


Abstract
Despite the established interindividual variability and ontogeny of the CYP3A enzymes, the most abundant phase I drug-metabolizing enzymes in human liver and intestine, the mechanisms that regulate basal expression remain poorly understood. Electrophoretic mobility shift assays using nuclear proteins extracted from human prenatal and postnatal liver samples identified multiple, developmentally distinct nuclear factor I (NFI)-containing protein complexes from human liver bound to sequences from the CYP3A4 (−243/−220) and CYP3A7 (−242/−219) proximal promoters. In addition, a hepatocyte nuclear factor (HNF) 3γ-containing complex from prenatal liver interacted with CYP3A7−242/−219 but not CYP3A4−243/−220. Cotransfection of HepG2 cells with a CYP3A4 proximal promoter construct and expression vectors for the NFI isoforms NFIA1.1, NFIB2, NFIC1, NFIC2, and NFIX1 enhanced the expression of luciferase activity. In contrast, cotransfection of NFIB2, NFIC1, NFIC2, NFIX1, and NFIX2 reduced the expression of luciferase under the control of the CYP3A7 gene promoter. Mutagenesis of the NFI/HNF3γ binding sites in the CYP3A7 and CYP3A4 proximal promoters suggests that regulation of basal promoter activity by members of the NFI transcription factor family occur via multiple mechanisms. These results demonstrate that members of the NFI transcription factor family regulate CYP3A4 and CYP3A7 basal expression in an isoform- and promoter-specific manner.
- P450, cytochrome P450
- C/EBP, CCAAT/enhancer binding protein
- Sp, specificity protein
- NFI, nuclear factor I
- HNF, hepatocyte nuclear factor
- NF-Y, nuclear transcription factor Y
- YY, yin-yang
- USF, upstream stimulatory factor
- EMSA, electrophoretic mobility shift assay
- MOPS, 3-(N-morpholino)propanesulfonic acid
- TNT, Tris/NaCl/Tween 20
- RT-PCR, reverse transcription-polymerase chain reaction
- PCR, polymerase chain reaction
- RXR, retinoid X receptor
- luc, luciferase
- DMEM, Dulbecco's modified Eagle's medium
- FBS, fetal bovine serum.
The cytochrome P450 (P450) superfamily of enzymes are mixed function oxidases responsible for the metabolism of both xenobiotic and endogenous compounds. Members of the CYP3A subfamily are the most abundant phase I drug/steroid-metabolizing enzymes expressed in human liver and intestine and are responsible for the metabolism of approximately 50% of drugs (Shimada et al., 1994; de Wildt et al., 1999; Guengerich, 1999). In humans, the CYP3A family consists of four members, CYP3A4, CYP3A5, CYP3A7, and CYP3A43, encoded on a 230-kilobase locus on chromosome 7q22.1 (Gellner et al., 2001).
Marked interindividual variability and distinct developmental profiles are characteristic of the expression of members of the CYP3A subfamily. From a developmental perspective, CYP3A7 is predominantly expressed in prenatal tissues, making up 50% of total P450 content in fetal liver (Lacroix et al., 1997). CYP3A7 expression is detected as early as 50 to 60 days of gestation and persists until after birth, when it decreases over the first few months of life (Lacroix et al., 1997; Stevens et al., 2003). Conversely, CYP3A4 is not detectable before birth, but it gradually increases in expression with postnatal age (Stevens et al., 2003). Although closely related, CYP3A4 and CYP3A7 display distinct metabolic capabilities (Williams et al., 2002) that result in developmentally related changes in the capacity to metabolize steroids and xenobiotics in humans (de Wildt et al., 1999; Hines and McCarver, 2002; Hines, 2008). CYP3A5 is polymorphically expressed in both fetal and adult tissues, whereas CYP3A43, the most recently identified family member, is expressed at low levels in adult liver, prostate, and testis (Gellner et al., 2001), and its contribution to xenobiotic metabolism is not fully understood.
In spite of the established interindividual variability and ontogeny of the CYP3A enzymes, the mechanisms that regulate basal expression remain poorly understood. Previous studies have suggested a role for transcription factor interactions with the proximal promoters of CYP3A7 and CYP3A4 to be important for differential regulation. Alignment of the CYP3A4 and CYP3A7 promoters has revealed that they are >90% identical up to approximately 9 kilobases upstream of exon 1 and then fall to approximately 25% identity farther upstream (Bertilsson et al., 2001). Functional studies of the proximal promoter regions of the CYP3A4 and CYP3A7 genes have shown that they are regulated by overlapping sets of transcription factors in vitro; however, differences in confirmed and putative DNA binding sites for transcription factors exist between the two promoters (Itoh et al., 1992; Hashimoto et al., 1993; Saito et al., 2001; Rodríguez-Antona et al., 2003; Bombail et al., 2004). For example, multiple sequence differences between the CYP3A4 and CYP3A7 gene promoters occur around the nifedipine-specific response element of CYP3A4 and the human fetal liver-specific element of CYP3A7, which are regulated by as yet unidentified protein(s).
