


Abstract
Voltage-gated sodium (Na+) channels are targets for local anesthetic (LA) drugs that bind in the inner pore of the channel with affinities related to the channel gating states. Our core model of the sodium channel (P loops and S5 and S6 segments from each of the four domains) was closed because it was developed using coordinates from the KcsA channel crystallographic structure. We developed a model of the activated, open channel based on the structure of the open MthK channel, which was characterized by bends at the S6 glycine or serine residues. This created a conformation that allowed energetically appropriate docking of the LA drugs. The alkylamino head of ionizable LA molecules was docked closer to the selectivity filter and in association with Phe-1579 of IVS6 and Leu-1280 of IIIS6 (Nav1.4), and the aromatic ring interacted with Tyr-1586 of IVS6 and Asn-434 of IS6. Comparison of multiple LA drugs showed relative binding affinities in the model consistent with experimental studies. The ionizable LA alkylamino heads interact primarily by van der Waals forces that position the charge so as to create a positive electrostatic barrier for cation permeation. Permanently uncharged benzocaine could be docked in the closed conformation as well, stabilizing the closed conformation. The structurally different anticonvulsant lamotrigine and one of its derivatives have a binding site that fully overlaps with that of the LA drugs. The open, activated channel creates the high-affinity binding site for these sodium channel blocker drugs, and block may be mainly electrostatic.
Local anesthetic (LA) drugs prevent and relieve pain by interrupting nerve excitation and conduction by direct interaction with voltage-gated Na+ channels to block the Na+ current (Butterworth and Strichartz, 1990; Catterall and Mackie, 1996; Hille, 2001). There are at least three unique human central nervous system Na+ channel isoforms, two peripheral nerve isoforms, an adult skeletal muscle isoform, and a cardiac isoform that is also expressed in some areas of the brain and in embryonic skeletal muscle (Catterall et al., 2003). The Na+ channel is a multimolecular complex. A large α-subunit, composed of approximately 2000 amino acids, forms the channel's pore and gating machinery and contains the principal drug binding sites. This α-subunit is composed of four highly homologous domains (I–IV), each with six presumably transmembrane α-helical segments (S1–S6) (Fozzard and Hanck, 1996; Catterall, 2000), assembled in a clockwise pattern around the central pore (Dudley et al., 2000). The pore itself has a fairly large outer vestibule, formed by the four extracellular P loop segments between each S5 and S6. These P loops fold back into the membrane to make a shallow funnel, with the innermost four residues (the DEKA motif) located at the turn-strand junction forming the selectivity filter. The filter is the narrowest region of the pore and is approximately 4 × 6 Å (Hille, 2001). The remainder of the pore below the filter is lined with residues of the converging S6 segments of the four domains.
The core S5-P-S6 part of the Na+ channel has been modeled with presentation of the P loop as a α-helix-turn-β-strand motif by Lipkind and Fozzard (2000), based on the X-ray crystal structure of the KcsA channel (Doyle et al., 1998). The channel's activation gate in this closed channel structure is located near the inner mouth of the pore, and it includes hydrophobic residues from the four S6 segments. Gating of the channel is initiated by movement of the four charged S4 segments in the membrane electric field, leading to opening of the activation gate (Hille, 2001).
LA drugs block the Na+ channel pore, accessing their binding site in nerve and skeletal muscle mainly or entirely from the cytoplasm through the inner pore (Narahashi and Frazier, 1971). They contain hydrophilic and hydrophobic domains that are separated by an intermediate linker containing an amide or an ester group (Fig. 1). The hydrophobic part includes an aromatic ring, whereas the hydrophilic part is usually a tertiary amine, sometimes alone and sometimes in a piperidine ring (Courtney and Strichartz, 1987). The presence of 2,6-methyl groups at the aromatic ring, aliphatic substituents at the amine group, and short intermediate chains enhance LA affinity (Ehring et al., 1988; Sheldon et al., 1991). The pKa of the LA tertiary amines range from 8 to 9. At physiological pH (7.4), they are mostly protonated and soluble in lipid membranes (Catterall and Mackie, 1996), so they can access the inner pore from the outside solution. The most commonly used LA drugs today are lidocaine, tetracaine, and bupivacaine, with some others specially suited to ophthalmic or dental use.
The Catterall laboratory has systematically substituted the residues of the four S6 segments in the Nav1.2 α-subunit isoform by alanine, which should function as a simple spacer (Ragsdale et al., 1994; Yarov-Yarovoy et al., 2001, 2002), and determined the effects on etidocaine block using three protocols to determine rested state, inactivated state, and use-dependent block (UDB). Etidocaine differs from lidocaine in having an ethyl side chain in the intermediate linker (Fig. 1), and it also has a higher affinity. Domain IV S6 mutations showed the largest changes in blocking affinity. Etidocaine had an inactivated state blocking IC50 of approximately 1 μM. In the resting state, some mutations showed a modest increase in block, but the affinity was more than 100-fold less than for open/inactivated block. I1760A, F1764A, and Y1771A showed reduced block in the inactivated state. Upon further study, I1760A had an increase in both onset rate and offset rate, leaving the affinity unchanged. F1764A had an IC50 of approximately 130 μM and a 20-fold increase in offset rate, which resulted in loss of UDB. Y1771 also had reduced block and an IC50 of approximately 35 μM (Ragsdale et al., 1994). Consequently, for Nav1.2, Phe-1764 and Tyr-1771 seem to be the important residues for open/inactivated state block. After a prolonged depolarizing pulse to test for inactivated state block, the mutations L1465A, N1466A, and I1469A of domain III S6 reduced block by 6- to 8-fold, but only L1465A and I1469A reduced UDB. No alanine mutations in IS6 and IIS6 affected the low-affinity rested state block, and only I409A (Nav1.2 numbering) had a small effect on inactivated state block (6-fold). Use dependence was evaluated by a 2-Hz train of 20-ms steps to 0 mV. The trains showed reduced block for I409A and abolition of UDB by N418A. Therefore, Asn-418 is important for interaction of etidocaine in the open/inactivated state.

Chemical structures of local anesthetics of the lidocaine family. The hydrophobic part contains the 2,6-dimethyl-substituted aromatic ring, whereas the hydrophilic part is a tertiary amine alone or a piperidine ring.
The lidocaine class of LA drugs have low affinity for Na+ channels in their closed, resting state, and much higher affinity in the open and/or inactivated states (Li et al., 1999; Hille, 2001). To consider the docking energies of a series of LA drugs, we first developed a model of the open Na+ channel, based on the MthK X-ray crystal structure, which is an open K+ channel (Jiang et al., 2002a). We then compared the docking energies of a series of structurally similar LA drugs, which differed only by alkyl substitution in the amino heads—lidocaine, etidocaine, tocainide, benzyl-tocainide, and the more conformationally restricted mepivacaine and bupivacaine (Fig. 1). This comparative analysis allowed us to identify nonbonded and electrostatic interactions with the Na+ channel pore that corresponded to the differences in drug affinities. We also found that the structurally different anticonvulsant lamotrigine and its derivative 227c89 were bound by the inner pore in a manner similar to the LA drugs. Some of these data have been previously reported in abstract form (Lipkind and Fozzard, 2005).
Materials and Methods
Modeling was accomplished in the Insight and Discover graphical environment (MSI, Inc., San Diego, CA), as described previously (Lipkind and Fozzard, 2000). Molecular mechanics energetic calculations used the consistent valence force field approximation. For minimization procedures, the steepest descents and conjugate gradients were used. Calculations of electrostatic potentials inside the pore of the Na+ channel core model was accomplished by the DelPhi model of Insight II, as described previously (Khan et al., 2002). The dielectric constants were set to 10 for the protein interior and to 80 for the solvent water. Electrostatic potentials are given in units of kT/e, where k is Boltzmann's constant, T is temperature in Kelvin, and e represents the elementary charge.
