All high-affinity PMAT substrates (e.g., MPP+, tyramine, dopamine, serotonin) possess at least one aromatic ring. On the other hand, cations without a planar hydrophobic surface (e.g., TEA, guanidine, choline) are either low-affinity substrates or do not interact with PMAT (Table 2, Figs. 7 and 8). These data suggest that high-affinity binding to the PMAT protein may involve π-π interaction with aromatic residues (e.g., tryptophan or tyrosine) of the transporter. Although hydrophobicity is important for binding (Fig. 8), a balance between hydrophobicity and hydrophilicity seems to be critical for subsequent substrate translocation and dissociation. Highly hydrophobic cations may bind to the transporter too tightly to be translocated and/or released efficiently. For example, type II cations such as verapamil are potent inhibitors but are not substrates of PMAT (Figs. 2 and 4). The apparent affinity of PMAT for tyramine (Km = 283 μM) is comparable with that of dopamine (Km = 329 μM) suggesting that the 3-hydroxy-group on the phenyl ring is not essential for substrate recognition. Although both histamine and TEA are low-affinity substrates, histamine is transported with much higher Vmax than TEA (Table 1). This may reflect a difference of histamine and TEA translocation by the PMAT transporter, and the smaller histamine may be more efficiently translocated than the bulkier TEA.
trans-Stimulation phenomena have been observed for a number of transporters including the OCTs (Busch et al., 1998; Zhang et al., 1999; Wright and Dantzler, 2004), where the presence of a substrate on the opposite side (trans) of the membrane stimulates the transport of a probe substrate. After binding, translocation and release of its substrate at the intracellular side, the transporter needs to reorient its substrate binding site toward the extracellular side for another round of transport. trans-Stimulation occurs if countertransport of a substrate from the trans-face accelerates the in-to-out reorientation of the substrate binding site. Our data showed that preloading cells with PMAT substrates (MPP+, dopamine, serotonin, histamine, and tyramine) significantly stimulated the uptake of MPP+ by PMAT (Fig. 3). TEA and guanidine, which are PMAT substrates as revealed by the tracer flux data, exhibited no trans-stimulatory effect, suggesting that the TEA- or guanidine-loaded transporter does not reorient any faster than the unloaded transporter. This is consistent with our kinetic studies, which revealed that TEA is transported at a lower efficiency than substrates that trans-stimulate PMAT (Table 1). Quinidine, quinine, and verapamil, which are high affinity PMAT inhibitors, strongly trans-inhibited MPP+ uptake by PMAT. These type II cations are also potent inhibitors of the OCTs and similar trans-inhibitory effects have been observed for type II cations on OCT1 (Zhang et al., 1998, 1999) and OCT3 (Martel et al., 2001). The trans-inhibitory effect may suggest that when loaded with bulky, hydrophobic compounds, the transporter undergoes a conformational change resulting in no or a much slower translocation than the unloaded transporter. Alternatively, these hydrophobic compounds may bind tightly to the transporter and were not washed off during the experimental procedures, or the preloaded compounds may diffuse across the membranes, resulting in cis-inhibition. The finding that PMAT substrates trans-stimulate MPP+ uptake indicates that PMAT is able to transport substrate in both directions. Our efflux study further demonstrated that PMAT could mediate cellular efflux of its substrate (Fig. 9). Together, our current and previous investigations suggest that PMAT operates as a facilitated carrier and that its transport direction is determined by the combined effects of transmembrane substrate concentration gradient and the physiologic inside negative membrane potential.
In summary, our study revealed that the newly cloned PMAT can function as a polyspecific transporter that interacts with many structurally diverse organic cations. The general functional characteristics (i.e., substrate and inhibitor profiles and mechanism of transport) are strikingly similar to those of the OCTs. PMAT may play a significant role in organic cation transport in vivo.
Acknowledgments
We thank Dr. Wendel L. Nelson and Thomas F. Kalhorn for valuable discussion on the physiochemical properties of compounds used in this study and the use of the SciFinder Scholar Program.
Footnotes
-
This study was supported by National Institutes of Health grants GM66233 and CA099026.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.105.016832.
-
ABBREVIATIONS: OCT, organic cation transporter; ENT, equilibrative nucleoside transporter; MPP+, 1-methyl-4-phenylpyridinium; TEA, tetraethyl ammonium; PMAT, plasma membrane monoamine transporter; KRH, Krebs-Ringer-Henseleit buffer; MDCK, Madin-Darby canine kidney.
- Received July 14, 2005.
- Accepted August 11, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}