Previous studies using the HepG2 human hepatoma cell line have demonstrated the interaction of the C/EBPα, Sp1, HNF3α, YY1, USF1, Sp1/Sp3, NFI, and HNF3β transcription factors with sequences from the CYP3A4 and CYP3A7 proximal promoter regions (Saito et al., 2001; Rodríguez-Antona et al., 2003; Bombail et al., 2004). Results reported by Saito et al. (2001) suggest a role for NFI and HNF3β transcription factors in the regulation of the proximal promoter of CYP3A7, but not CYP3A4, in HepG2 cells. Although HepG2 cells have been used extensively as an in vitro model to investigate the mechanisms of basal and inducible regulation of the human CYP3A4 and CYP3A7 gene promoters, HepG2 cells are limited in delineating the role of these transcription factors and response elements in the expression of human CYP3A enzymes throughout development. The results of the present study are based on the hypothesis that developmental changes in the interactions of transcription factors with sequences of the CYP3A4 and CYP3A7 gene promoters contribute to the prenatal-postnatal transition of these enzymes in humans. The purpose of the current study was to identify nuclear proteins from human prenatal and postnatal liver that interact with previously reported overlapping response elements for NFI and HNF3β in the proximal promoters of human CYP3A4 and CYP3A7. The data provide evidence that differentially expressed isoforms of the NFI transcription factor family are found in human prenatal and postnatal liver and differentially regulate the proximal promoters of CYP3A4 and CYP3A7 in HepG2 cells, implying that these proteins may play a role in developmental regulation of human CYP3A enzymes in liver.
Materials and Methods
Liver Samples.
Human fetal and pediatric liver samples were obtained from the University of Maryland Brain and Tissue Bank for Developmental Disorders (Baltimore, MD) or from the Center for Birth Defects Research at the University of Washington (Seattle, WA). All tissues were maintained at −80°C before preparation of nuclear extracts or RNA. The use of these tissues was approved by the University of Missouri-Kansas City Pediatric Health Sciences Review Board.
Preparation of Nuclear Extracts.
Nuclear extracts were prepared from fetal and postnatal liver samples by homogenization in 50 mM Tris-Cl, pH 7.4, 150 mM KCl, and 2 mM EDTA followed by centrifugation at 800g for 15 min at 4°C to pellet cellular debris, including nuclei. The supernatant was removed and used for subsequent isolation of microsomal and cytosolic fractions by differential centrifugation. Nuclear proteins were extracted from the pellet by resuspension in high-salt nuclear extraction buffer [10 mM HEPES, pH 7.9, 1 mM EDTA, 0.5 M KCl, 6% glycerol, 1 mM dithiothreitol, and protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO)] followed by incubation on ice for 1 h with intermittent mixing. The extracts were centrifuged at 25,000g for 30 min at 4°C. The supernatants containing nuclear proteins were flash-frozen in liquid nitrogen and stored in small aliquots at −80°C. Protein concentrations of nuclear extracts were determined using the Micro BCA protein assay kit from Pierce Chemical (Rockford, IL).
Electrophoretic Mobility Shift Assays.
Complementary oligonucleotide probes (Sigma-Genosys, The Woodlands, TX) containing the following regions were synthesized: CYP3A7−242/−219 and CYP3A4−243/−220 (Table 1, base pairs numbered relative to the ATG translation start site). The oligonucleotides were annealed and then end-labeled with [γ-32P]ATP and T4 polynucleotide kinase. Binding reactions contained 15 μg of nuclear extract, 0.005 pmol of 32P-labeled oligonucleotides, and 1.25 μg of poly[dI:dC] (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK) in 1× binding buffer (20 mM HEPES, pH 7.9, 5 mM MgCl2, 1 mM EDTA, 4% glycerol, 1 mM dithiothreitol, and 100 mM NaCl) and were incubated on ice for 5 min before the addition of unlabeled competitor oligonucleotides. After the addition of competitor oligonucleotides, reactions were incubated at room temperature for 10 min followed by the addition of 32P-labeled oligonucleotides and incubation at room temperature for an additional 15 min. Immunodepletion assays were performed by the addition of specific antibodies [NFI (H-300), HNF3α (H-120), HNF3β (M-20), and HNF3γ (N-19); Santa Cruz Biotechnology, Inc., Santa Cruz, CA] after the addition of and incubation with 32P-labeled oligonucleotides, as indicated. Immunodepletion reactions were incubated for 15 min after the addition of antibodies. Bound and unbound DNA were separated on 6% polyacrylamide gels run in 50 mM Tris, 45 mM boric acid, and 0.5 mM EDTA for 1.5 h at 125 V. DNA-protein complexes were visualized by autoradiography.
Primer sequences
Immunoblotting.
Human liver or HepG2 nuclear proteins were separated on 10% NuPage Bis-Tris gels in NuPage MOPS-SDS running buffer (Invitrogen, Carlsbad, CA). Proteins were transferred to Hybond ECL membranes (GE Healthcare), and membranes were blocked for 1 h at room temperature in 10 mM Tris, 150 mM NaCl, and 0.2% Tween 20, pH 8.0 (TNT) containing 4% skim milk powder. Blocked membranes were incubated for 18 h with NFI antibody (H-300; Santa Cruz Biotechnology, Inc.), HNF3γ antibody (N-19; Santa Cruz Biotechnology, Inc.), or RXRα (D-20; Santa Cruz Biotechnology, Inc.) diluted 1:10,000-fold in 4% skim milk powder-TNT at 4°C. To detect bound antibody, blots were incubated with horseradish peroxidase-conjugated anti-rabbit (GE Healthcare) or anti-goat antibodies (Santa Cruz Biotechnology, Inc.) diluted 1:25,000 in 4% skim milk powder-TNT for 60 min, washed (six times for 5 min each), incubated with ECL Plus chemiluminescence reagents (GE Healthcare) according to the manufacturer's directions, and exposed on Hyperfilm ECL (GE Healthcare).
Quantitative RT-PCR.