Results
Modeling the Na+ Channel Open State. Our previous model of the S5-P-S6 Na+ channel pore-forming component of Nav1.4 (Lipkind and Fozzard, 2000) used the backbone coordinates of the closed KcsA channel (Doyle et al., 1998) and an α-helix-turn-β-strand motif for the P loops to compose an energetically appropriate outer vestibule for binding of the guanidinium toxins and μ-conotoxin. This Na+ channel vestibule/selectivity region is wider than that of the KcsA P loop structure because of the need to accommodate these large toxin molecules within the vestibule itself and because of the roles of side chains instead of the main chain in forming the selectivity site. As a result, the structural motifs of the P loop main chains of Na+ and K+ channels are different. For Na+ channels, the P loops have inner turns exactly at the DEKA selectivity filter residues, and then they ascend back toward the extracellular side of the pore (Yamagishi et al., 1997; Lipkind and Fozzard, 2000). This is not the case for K+ channels, where the turn residues are located deeper than the extended selectivity filter itself. Docking of this vestibule structure into the S5–S6 teepee structure, which was based on the M1–M2 main chain coordinates, resulted in location of the Na+ channel selectivity ring at the level of Ile-1575 in IVS6 and between the side chains of Lys-1237 and Ala-1529 of the selectivity filter, so that the inner pore vestibule began at a higher level in the Na+ channel than in the K+ channel. The Protein Data Bank coordinates of the vestibule are included in the Supplemental materials.
The KcsA channel crystal structure is likely to be of a closed channel. A different structure has been determined for the homologous Ca2+-gated MthK channel that was crystallized in the presence of Ca2+, so that it was in the open state (Jiang et al., 2002a). In that structure the M2 helix is not straight. Rather, the lower half of the M2 helix is bent outward at a highly conserved glycine (Gly-83 in MthK or Gly-99 in KcsA). This glycine is a hinge that allows bending of M2 (Jiang et al., 2002b), resulting in a substantial rearrangement of the relationships between M1 and M2 residues below the hinge. The cloned Na+ channels have a glycine in similar locations in the S6 segments of domains I, II, and III, but in domain IV the equivalent residue is a serine. It is likely that serine would also allow hinging of the S6 similar to glycine. As we suggested previously (Sunami et al., 2004), the side chain hydroxyl of Ser-1578 (IVS6) is able to compensate for the broken main chain hydrogen bond inside of the straight closed conformation of the IVS6 α-helix by formation of a new hydrogen bond with the main chain carbonyl of Tyr-1574 in the open state.
LA drugs bind preferentially to the open state of the activation gate; consequently, we needed to modify our Na+ channel core S5-P-S6 model to correspond to an open conformation. Critical to formation of an LA binding site is the alignment of S6 residues from the four Na+ channel domains. To align the M2 transmembrane sequence of the MthK channel with the I–IVS6 segments of the Nav1.4 channel, we propose that residues interacting with LA drugs are directed inside the inner pore. Such an alignment maximizes the coincidence of the LA-sensing residues with the pore-facing residues of MthK (Ile-84, Ala-88, and Val-91, located below the hinge Gly-83). Based on homology, we explored the consequences of the alignment in Table 1. The MthK hinge location Gly-83 aligns with glycines in IIS6 and IIIS6, and with a serine in IVS6, which also aligns Phe-1579 with Ile-84 and Leu-1280 with Ala-88. These amino acid residues of Nav1.4 correspond to Phe-1764 and Leu-1465 of Nav1.2 (Ragsdale et al., 1994; Yarov-Yarovoy et al., 2001). The alignment for IVS6 is also supported experimentally by cysteine scanning and methanethiosulfonate reagent interaction (Sunami et al., 2004), where the cysteine mutant of Phe-1579, F1579C, was accessible to the permanently charged MTSET from the inside. If Gly-430 in IS6 is also aligned with the MthK Gly-83, then Asn-434, a residue thought to be part of the LA binding site would face into the protein. We therefore chose to align Ser-429 with the MthK hinge residue, so that Asn-434 (corresponding to the position of Val-91 of MthK) would be available within the pore for interaction with LA. In this arrangement, the hydroxyl side chain of Ser-429 compensates for its broken main chain hydrogen bond by a new one with the main chain carbonyl of Ile-425. The IS6 alignment was the only feature of the open channel model that was chosen because of reported interactions of inner pore residues with LA.
Alignment of S6 segments of Nav1.4 with M2 segments of MthK
In combination with our prior model (Lipkind and Fozzard, 2000), we used the main chain M2 MthK coordinates for the location of S6 segments, populated with the Nav1.4 residues, and energy optimized the residue interactions. As before, the separation of the P loops necessary to accommodate the guanidinium toxins resulted in a vestibule wider than those of K+ channels. This restricted the depth of the outer vestibule into the S5–S6 teepee, so that the selectivity ring of the Na+ channel is located at the level of Ile-1575. If it were as low as in the K+ channels, then the critical LA binding residue Phe-1579 would have been covered by the P loops.
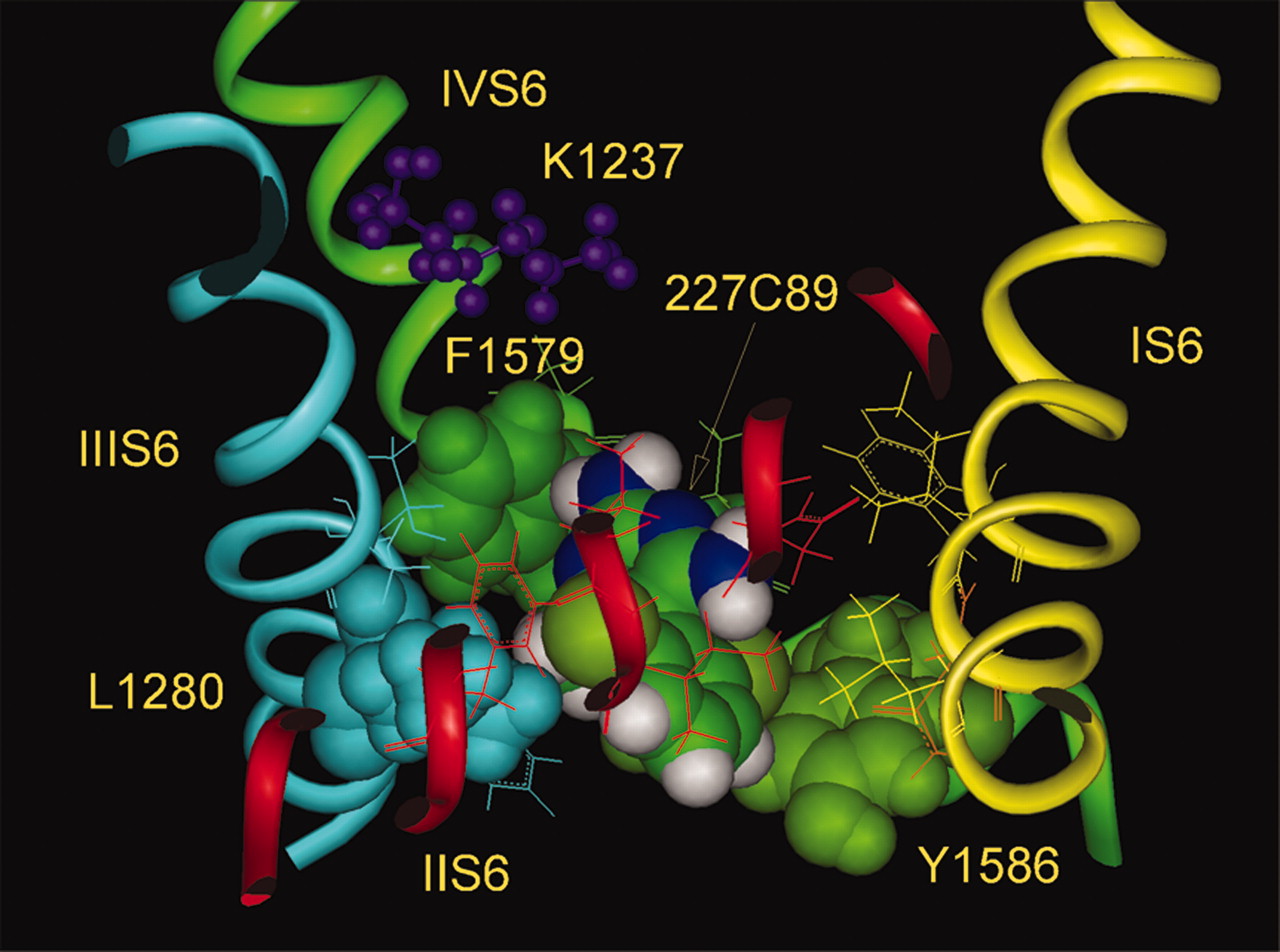
The LA Binding Site. Although some uncertainly remains for several residues, the main evidence favors the involvement of four residues in LA binding during activity: Phe-1579 and Tyr-1586 in IVS6, Leu-1280 in IIIS6, and Asn-434 in IS6, with Phe-1579 contributing the greatest amount of energy to the interaction (Ragsdale et al., 1994; Yarov-Yarovoy et al., 2001, 2002). Moreover, substitution of all four residues by alanine practically abolished UDB, underlining their participation in the LA open/inactivated state block. Location of these amino acid residues inside the inner pore is shown in Fig. 2. Additional amino acid residues are considered by Nau and Wang (2004), but they are not likely to interact directly with the LA drugs. For now, we assume that those mutations have their effect indirectly and that they are not part of the LA binding site.