Total RNA was extracted from HepG2 cells or 20 to 30 mg of human liver using the Illustra RNAspin Mini RNA isolation kit (GE Healthcare) according to manufacturer's recommendations, with an on-column DNase I treatment. RNA quantity and quality were verified using an Experion automated electrophoresis system (Bio-Rad Laboratories, Hercules, CA). One-step, quantitative RT-PCR reactions were performed to detect CYP3A4 and CYP3A7 as described previously (Leeder et al., 2005) or the NFI isoforms (primers in Table 1) on 15 ng of total RNA with the QuantiTect SYBR Green OneStep RT-PCR kit (QIAGEN, Valencia, CA) on a DNA Engine Opticon 2 instrument (MJ Research, Watertown, MA). Serial dilutions of PCR amplicons (CYP3A4 and CYP3A7) or cDNA plasmids (NFI isoforms) were used to generate standard curves ranging from 100 to 107 copies. Transcript numbers were calculated from linear regression analysis of the respective standard curves. Quantitative RT-PCR for 18S ribosomal RNA was performed using the TaqMan ribosomal RNA control reagent kit (Applied Biosystems, Foster City, CA). Expression levels are expressed as copies per nanogram of total RNA corrected for 18S rRNA.
Plasmid Constructs.
The reporter constructs CYP3A4*1A−694/−48-luc (numbering given relative to the ATG start codon), CYP3A7*1A−685/−56-luc, and CYP3A7*1C−685/−56-luc were generated by PCR amplification of the corresponding sequences from genomic DNA using primers CYP3A4-869 F and CYP3A4-48 R or CYP3A7-856 F and CYP3A7-56 R (Table 1). The resulting amplicons were digested with SacI to cut at naturally occurring sites at −694 of CYP3A4 or −685 of CYP3A7 and HindIII to cut at sites incorporated into the reverse primers. The CYP3A7 and CYP3A4 promoter sequences were then ligated upstream of the luciferase gene in the pGL3-basic reporter plasmid (Promega, Madison, WI). Using these reporter constructs as templates, the following variants of CYP3A4 and CYP3A7 were generated. The cytosine at position −233 of CYP3A4*1A was replaced with an adenine, the same nucleotide that occurs at position −232 in the CYP3A7*1A promoter, using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) with the primers CYP3A4-233mut F and CYP3A4-233mut R (Table 1) as follows. Reactions were incubated at 95°C for 1 min followed by 30 cycles of a denaturation at 95°C for 1 min, an anneal at 55°C for 1 min, and an extension at 65°C for 11 min. Upon completion of cycling, DpnI was added to the reaction to digest the parental DNA template, and this digested product was then transformed into DH5α cells. Similar procedures were used to modify sequences of CYP3A7*1A−685/−56-luc using the primers listed in Table 1. All plasmid constructs were verified by sequencing.
Expression constructs encoding NFIA1.1, NFIB2, NFIC2, and NFIX2 in the pCH expression vector were kindly provided by Dr. Richard Gronostajski (State University of New York at Buffalo, Buffalo, NY) (Chaudhry et al., 1997; Gründer et al., 2003). The expression vector for NFIX1, which differs from NFIX2 by the inclusion of exon 7, was prepared by PCR amplification using NFIX1 for and NFIX1 rev (Table 1) from pooled human liver cDNA. The resulting amplicon was digested with ApaI and AgeI and ligated into pCH-NFIX2 digested with the same enzymes. NFIC1 cDNA was amplified from pooled human liver cDNA using NFIC for and NFIC rev. The resulting amplicon was cloned into pCR4-TOPO using the TOPO TA cloning kit (Invitrogen) according to manufacturer's recommendations. The expression vector for NFIC1, which differs from NFIC2 by alternative splicing at the 3′ end of the transcript, was prepared by cutting the alternatively spliced portion from pCR4-NFIC1 using SacI and NheI and ligating into pCH-NFIC2 digested with the same enzymes.
Transient Transfection and Luciferase Assays.
HepG2 cells were obtained from American Type Culture Collection (Manassas, VA) and maintained in Dulbecco's modified Eagle's medium (DMEM) (Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals, Norcross, GA). The cells were grown at 37°C and 5% CO2. For transient transfection assays, HepG2 cells were plated in 12-well plates in DMEM without phenol red (Sigma-Aldrich) supplemented with 5% FBS to result in a density of 60 to 80% at the time of transfection 24 h later. The cells were transfected with 500 ng of the indicated firefly luciferase reporter construct and 5 ng of pCMV-RL (Promega) with or without NFI expression constructs using Lipofectamine LTX with Plus reagent (Invitrogen) according to manufacturer's recommendations. Three hours after transfection, medium was replaced with DMEM without phenol red supplemented with 5% FBS for 40 to 48 h. The cells were harvested in 1× passive lysis buffer (Promega), and luciferase activity was detected using the Dual-Luciferase reporter assay system (Promega) on 10 μl of the cell lysate. Luciferase activity was quantified using a Lumat LB 9507 tube luminometer (Berthold Technologies, Bad Wildbad, Germany).
Results
Developmentally Regulated Protein Complexes from Human Liver Interact with the CYP3A4 and CYP3A7 Promoters.