The side chain of the key residue Phe-1579 forms a cover to the LA binding region and is located close to the side chain of Leu-1280. These two residue side chains are separated by a narrow cavity approximately 6 Å in width, which would be enough for docking of the alkylamine head of LA drugs. The bottom of the site is formed by the side chains of Tyr-1586 and Asn-434, and the distance from the top Phe-1579 to the bottom of the binding cavity is ∼10 Å. Despite their proximity to the proposed binding site, two amino acid residues, Val-1583 and Thr-1279, have been shown by alanine substitutions to produce no change in binding affinity. In this model, the side chain of Phe-1579 prevents immediate contact of LAs with the selectivity ring itself. Indeed, the selectivity ring residues seem to be protected from the inner pore: cysteine mutants of the selectivity filter residues interact with external Cd2+ or MTSEA, but they are not accessible to inside Cd2+ or MTSEA (Yamagishi et al., 1997).
Further modification was made to allow docking of LAs to be more straightforward. No evidence has been found for involvement of residues in IIS6 in LA binding. As with alanine-scanning mutagenesis, substitutions of lysine in IIS6 (N784K, L785K, V787K, and L788K) failed to affect binding of bupivacaine (Wang et al., 2001), allowing us to exclude IIS6 from the site. The lack of effect of these lysine mutations on affinity is a puzzling finding, and it may imply that the Na+ channel is asymmetrical, with IIS6 displaced away from the center of the pore. In our symmetrical model, we avoided any spurious interactions with the symmetrically placed IIS6 during the docking procedure by changing all of these inner pore residues to alanine. Restoration of the correct residues to IIS6 after the docking procedure was completed did not result in any change in the location of the LA molecules. In addition, Nau et al. (1999) reported that the mutant N434F improved binding of bupivacaine 6-fold, although its replacement by alanine had no effect (Yarov-Yarovoy et al., 2002), so IS6 Asn-434 was changed to phenylalanine in the model to decrease the conformational space for docking of the LA molecules.

Top view of the modeled binding site of local anesthetics inside the open pore of the Na+ channel (Nav1.4). The backbones for S6 helices of domains I–IV are shown as yellow, red, blue, and green ribbons, respectively. Amino acid residues distinguished by alanine scanning mutagenesis (Ragsdale et al., 1994; Yarov-Yarovoy et al., 2001) for direct interactions with molecules of local anesthetics—Phe-1579 and Tyr-1586 of IVS6, Leu-1280 of IIIS6, and Asn-434 of IS6, and some neighboring residues (Val-1583, Thr-1279, and Phe-1283) are shown by space-filled images. Ball and stick representations are shown for the four selectivity filter residues—Asp-400, Glu-755, Lys-1237, and Ala-1529 of the domains I–IV P loops.
For docking, we followed the original proposal of Ragsdale et al. (1994) that the alkylamine group is located higher, near Phe-1579 and close to the selectivity filter, and the aromatic ring is lower and close to Tyr-1586, near the activation gate in the open state. A number of experimental observations support this orientation. For example, the permanent charge on QX-314 is located deep in the membrane field, 70% of the transmembrane voltage from the inner mouth (Gingrich et al., 1993). Sunami et al. (1997) showed that lidocaine was electrostatically affected by the positively charged lysine in the selectivity filter, but neutral analogs were not affected, placing the amine end of lidocaine within approximately 10 Å of the domain III lysine of the selectivity ring. Substitution of lysine for Tyr-1586 and Asn-434 of Nav1.4 at the bottom of the proposed binding site produced rather small reductions in affinity (Wang et al., 1998b; Wright et al., 1998; Nau et al., 1999), whereas substitution by lysine for Phe-1579 and Leu-1280 at the top reduced affinity by approximately 25-fold (Wright et al., 1998; Nau et al., 2003). Two ortho-methyl groups increase the hydrophobicity of the aromatic ring of etidocaine, so the ring probably behaves more like an aliphatic than an aromatic group (Courtney and Strichartz, 1987), and it may not participate in aromatic-aromatic interactions with Tyr-1586. Mutational studies by Li et al. (1999) support this conclusion. They found that at the tyrosine site of Nav1.3 (1586 in Nav1.4) hydrophobic substitutions (by cysteine, isoleucine, or phenylalanine) had minimal effect on UDB by tetracaine. In contrast, the aromatic ring at the phenylalanine site (1579 in Nav1.4) seems to be essential, becauseit may be replaced by tyrosine or tryptophane without change in affinity but not by other amino acid residues (Li et al., 1999). This points to the aromatic ring of Phe-1579 as the most important interaction. One scenario is that the alkylamine head interacts with the aromatic ring of Phe-1579 in a cation-π electron relationship (Dougherty and Stauffer, 1990; Heginbotham and MacKinnon, 1992), but the role of aliphatic chains on the amine also suggests a hydrophobic interaction.
Docking of Mepivacaine. We begin by docking mepivacaine, because inclusion of its amino group within a bulky piperidine ring makes it the most conformationally restricted structure among the LA drugs (Fig. 3). The presence of bulky cyclic structures (with ortho-substituents) on both sides of mepivacaine practically excludes their rotation about the central amide bond of the linker. In contrast to lidocaine, mepivacaine (see also bupivacaine) contains an asymmetric center at C1 of the piperidine ring, forming two stereoisomers (enantiomers) R and S. Because rotation about the central amide bond cannot occur, each stereoisomer adopts a single conformation. If the C-H at C1 of the piperidine ring is directed downward, then substituents at C1 (amine, amide and alkyl groups) can be located clockwise (R-stereoisomer) or anticlockwise (S-stereoisomer). Their optimized structures are shown in Fig. 3. Docking of (R)- and (S)-mepivacaine produces very similar arrangements of the piperidine ring inside the groove between the side chains of Phe-1579 and Leu-1280, and the orientations are indistinguishable energetically (Fig. 4). During energetic minimization of the mepivacaine-pore complex, the locations of the side chains of Phe-1579, Tyr-1586, and Leu-1280 were also optimized. In the orientation shown, the methyl substituent of the piperidine ring faces the pore, but it also can fit into the groove. Alternative orientations are excluded for bupivacaine, which has the long butyl substituent on the ring. Usually the affinities of binding of pure stereoisomers of LAs are not significantly different. Although the data are not available for mepivacaine, affinities for the two enantiomers of bupivacaine are practically the same (Nau et al., 2003). Energy optimization determined that the energies of van der Waals (nonbonded) interactions between mepivacaine and the side chains of Phe-1579 and Leu-1280 yielded -3.7 and -2.6 kcal/mol, respectively. The optimal docking of the piperidine ring in the groove between these side chains automatically locates the dimethyl-substituted aromatic ring in the immediate proximity of the side chains of Tyr-1586 of IVS6 and Phe-434 (which replaced Asn-434) of IS6. The location identified for the aromatic ring using the rigid structure of mepivacaine was then used as an initial approximation of the location of this ring for the conformationally flexible alkylamine heads of etidocaine and lidocaine. The Protein Data Bank coordinates for interaction of (R)- and (S)-mepivacaine with the inner pore are provided as Supplemental material.