To determine whether developmentally regulated transcription factors expressed in human liver interact with sequences from the CYP3A4 and CYP3A7 proximal promoters that have been shown to contribute to differential regulation of the two promoters in HepG2 cells (Saito et al., 2001), we performed EMSAs with nuclear extracts isolated from human prenatal and postnatal liver samples and sequences from the CYP3A4 (−243 to −220 base pairs relative to the ATG) and CYP3A7 (−242 to −219) gene promoters. Nuclear extracts from postnatal human liver resulted in the formation of two complexes with the sequence from the CYP3A4 proximal promoter that were similar in mobility to two protein complexes that bound to the CYP3A7 proximal promoter (Fig. 1, A and C, complexes I and II). EMSAs with prenatal liver nuclear extracts resulted in the formation of protein-DNA complexes that migrated more slowly than those observed with postnatal extracts (Fig. 1, B and D). In contrast to postnatal protein-DNA complexes, which were similar with both promoters, binding of prenatal nuclear proteins to CYP3A7 differed from that to CYP3A4 by the presence of an additional band with slightly faster mobility (Fig. 1D, complex IV) than the major retarded band observed in EMSAs with both CYP3A4 and CYP3A7 promoter sequences (Fig. 1, B and D, complex III). These data demonstrate binding of human liver nuclear proteins to CYP3A4 and CYP3A7 proximal promoter elements that differ with respect to developmental stage and promoter preference. It is interesting to note that nuclear proteins from HepG2 cells that form complexes with this region of CYP3A4/7 have a mobility in EMSAs most similar to prenatal liver (Fig. 1E).
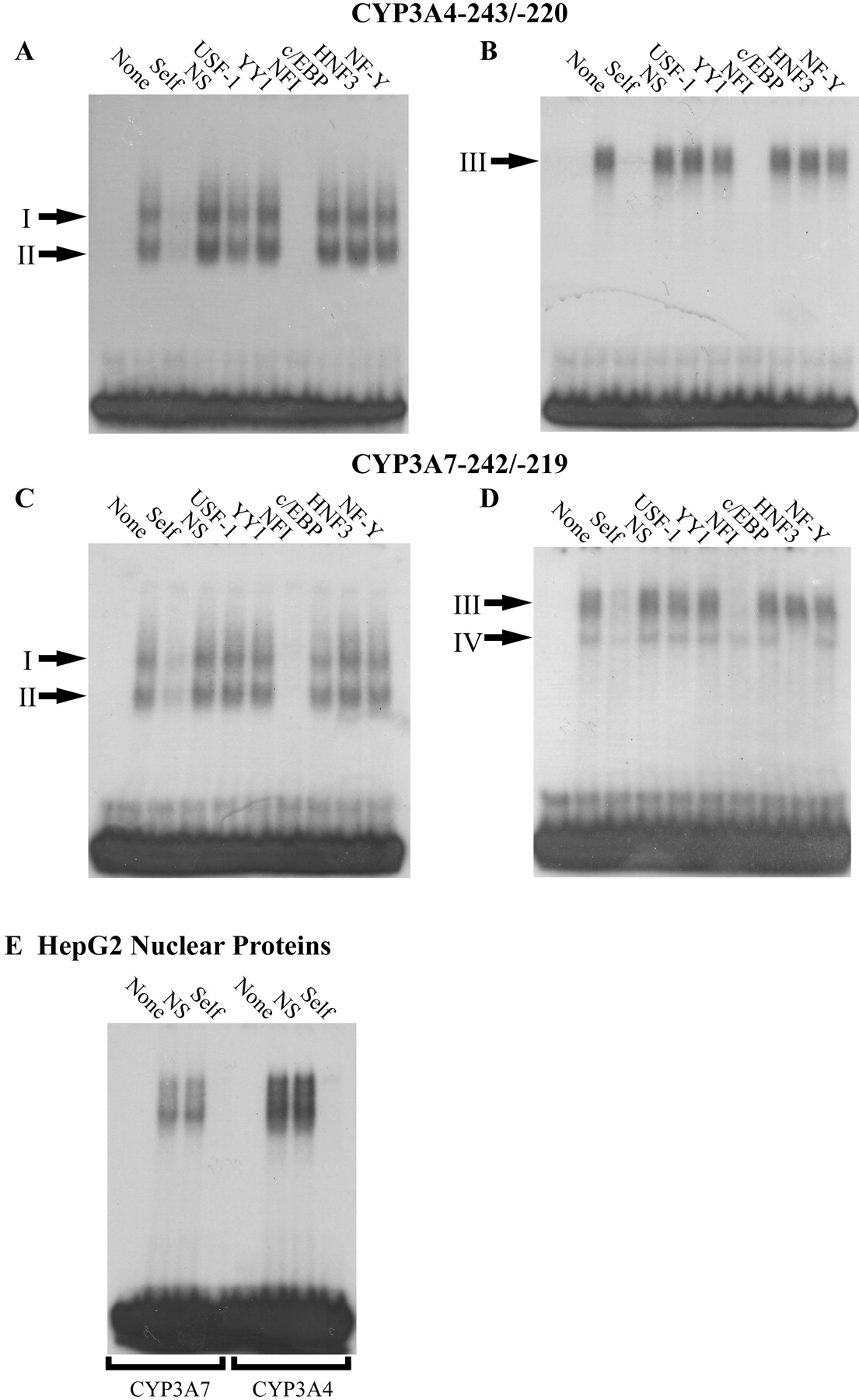
Developmentally distinct protein complexes bind to the −220 to −243 regions of the CYP3A4 and CYP3A7 promoters. Electrophoretic mobility shift assays were performed as described using 15 μg of human postnatal liver (A and C), prenatal liver (B and D), or HepG2 (E) nuclear extracts and labeled CYP3A4−243/−220 (A and B; E, lanes 5–8) or CYP3A7−242/−219 (C and D; E, lanes 1–4). Unlabeled, competitor oligonucleotides were added in 100-fold molar excess as indicated above the lanes (NS, nonspecific) and suggest that members of the NFI transcription factor family are part of both prenatal and postnatal complexes.
To determine the identity of proteins within the complexes bound to the CYP3A4−243/−220 and CYP3A7−242/−219 sequences, excess unlabeled competitor oligonucleotides containing consensus sequences for USF1, YY1, NFI, C/EBP, HNF3, or NF-Y were added to binding reactions before the addition of labeled DNA. The NFI consensus sequence was able to completely deplete binding of both complexes (Fig. 1, A and C, complexes I and II) formed by postnatal nuclear extracts and the CYP3A4−243/−220 and CYP3A7−242/−219 oligonucleotides. No effect on binding was observed with the other consensus recognition sites tested. The NFI consensus sequence was also able to compete for binding with complex III formed between nuclear extracts from prenatal liver and CYP3A7−242/−219 and CYP3A4−243/−220 (Fig. 1, B and D). The faster migrating protein-DNA complex IV observed with CYP3A7−242/−219 was depleted by the addition of a consensus binding sequence for HNF3 (Fig. 1D).
Antibodies against NFI and HNF3 proteins were added to the binding assays to confirm the presence of these transcription factors in the shifted complexes bound to CYP3A4 and CYP3A7 as suggested by competition assays. An antibody that recognizes all human isoforms of NFI reduced binding of complexes I and II (Fig. 2, lanes 3 and 9) from postnatal liver and depleted binding of complex III from prenatal liver (Fig. 2, lanes 6 and 12). Complex IV was depleted by the addition of an antibody against HNF3γ (Fig. 2B, lane 15) but not HNF3α (lane 13) or HNF3β (lane 14), suggesting that this complex contains HNF3γ. In summary, multiple, developmentally distinct NFI-containing protein complexes from human liver are capable of interacting with the CYP3A4 and CYP3A7 proximal promoters, whereas an HNF3γ-containing complex from prenatal liver interacts with the CYP3A7 proximal promoter but not CYP3A4.
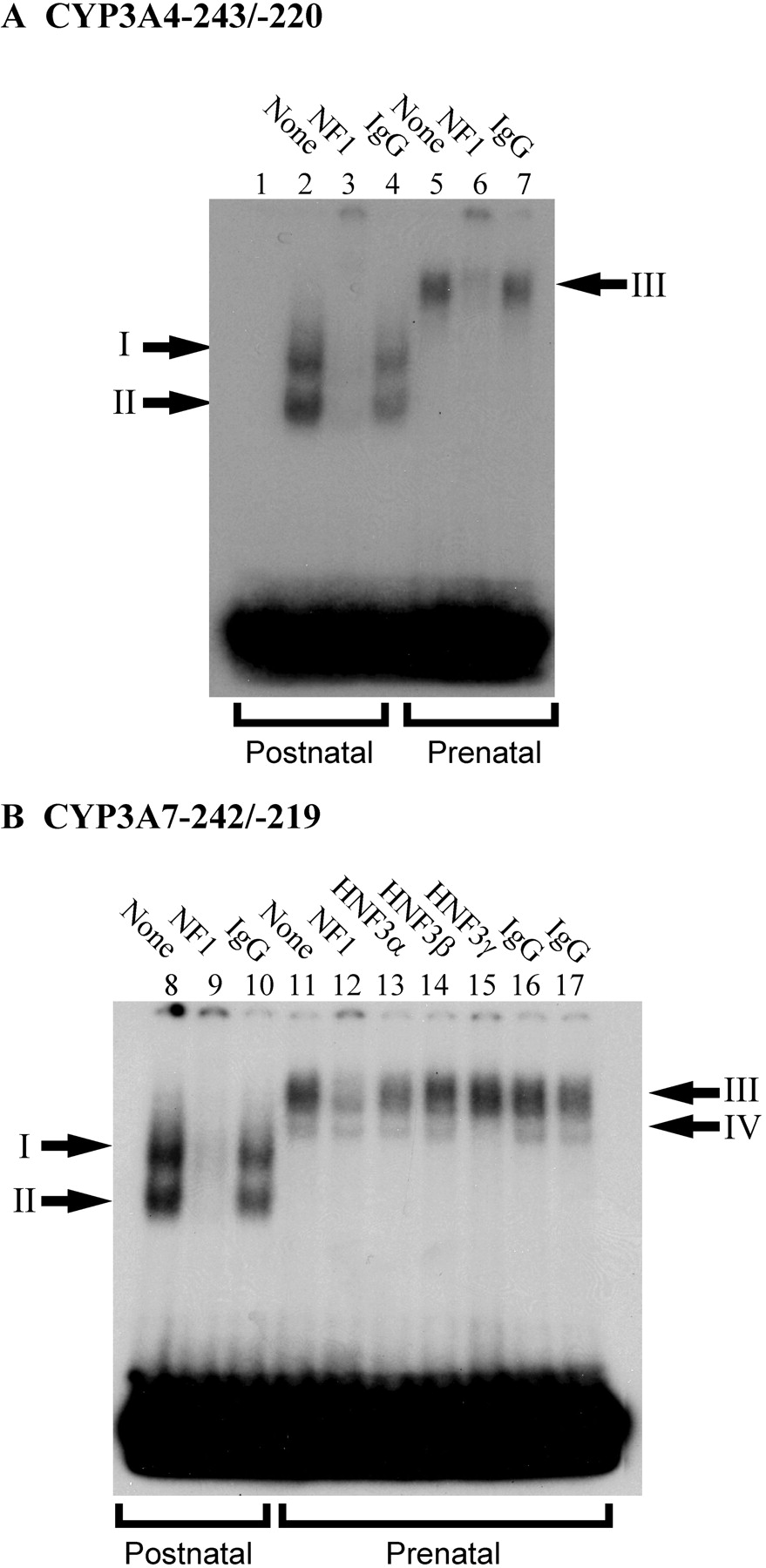
Immunodepletion of protein-DNA complexes with antibodies for NFI and HNF3γ. Addition of an antibody that recognizes all human NFI isoforms (A) to EMSA reactions resulted in depletion of complexes I and II from postnatal liver (lane 3) and complex III from prenatal liver (lane 6) capable of binding to the CYP3A4−243/−220 probe. Addition of the NFI antibody to EMSA reactions reduced binding of complexes I and II from postnatal liver and complex III from prenatal liver capable of binding to the CYP3A7−242/−219 probe but did not affect formation of complex IV (B). Only addition of an antibody that recognizes human HNF3γ and not HNF3α or HNF3β was able to block binding of complex IV to the CYP3A7 probe.