Docking of Etidocaine and Lidocaine.Figure 5 (top) shows the side view of the open channel model after docking of etidocaine into the pore. In the optimal arrangement, the ethyl branch at the inner linker of etidocaine (Fig. 1) was directed to the bottom of the groove between the side chains of Phe-1579 and Leu-1280, so that the hydrophobic segment faced away from the pore. In the initial arrangement, the dimethyl-substituted aromatic ring was located in proximity to the side chains of Tyr-1586 and Phe-434, similar to that found for mepivacaine. Energy minimization was made with this orientation. As a further check of this interaction, a molecular dynamics run failed to change the optimal location of etidocaine. In this orientation, the aromatic ring of Phe-1579 forms immediate van der Waals contacts with the surface of etidocaine that includes the N-H bond and the ethyl substituent of the amine group, and also the ethyl at the linker branch, corresponding to a van der Waals energy of -5.1 kcal/mol. The side chain of Leu-1280 interacts with the latter ethyl and the propylalkyl chain at the amine head. The dimethyl-substituted aromatic ring is located near Tyr-1586, but the location of the Asn-434 (replaced by phenylalanine in the calculation) did not constrain etidocaine. Interactions with Leu-1280 and Tyr-1586 were weaker, with estimated energies of approximately -2.5 to -3 kcal/mol. Substitution in the model of alanine for Phe-1579 reduced the interaction with etidocaine by ∼3 kcal/mol, as expected for the experimental F1764A mutation (Nav1.2) of 135-fold (Ragsdale et al., 1994). In contrast, alanine substitution in the model for the other interacting residues (Leu-1280 and Tyr-1586) reduced etidocaine binding by ∼1 to 2 kcal/mol each, corresponding to the experimental loss of affinity of 10- to 30-fold (Ragsdale et al., 1994; Yarov-Yarovoy et al., 2001). It is important to note that the model of the inner pore was made independently of known LA interactions. The model predicted accurately the changes in energy of interaction from mutations in the site, but note that LA affinities are rather low and that there are also multiple small interactions that contribute to its binding.

Three-dimensional molecular structures of (R)- and (S)-mepivacaine, displayed by stick and space-filled images.
Lidocaine structure resembles that of etidocaine in important ways. The dimethyl-substituted aromatic ring and the amide linker are the same. Retaining their locations found for etidocaine, we rotated the diethyl amine head around its C-N linker bond. The optimal arrangement, excluding prohibited contacts within the groove, was derived (Fig. 5, middle). The groove itself easily adapts to the interposition of the diethylamine head, producing van der Waals contacts with both ethyl groups, whereas the N-H bond is directed into the pore. This is in accord with the report of Sheldon et al. (1991), who showed that alkyl substitutions of the amine group improved binding of LAs. At the same time, the energy of nonbonded interactions of lidocaine with the side chain of Phe-1579 is less (-4.1 kcal/mol) than that for etidocaine. Possible electrostatic interactions of the amino heads with small partial charges on the aromatic ring side chains are the same for both LAs (approximately -0.5 kcal/mol in this estimate). We suggest that the higher affinity of etidocaine is the result of additional stabilizing energy of van der Waals interactions primarily between the side chain of Phe-1579 (and also with Leu-1280) and the ethyl branch at the inner linker of etidocaine. Comparison of lidocaine in the binding site with mepivacaine, which provided the initial coordinates for docking, is shown in Fig. 5 (middle and bottom). The Protein Data Bank coordinates for etidocaine and lidocaine, and other docking conformations discussed subsequently, can be provided on request.

Mepivacaine. Optimal arrangement for (R)-mepivacaine (top), shown by space-filled image, inside the inner open pore of the Na+ channel (top view). The rigid piperidine ring of (R)-mepivacaine fills the groove between the side chains of Phe-1579 (IVS6) and Leu-1280 (IIIS6) (also shown by space-filled images), whereas the dimethyl-substituted aromatic ring is in proximity to the side chain of Tyr-1586. The side chain of Phe-1579 forms the top of the binding site and its location provides the most optimal interaction (van der Waals and electrostatic) with mepivacaine. Optimal arrangement of S-mepivacaine (bottom) inside the pore (top view) is energetically indistinguishable from the arrangement found for (R)-mepivacaine. Ball and stick representation (violet) is given for Lys-1237 of the selectivity filter.
Test of Etidocaine-Lidocaine Prediction. There are published data on the WT Nav1.2 binding affinities of both etidocaine and lidocaine and for the mutants F1764A and Y1771A (Ragsdale et al., 1994, 1996). Lidocaine has an IC50 of approximately 11 μM for the wild-type channel. F1764A showed a 25-fold increase in IC50, and Y1771A showed a 13-fold increase (Ragsdale et al., 1996). Lidocaine is different from etidocaine only by the absence of the ethyl substituent in the linker. This could be considered as a mutant of etidocaine, permitting calculation of mutant cycles (Fig. 6). The coupling energy (ΔG = RTlnΩ, where Ω is the coupling constant; Hidalgo and MacKinnon, 1995) for interaction with Tyr-1771 is very small (0.5 kcal/mol), whereas for Phe-1764 the coupling energy is 1 kcal/mol (Fig. 6). Therefore, Phe-1764 (Phe-1579 of Nav1.4) is sensitive to the structural differences between etidocaine and lidociane, whereas Tyr-1771 (Tyr-1586 of Nav1.4) is not. This suggests that the identical aromatic rings of the two drugs are closer to Tyr-1771 and the differing alkylamine heads are closer to Phe-1764, consistent with the original suggestion of Ragsdale et al. (1994) and the arrangement shown in Fig. 5.