Developmental Expression of NFI and HNF3γ Transcription Factors in Human Liver.
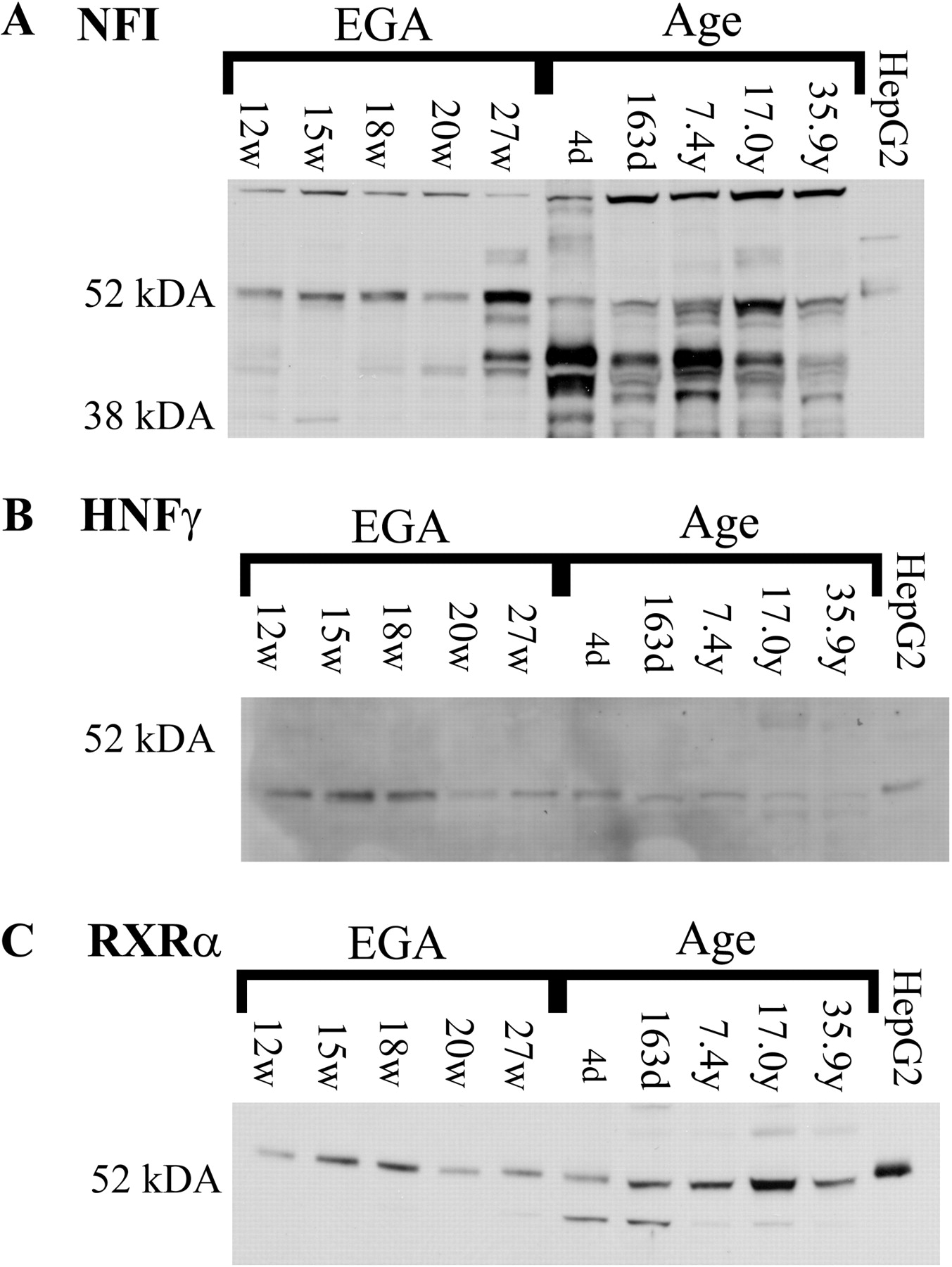
The differing mobility of NFI-containing complexes between prenatal and postnatal liver nuclear extracts may be due to differences in the expression of NFI isoforms or to the presence of differing auxiliary factors in the protein-DNA complexes. Immunoblots were performed with nuclear extracts from human prenatal and postnatal liver samples to determine developmental changes in NFI expression. In prenatal liver nuclear extracts, multiple immunoreactive NFI proteins were observed with the predominant protein approximately 50 kDa and a minor band of approximately 40 kDa (Fig. 3A). In contrast, multiple proteins of approximately 40 kDa were the major immunoreactive products observed in postnatal liver. In addition, a doublet is observed at 50 kDa in samples more than 7 years of age. To control for potential differences in nuclear fractionation of frozen liver samples, immunoblots of the nuclear protein RXRα were performed (Fig. 3C). Previous studies from our laboratory indicate that RXRα expression in human liver does not change during development (Vyhlidal et al., 2006). The changes seen on the NFI immunoblot are consistent with a faster migrating complex from postnatal liver bound to CYP3A4−243/−220 and CYP3A7−242/−219 versus the slower migrating complex from prenatal human liver; however, they do not preclude the presence of additional proteins in protein-DNA complexes observed in EMSAs. In addition, similar to results from EMSAs, immunoreactive NFI proteins in HepG2 cell nuclear extracts most closely resemble the pattern observed with human prenatal liver nuclear extracts.

Developmental expression of NFI and HNF3γ transcription factors in human liver and HepG2 cells. To determine the existence of potential changes in the developmental expression of NFI isoforms (A) and HNF3γ (B), immunoblotting was performed with liver nuclear extracts from prenatal (n = 5) and postnatal livers (n = 5) and from HepG2 cells as described. Immunoblotting was performed to detect RXRα (C) to control for differences in loading and efficiency of extraction. Estimated gestational age (EGA) and postnatal ages are indicated above each lane.
Immunoblotting of nuclear proteins isolated from human prenatal and postnatal liver samples suggests developmental expression of NFI proteins. However, it is not possible to determine which NFI isoforms or variants are expressed at each stage due to the unavailability of antibodies specific for the individual isoforms. Therefore, to determine the relative expression of the NFI isoforms, real-time RT-PCR was performed using isoform-specific primers on RNA isolated from prenatal (n = 27) and postnatal (n = 25) liver samples. These primers detect total mRNA expressed from each of the four genes but do not detect alternative splicing that may be present. Total NFIA (Fig. 4A, ○), NFIB (Fig. 4A, ▪), and NFIX (Fig. 4C) mRNAs were expressed at similar levels in prenatal and postnatal liver after correction for 18S rRNA levels. In contrast, NFIC mRNA levels were statistically (p < 0.001, two-tailed t test) lower in prenatal liver samples (7.9 ± 4.6 molecules/ng total RNA after 18S rRNA correction) than in postnatal liver samples (28.6 ± 20.8 molecules/ng total RNA after 18S rRNA correction). The difference in protein expression in prenatal versus postnatal liver may be due to developmental expression of NFIC; however, these results do not eliminate alternative splicing as a source of developmental variation for all members of the NFI transcription factor family.

Developmental expression of NFI mRNA in human liver. Quantitative RT-PCR was performed as described to determine mRNA expression levels of total NFIA (A, ○), NFIB (A, ▪), NFIC (B), and NFIX (D). Relative expression levels are expressed as molecules per nanogram of total RNA after correction for 18S rRNA and are plotted relative to estimated gestational age (EGA) or postnatal age.
Data from in vitro binding studies with nuclear proteins isolated from prenatal and postnatal human liver suggest that expression of HNF3γ is developmentally regulated. Immunoblots were performed with nuclear extracts from human prenatal and postnatal liver samples from individuals of increasing age. HNF3γ was detected in all prenatal liver nuclear extracts (12.4–27 weeks estimated gestational age) and seemed to decrease with increasing gestational age (Fig. 3B, lanes 1–5). In contrast, HNF3γ was not detected in any postnatal liver samples tested ranging from 4 days after birth to 35 years of age (Fig. 3B, lanes 6–10). Similar to in vitro binding assays and NFI expression patterns, HNF3γ was detected with HepG2 cells (Fig. 3B, lane 11).
CYP3A4 and CYP3A7 Proximal Promoter Activity in HepG2 Cells.
HepG2 cells have been used extensively as an in vitro model to investigate the mechanisms of basal and inducible regulation of the CYP3A4 and CYP3A7 gene promoters. However, HepG2 cells, at densities commonly used for reporter assays, possess a phenotype similar to fetal liver (Kelly and Darlington, 1989), and data from in vitro binding assays (Fig. 1E) and immunoblotting (Fig. 3) with nuclear proteins from HepG2 cells further support the conclusion that HepG2 cells have a phenotype comparable with that of fetal liver. Consistent with a fetal phenotype, we observed relative expression levels of endogenous CYP3A4 and CYP3A7 mRNA that are most similar to those observed in fetal liver (Fig. 5A) (Leeder et al., 2005). To determine whether differences in endogenous levels of CYP3A gene expression in HepG2 cells are reflected in differences in proximal promoter activity, transient transfections with reporter constructs in which sequences from either the CYP3A4 (base pairs −694/−48 relative to the ATG start codon) or CYP3A7 (base pairs −685/−56 relative to the ATG start codon) gene promoters direct the expression of firefly luciferase were performed. In transient transfections of HepG2 cells, the CYP3A7*1A−685/−56 promoter resulted in an approximately 13-fold increase in luciferase activity relative to the CYP3A4*1A−694/−48 proximal promoter (Fig. 5B), consistent with endogenous expression levels. It is interesting that the CYP3A7*1C−685/−56 promoter resulted in similar levels of luciferase activity compared with the CYP3A7*1A−685/−56 promoter (Fig. 5B), suggesting that aberrant expression associated with this polymorphism may be restricted to postnatal liver. These results are consistent with the hypothesis that at least some of the determinants underlying developmental changes in CYP3A gene expression lie within the proximal promoter regions of the CYP3A4 and CYP3A7 genes.

HepG2 cells possess a phenotype most similar to fetal liver. Quantitative RT-PCR was performed as described to detect endogenous expression levels of CYP3A4 and CYP3A7 mRNA in HepG2 cells (A). Bars represent the average number of molecules/50 ng total RNA of four replicate samples. Transient transfections of HepG2 cells were performed as described to determine whether basal promoter activity of the CYP3A4 and CYP3A7 proximal promoters reflects endogenous expression levels (B). Firefly luciferase activity was normalized to Renilla reniformis luciferase activity from the cotransfected control vector. Graphs represent average relative luciferase activity ± S.D. for four replicate samples from three independent transfections.