Etidocaine, lidocaine, and (R)-mepivacaine. Optimal arrangement of drugs inside the inner pore of the Na+ channel (side view). The initial approximation for location of the dimethyl-substituted aromatic ring of etidocaine (top) was taken from that found for the “rigid” mepivacaine (bottom). The alkylamine head and the ethyl group at the linker branch produced the most optimal interactions with the side chains of Phe-1579 and Leu-1280, and consequently the lowest observed values of Kd for local anesthetics (Ragsdale et al., 1994). A similar approximation was used for docking of lidocaine (middle). Compare docking of lidocaine and mepivacaine with their diethylamine and piperidine heads, respectively.
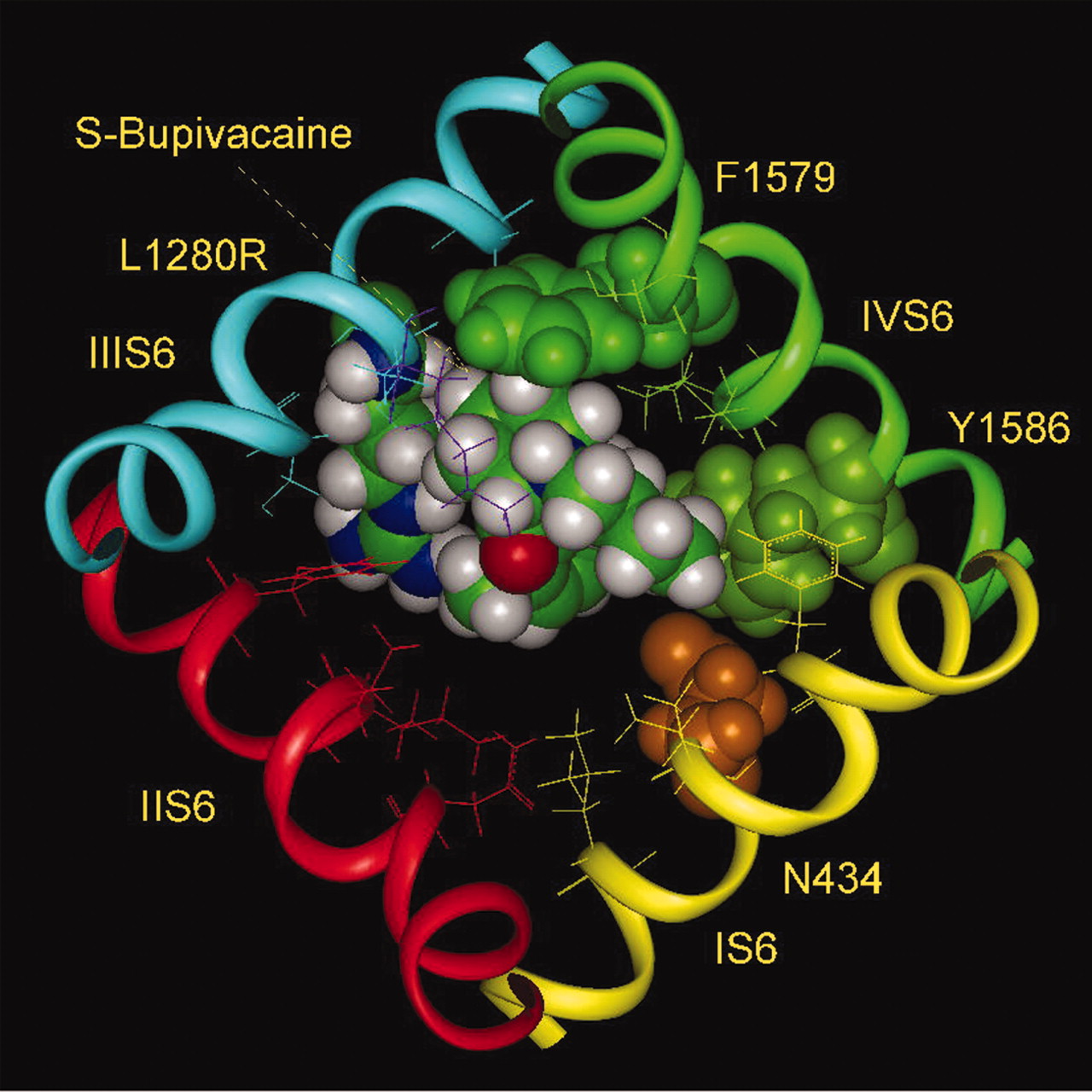
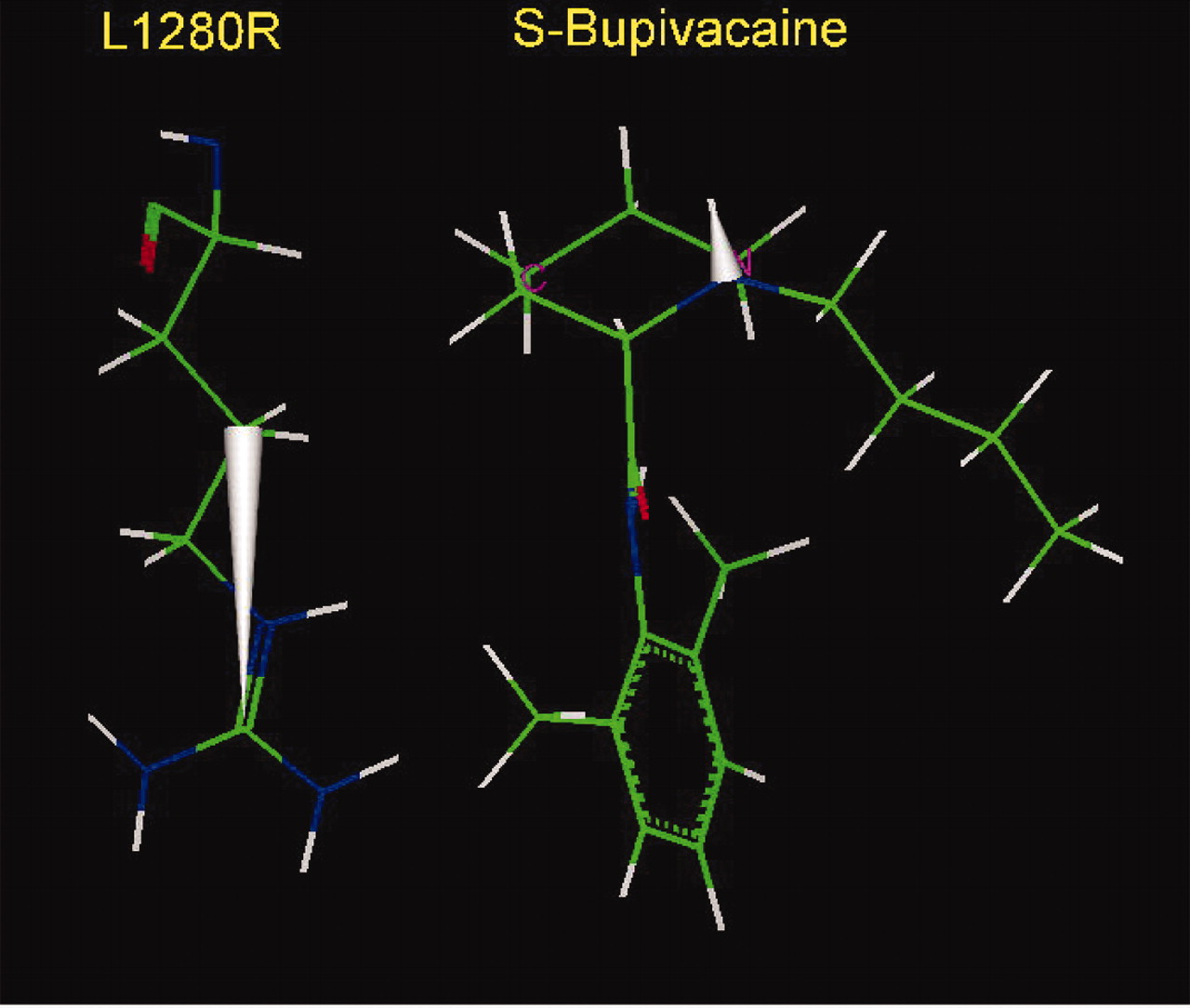
Docking of Bupivacaine. The model of bupivacaine binding is similar to that of mepivacaine, with the N-methyl group replaced by an N-butyl group. Although both R- and S-stereoisomers of bupivacaine bind with similar affinity to WT Nav1.4 channels, substitution of Leu-1280 by Arg introduced stereoselectivity. S-Bupivacaine interacted with the mutant channel with affinity similar to WT, but the R-stereoisomer affinity was reduced by 15-fold (Nau et al., 2003). At the same time, the L1280K mutant reduced binding affinity of both enantiomers by 20- to 25-fold. The lower affinity for lysine is plausibly the result of electrostatic effect on the positively charged amino group of the piperidine ring, but the difference in effect between lysine and arginine implies that arginine is different. Perhaps the guanidinium ring of arginine at position 1280 can have a specific interaction with the aromatic ring of bupivacaine, offsetting the electrostatic repulsion between the charges of arginine and the amino group of the piperidine ring.
The amino groups of the resonating side chains of asparagine and glutamine have a tendency to point to the center of aromatic rings (Levitt and Perutz, 1988), resembling a hydrogen bond. The amino group of the resonating guanidinium group of arginine may participate in a similar interaction. Protein structural analysis also shows that the rings of aromatic side chains have a high tendency to be located in close contact with the guanidinium group of arginine, adopting either perpendicular or parallel stacking (Flocco and Mowbray, 1994). Even the simple estimate of the energies of nonbonded interactions of the side chain at position 1280 with S-bupivacaine (Fig. 7) shows that the energy is lower by 2 kcal/mol for arginine than for the WT leucine. This easily compensates for the electrostatic repulsion with the amino group, which can be estimated also to be approximately 2 kcal/mol on the basis of experimental data on substitution of Leu-1280 by lysine (Nau et al., 2003). It is therefore not surprising that the affinities of binding of S-bupivacaine by WT and its mutant L1280R are very close. An alternative or supplementary explanation for the stereoselectivity of bupivacaine is the direction of the N-H dipole of the alkylamine head. In the S-stereoisomer optimal orientation the positive end of the dipole is directed upward, but for the R-stereoisomer it is directed downward, as also demonstrated in the docking model for mepivacaine (Fig. 4), which creates an antiparallel arrangement of dipoles of the guanidinium and N-H groups, in the first case (Fig. 8) and parallel arrangement for the second. The dipole-dipole interactions introduce electrostatic repulsion between a downward-directed N-H drug dipole relative to the guanidinium group at position 1280. In contrast, the dipoles would be stabilizing with the drug dipole in the upward direction (Figs. 7 and 8). The energy of different orientations of the N-H dipole could be estimated to be approximately 1 kcal/mol on the basis of the affinities of binding of (R)- and (S)-bupivacaine by the L1280R mutant (Nau et al., 2003).

Mutant cycle analysis of binding of etidocaine and lidocaine with the wild-type Na+ channel and its mutant F1764A (Nav1.2). The changes in the binding affinity with each mutation are shown at the upper and lower perimeter (Ragsdale et al., 1994, 1996). The ratio of changes determines the coupling constant Ω and the coupling energy ΔG(ΔG = RT lnΩ, in kilocalories per mole). This example of mutant cycle analysis shows a 1 kcal/mol improvement of binding of etidocaine relative to lidocaine by the side chain of Phe-1764, whereas for the side chain of Tyr-1771 the change is negligible.

S-Bupivacaine. The optimal arrangement of S-bupivacaine in the inner pore of the Na+ channel (Nav1.4) with the mutant L1280R of IIIS6 (top). The side chain of arginine at the position 1280 participates in stabilizing van der Waals and electrostatic interactions with the aromatic ring of bupivacaine, compensating for the electrostatic repulsion of arginine with the amino head of bupivacaine. Consequently, the binding affinities of S-bupivacaine with WT channel (Nav1.4) and its L1280R mutant are similar (Nau et al., 1999). Note the upward direction of the N-H dipole of the amino head of S-bupivacaine relative to the guanidinium group of the side chain of arginine.

S-Bupivacaine. Relative location of S-bupivacaine and the side chain of arginine, substituted for Leu-1280 of IIIS6. This figure underlines also an antiparallel arrangement of dipoles of the guanidinium group of arginine and the N-H bond of the alkylamino head of S-bupivacaine, shown by the solid silver cones.

Chemical structures of tocainide and benzyl-tocainide.
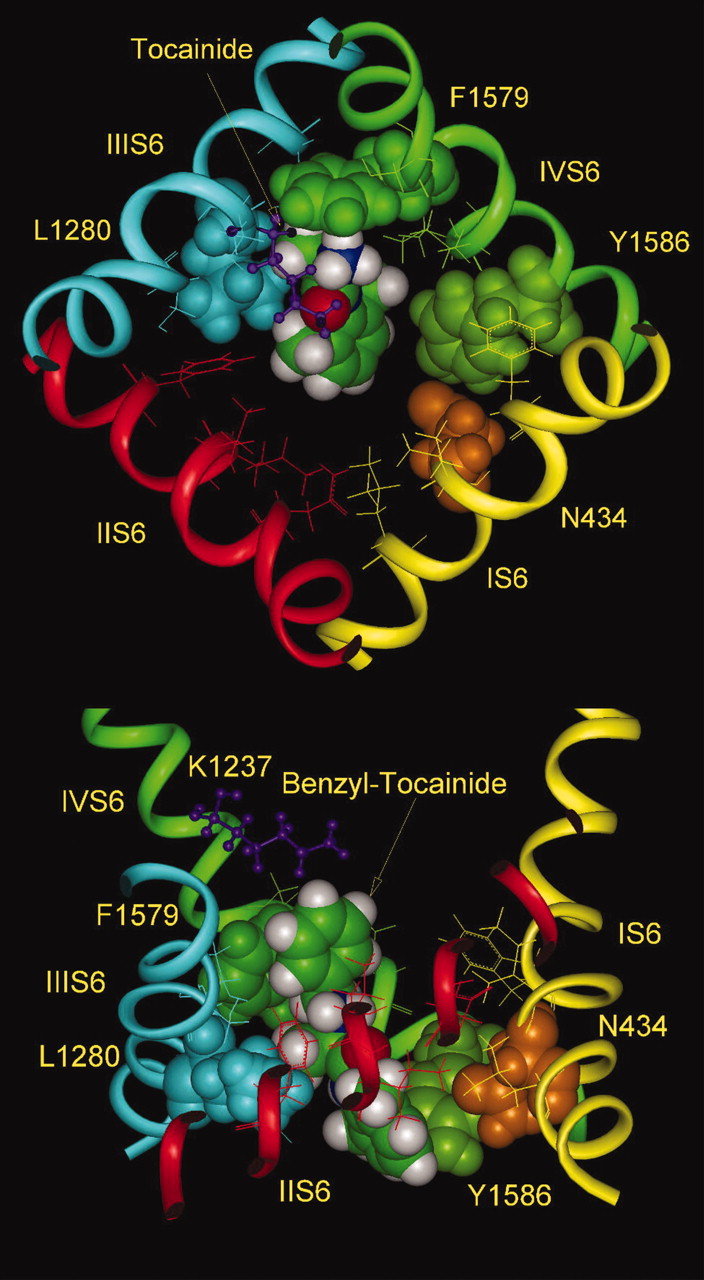
Tocainide and benzyl-tocainide. Optimal arrangement of tocainide (top view) and benzyl-tocainide (side view) inside the inner pore of the Nav1.4 channel. The absence of alkyl substituents at the amino head of tocainide led to reduction in the van der Waals interactions with the side chain of Phe-1579. In benzyl-tocainide, the benzyl group of LA adopts an off-centered parallel orientation relative to the aromatic ring of the side chain of Phe-1579. The additional energy of interaction provides approximately 3 kcal/mol, explaining the higher binding affinity of benzyl-tocainide by 100-fold, relative to tocainide (De Luca et al., 2003).
Docking of Tocainide and Benzyl-Tocainide. De Luca et al. (2003) have presented data for block of the inactivated Nav1.4 channel by tocainide and its benzyl derivative on the amine head (Fig. 9). Benzyl-tocainide was more than 100-fold more potent than tocainide (Ki = 1.1 and 115 μM, respectively). Comparison of docking of these two molecules suggests that the benzyl group interacts with Phe-1579 in an off-centered parallel orientation of the two aromatic rings (McGaughey et al., 1998) provided additional stabilization of binding through nonbonded interaction equal to approximately 3 kcal/mol (Fig. 10). It seems that the absence of alkyl substituents is the main factor in reduced affinity of tocainide. The energy of van der Waals interactions of benzyl-tocainide with Phe-1579 is -5.8 kcal/mol, the lowest one among the derivatives we studied (Fig. 1).
Docking of the Anticonvulsant Lamotrigine and a Derivative, 227c89. Voltage-gated sodium channels are also targets for chemically diverse anticonvulsant drugs such as lamotrigine, which show blocking properties similar to those of LA drugs (Ragsdale et al., 1996; Yarov-Yarovoy et al., 2001; Liu et al., 2003). Some overlap in binding sites is likely to exist despite the difference in drug structure, because some of the same channel residues are involved in binding of both classes of drug. Alanine scanning mutagenesis of segments IIIS6 and IVS6 identified Phe-1764 and Tyr-1771 (Nav1.2) as important residues for binding of lamotrigine and its derivative 227c89. An important difference from the pattern with LA drugs, however, is that the effect of F1764A is much smaller (7- to 8-fold) than for etidocaine or lidocaine, and the effect of the mutation Y1771A is greater (12- to 16-fold) (Liu et al., 2003). For IIIS6 the mutation L1465A (Nav1.2) the reduction in affinity for lamotrigine and 227c89 is approximately the same as for etidocaine and lidocaine (4- to 8-fold) (Yarov-Yarovoy et al., 2001). Consequently, the sites seem to overlap, but with significant differences in the binding interactions.
Both drugs consist of a dichlorophenyl ring, connecting to another nitrogen-containing aromatic ring (Fig. 11), and both the triazine ring of lamotrigine and the pyrimidine ring of 227c89 also contain an additional amino group in the para-position. The delocalization of the lone-pair electrons at the nitrogen of this group and the resonance with the π-cloud of the aromatic rings induces a positive charge at the nitrogens, which is much stronger because of the higher electronegativity of the nitrogens inside the aromatic rings. The special role of the resonating para-amino group is demonstrated by the observation that its substitution by a methyl group resulted in an inactive derivative of lamotrigine (Clare et al., 2000). Therefore, it is logical to arrange the partial positive charge of the para-amino group closer to the side chain of Phe-1579, whereas the dichlorophenyl moiety is in proximity to Tyr-1586 (Nav1.4). The alkyl groups of the LA drugs would then have a stronger interaction with the aromatic ring of Phe-1579, as demonstrated by the greater fall in affinities of the LA drugs upon mutation of the residues to alanine.
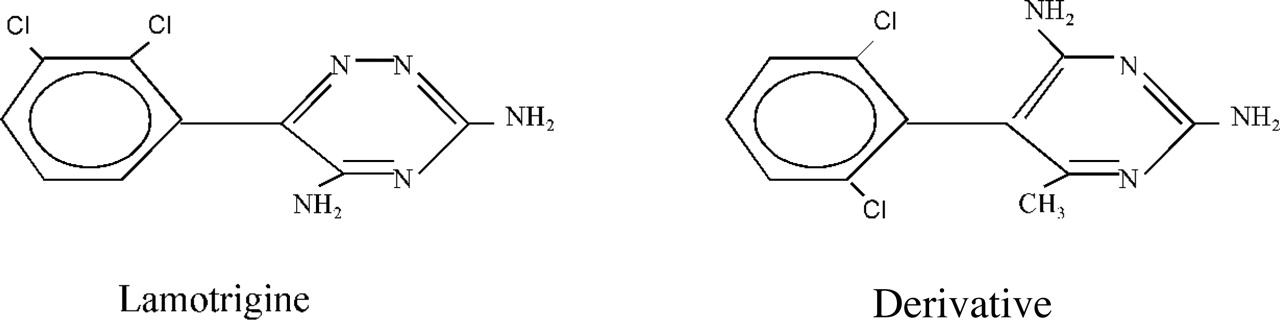
Chemical structures of the anticonvulsant lamotrigine and its derivative.
Symmetrical location of two chloro substituents on the phenyl group of 227c89 makes arrangement of this ring less questionable. The simultaneous presence of the ortho-substituents in both the phenyl and the pyrimidine rings results in the rings being perpendicular to each other and unable to rotate (Fig. 11). The optimal docking of 227c89 (Fig. 12) results in the para-amino group in immediate contact with the side chain of Phe-1579 at the top of the binding site, whereas the dichlorophenyl moiety interacts with the side chain of Tyr-1586 and the atom of chloride is closer to the partial positive charges of the aromatic ring hydrogens. In this arrangement the methyl substituent of the pyrimidine ring is located in the inner groove between the side chains of Leu-1280 and Phe-1579, whereas the amino group, also in the ortho-position of the same ring, faces the pore and could repel Na+ permeation. On the whole, the size of lamotrigine and 227c89 and their location are very similar to those found for the LA drugs, and the mechanism of block may be similar.
Discussion
It is important to consider whether the open/activated conformation model was developed independently of experimental information about LA binding interaction with specific residues. For our original model of the closed Na+ channel pore (Lipkind and Fozzard, 2000), the S6 helices were aligned with the KcsA crystallographic structure. Phe-1579 and Tyr-1586 in IVS6 were oriented toward the pore because of their presumed interaction with LA drugs.
Since then, we have obtained direct information using cysteine scanning and interaction with MTSEA and MTSET reagents that independently assigns these residues to the inner pore lining (Sunami et al., 2004). Rearrangement of the S5 and S6 helices of the closed model to make the open/activated channel was based on the MthK crystallographic main chain structure. Consequently, prior insight into the LA binding site was not required for our open channel binding site model.
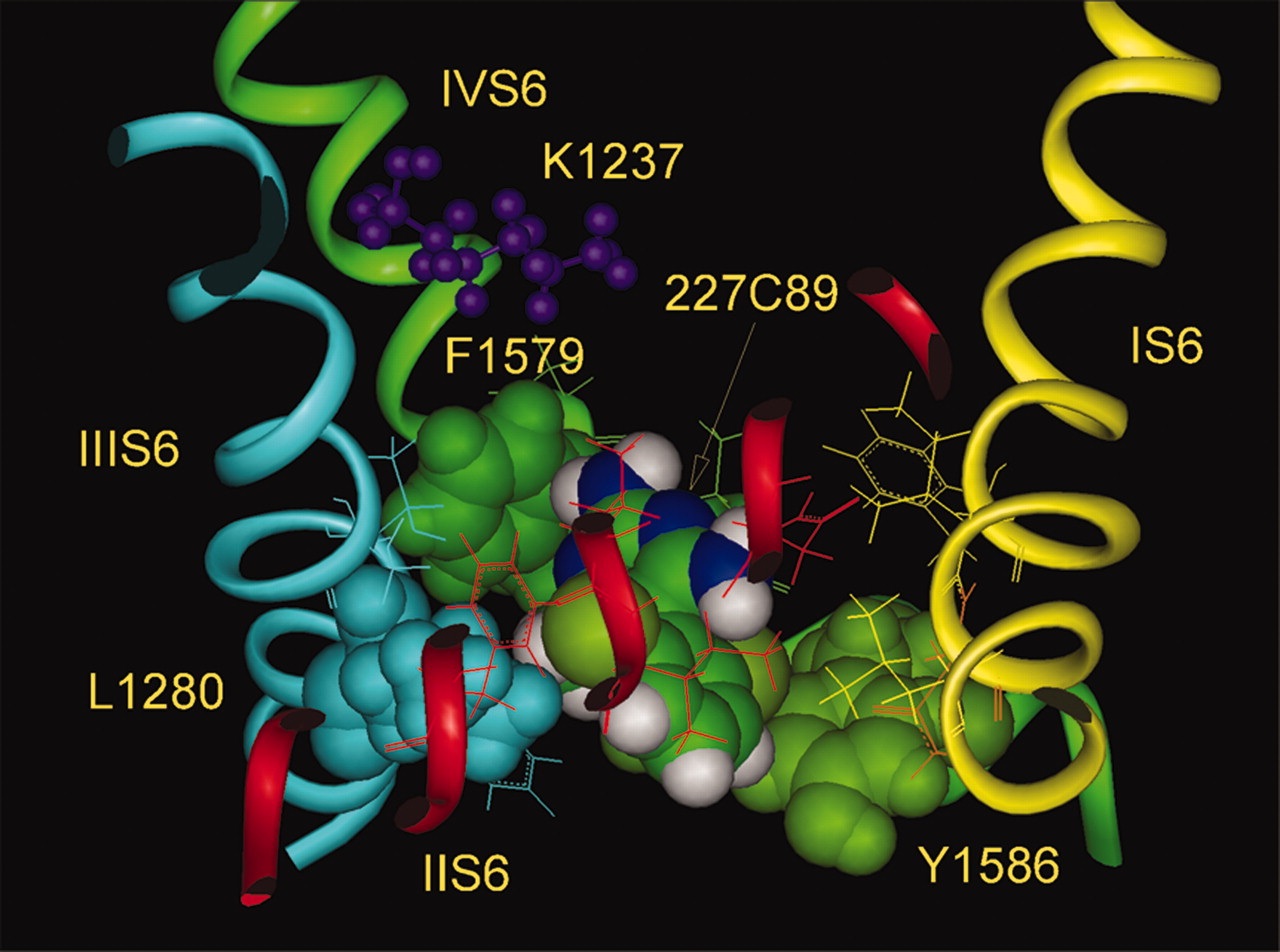
227c89. Docking of 227c89 inside the inner pore of the Nav1.4 channel (side view) with immediate contact of the para-amino group at the pyrimidine ring with the aromatic ring of Phe-1579. The partial positive charge and absence of alkyl substituents provide weaker interactions with this side chain. The methyl group of the pyrimidine ring is located in the inner groove between Leu-1280 of IIIS6 and Phe-1579 of IVS6. The dichlorophenyl moiety interacts with Tyr-1586 and the separation between the side chains of Phe-1579 and Tyr-1586 corresponds to the size of 227c89.
Modeling of binding of LA drugs inside the open channel pore requires additional comment. Recent experimental observations have questioned the traditional hypothesis that the LA drugs such as lidocaine stabilize fast inactivation (Catterall and Mackie, 1996; Hille, 2001). Vedantham and Cannon (1999) suggest that binding is to the open state and that the fast inactivation ball returns to its unblocking position at the same rate in the presence of lidocaine as in drug absence. Lidocaine may stabilize the open Na+ channel conformation, secondarily facilitating development of inactivation states. Several reports show that lidocaine can block the open state of the inactivation-deficient Nav1.4 channel with similar affinities to those seen with WT Nav1.2 (Ragsdale et al., 1994; Wang et al., 2004). Two other examples of open state block by LA are that flecainide requires that the channel open first, before block develops (Liu et al., 2003), and that QX-314 produces sustained block after fast inactivation is removed by the QQQ mutation and the block is removed by the F1579A mutation (Kimbrough and Gingrich, 2000).
Because of their size, lidocaine-like molecules do not fit into the closed pore properly when it is modeled on the basis of the KcsA structure. Converging S6 segments constrain space within the inner pore, especially around Tyr-1586. This residue is part of the densely packed S6 crossing formed also by Leu-437 of IS6, Phe-791 of IIS6, and Phe-1283 of IIIS6. After optimizing this crossover interaction to avoid nonbonded repulsions of these amino acid residues, the side chains of Tyr-1586 and Leu-1280 are screened from the inner pore by the side chains of Asn-434 and Phe-1283, which themselves were not identified by alanine scanning as parts of the binding site. The narrowing walls of the inner pore close to the S6 crossing significantly restrict the space for binding and the accessibility of residues important for binding, perhaps explaining the low resting state binding affinities. The mutation F1579A (and F1764A in Nav1.2) reduces only slightly the potency of etidocaine block of the resting state (Ragsdale et al., 1994; Wang et al., 1998a), whereas alanine substitution for Tyr-1586 had no effect. However, several substitutions of bulky amino acid residues between Phe-1579 and Tyr-1586 (for example, Val-1766 and Val-1767 in Nav1.2) significantly enhanced resting state etidocaine block, perhaps by providing a larger space and access to Phe-1579. Activation opens the overlapping S6 helices to create a binding site that includes the groove between Phe-1579 and Lys-1280 on the top and the now-accessible side chain of Tyr-1586 on the bottom, converting a binding site from low- to high-affinity conformation. In turn, the binding of LA molecules stabilizes the open pore conformation and by steric interference it opposes deactivation to the closed state.
In the open configuration model, the bends in the S6 helices were readily produced at the glycine/serine sites homologous to those in the MthK open channel structure. Residues closest to the hinge, such as Phe-1579, move minimally, whereas those more distal move long distances. The bent inner S6 segments overlap S5 of the adjacent domain. A consequence of this overlap is that one S6 segment cannot bend to a stable open position without its neighboring S6 also bending, so the opening step requires a concerted event of all four S6 segments. During hinging the S6 helix not only bends, but it also twists, so that the orientations of the distal hinged residues are changed. Tyr-1586 becomes available for LA drug interaction, and the important groove between Phe-1579 and Lys-1280 develops.
A strong argument supporting the model of an open conformation is that it created energetically appropriate binding sites for multiple members of the LA family. The shape and size of these molecules are stereochemically complimentary to this model of the binding site in the open pore, and the alkylamine heads fit into the cavity formed by side chains of Leu-1280 and Phe-1579. Docking of analogs with significant differences in structure yielded differences in energetic interactions similar to those found experimentally, including differences between optical isomers. In the series of LA drugs that we studied, the differences in binding affinities are determined primarily by van der Waals nonbonded interactions of the tertiary amino group alkyl chains with the aromatic ring of Phe-1579. In the hierarchical sequence of tocainide, lidocaine, etidocaine, and benzyl-tocainide, the energies of nonbonded interactions with Phe-1579 are higher by ∼1 kcal/mol for each step (Table 2), consistent with the experimental data for binding affinities. In that sense, Phe-1579 represents the “hot spot” of the LA binding site, and extent of saturation of its aromatic ring by van der Waals attractive forces determines the affinity differences. Saturation of this aromatic ring by nonbonded interactions also determines the limits of binding affinity to Phe-1579. In the case of the lidocaine homologs, four to five carbons in the two amino group aliphatic chains produce optimal binding, and added length did not change IC50 values (Sheldon et al., 1991). LA molecules do not interact directly with selectivity filter residues in this model, and the charged amino group is located 9 Å below the lysine in the selectivity filter.
Interactions with the IVS6 Phe-1579 site Unb is the energy of van der Waals interaction, and Uel is energy of electrostatic interaction.
Practically all clinically useful LA drugs contain protonated tertiary amines at physiological pH, with benzocaine as the exception. In contrast to the lidocaine family, the permanently uncharged benzocaine blocks preferentially the resting state, and with very low affinity of 800 μM (Wang et al., 1998a), which then decreases significantly (by 6-fold) at depolarizing potentials (Wang and Wang, 1994). So, the mechanism of block by benzocaine might be different. Mutation of the critical Phe-1579 reduces etidocaine UDB by ∼130-fold (Ragsdale et al., 1994), but it reduces the resting benzocaine block by only 2- to 3-fold (Wang et al., 1998a). We suggest that benzocaine may produce block by stabilizing the resting state. Benzocaine is smaller, and it is possible to fit it into the pore in the closed state, possibly explaining the lack of use dependence, because activation might disrupt its binding site.
If the mechanism of LA block is not stabilization of the inactivated state (Vedantham and Cannon, 1999), then what is the mechanism? Some recent experimental data point to an effect on recovery of gating charge in IIIS4 (Sheets and Hanck, 2003). Our model offers another factor that may contribute to block. Because LAs do not interact significantly with all of the widely separated S6 segments, specifically IIS6, they may not be able to block the channel by simple steric hindrance. Calculation of electrostatic fields within the inner pore offers the tantalizing suggestion that the alkylamine positive charge is located by the binding process so that it creates an electrostatic barrier to movement of Na+ ions. For all but benzocaine, it is likely that only the charged form of the drugs produce block (Catterall and Mackie, 1996), consistent with the idea of an electrostatic barrier. Supporting this idea is the report of Zamponi et al. (1993) that values of unblocking rates and the voltage dependence of unblock rates were identical for lidocaine and its permanently charged derivative QX-314, allowing the conclusion that the electrostatic mechanism of block was identical for tertiary and quaternary amines. Zamponi and French (1993) also showed that fast, open channel block is associated with the charged primary amine in the LA structure.

Electrostatic potentials in the pore of the modeled Nav1.4 channel without drug (top) and with drug (bottom). The contour of the negative isopotential surface in the absence of LA is shown at the level of -2kT (red solid surface), which fills the volume of both the vestibule and the inner pore. Electrostatic potentials in the pore of the Nav1.4 channel with lidocaine in its binding site (bottom). The contours of the isopotential surfaces are shown at the level -2kT (red) and +2kT (blue solid surface). The positive charge of the alkylamine head of lidocaine (presented here by the equipotential +2kT) sharply interrupts the negative electrostatic potential of the outer vestibule at the level of the selectivity filter (here shown by stick and ball images), hindering Na+ permeation. The channel is shown in side view. The backbones of the pore and the outer vestibule are shown by green and yellow ribbons, respectively.
Negatively charged amino acids in the selectivity filter (Asp-400 and Glu-755) and in the vestibule outer ring (Glu-403, Glu-758, Asp-1241, and Asp-1532) create a negative electrostatic potential not only in the outer vestibule (Khan et al., 2002) but also inside the inner pore below the selectivity filter. This allows permeating Na+ ions to reach the cytoplasmic side of the selectivity filter (Fig. 13, top). In the region of Lys-1237 of the selectivity filter the negative electrostatic field is weaker, and the positively charged amino heads of LA molecules easily overcome the negative field, producing an independent positive electrostatic potential (as much as 100 mV) that spans the inner pore below the selectivity filter (Fig. 13, bottom), where it could interrupt Na+ permeation. Although charge plays a role in the binding interaction itself, the key interaction of the alkylamino head to Phe-1579 is hydrophobic, and this hydrophobic interaction determines the location of the positive charge, placing it in the appropriate position inside the pore to create an electrostatic barrier. In turn, the dimethyl-substituted aromatic ring of the LA expels water from the inner pore and creates an environment with low dielectric constant around the charge. This strengthens the magnitude of the positive electrostatic potential that may interfere with Na+ current.
We recently found experimental evidence that μ-conotoxin blocks the outer mouth of the Nav1.4 channel by an electrostatic mechanism produced by positioning of the positive charge of the side chain of the toxin Arg-13 (Hui et al., 2002). Another experimental observation that also supports the idea of an electrostatic barrier to Na+ permeation produced by the charge on the LA molecule is a reduction in current amplitude resulting from covalent modification of cysteine mutants of Phe-1579 and Val-1583 by the positively charged MTSEA and MTSET reagents (Vedantham and Cannon, 2000; Sunami et al., 2004). The report of Wright et al. (1998) that the Nav1.4 mutant F1579K expressed whole cell currents seems to conflict with the idea of electrostatic block from a positive charge at this position. However, Sunami et al. (2004) found that introduction of a positive charge by modification of F1579C by MTSEA or MTSET resulted in approximately 50 to 60% reduction in current. The mechanism of this partial block could not be evaluated by whole cell currents. However, single channel conductance of the F1579K mutant was approximately 50% of WT (R. Shah and D. A. Hanck, personal communication). These studies suggest that the alkylamine head of the LA molecules positions a positive charge near Phe-1579 in a fixed orientation near the center of the pore, creating an electrostatic component that may contribute to block, in addition to steric and gating components.
Footnotes
-
This study was supported by United States Public Health Service grant R01-HL65661.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.105.014803.
-
ABBREVIATIONS: LA, local anesthetic; Nav1.4, skeletal muscle isoform of the Na+ channel; Nav1.2, brain II isoform of the Na+ channel; DEKA, the four amino acids thought to form the selectivity filter of the Na+ channel: aspartate, glutamate, lysine, and alanine; UDB, use-dependent block; KcsA, K+ channel from Streptomyces lividans; MthK, Ca2+-activated K+ channel from Methanobacterium thermoautotrophicum; S5 and S6, fifth and sixth transmembrane segments of each domain of the Na+ channel α-subunit; M1 and M2, first and second transmembrane segments of the KcsA and MthK channels; P loop, extracellular loop connecting S5 and S6 and containing the outer vestibule and selectivity filter; MTSET, methanethiosulfonate ethyltrimethylammonium; MTSEA, 2-(aminoethyl)-methanethiosulfonate hydrobromide; WT, wild-type; QX-314, lidocaine N-ethyl bromide.
-
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.

- Received May 13, 2005.
- Accepted September 16, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}