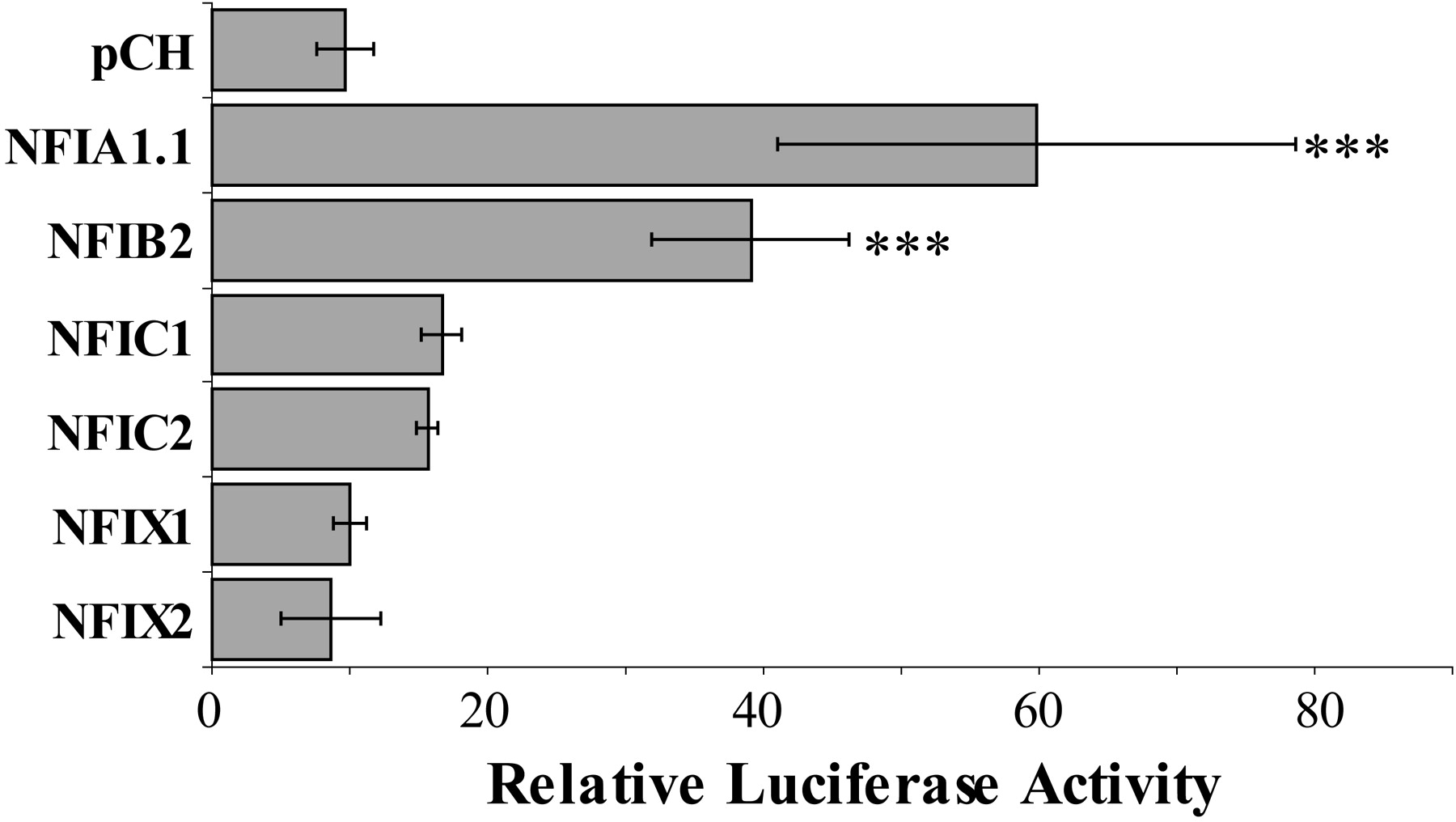
Differential Regulation of CYP3A4 and CYP3A7 Proximal Promoters by Isoforms of the NFI Transcription Factor Family.
EMSAs and immunoblotting with nuclear proteins from human prenatal and postnatal liver are consistent with the hypothesis that developmentally regulated NFI transcription factor family members in human liver are involved in the regulation of CYP3A4 and CYP3A7 expression. To determine whether members of the NFI transcription factor family might contribute to differential regulation of the CYP3A4*1A and CYP3A7*1A proximal promoters, the CYP3A4*1A (−694/−48) or CYP3A7*1A (−685/−56) promoter-reporter constructs were cotransfected into HepG2 cells with expression vectors for NFIA1.1, NFIB2, NFIC1, NFIC2, NFIX1, or NFIX2. Cotransfection of the NFIA1.1, NFIB2, NFIC1, NFIC2, and NFIX1 variants enhanced the expression of luciferase under the control of the CYP3A4*1A proximal promoter, whereas NFIX2 had no effect on CYP3A4*1A promoter activity (Fig. 6A). In contrast, cotransfection of NFIB2, NFIC1, NFIC2, NFIX1, and NFIX2 reduced the expression of luciferase under the control of the CYP3A7*1A gene promoter, whereas NFIA1.1 had no effect on CYP3A7*1A promoter activity (Fig. 6B). These data demonstrate that members of the NFI transcription factor family are capable of differential regulation of the CYP3A4 and CYP3A7 proximal promoters in a promoter- and isoform-specific manner.
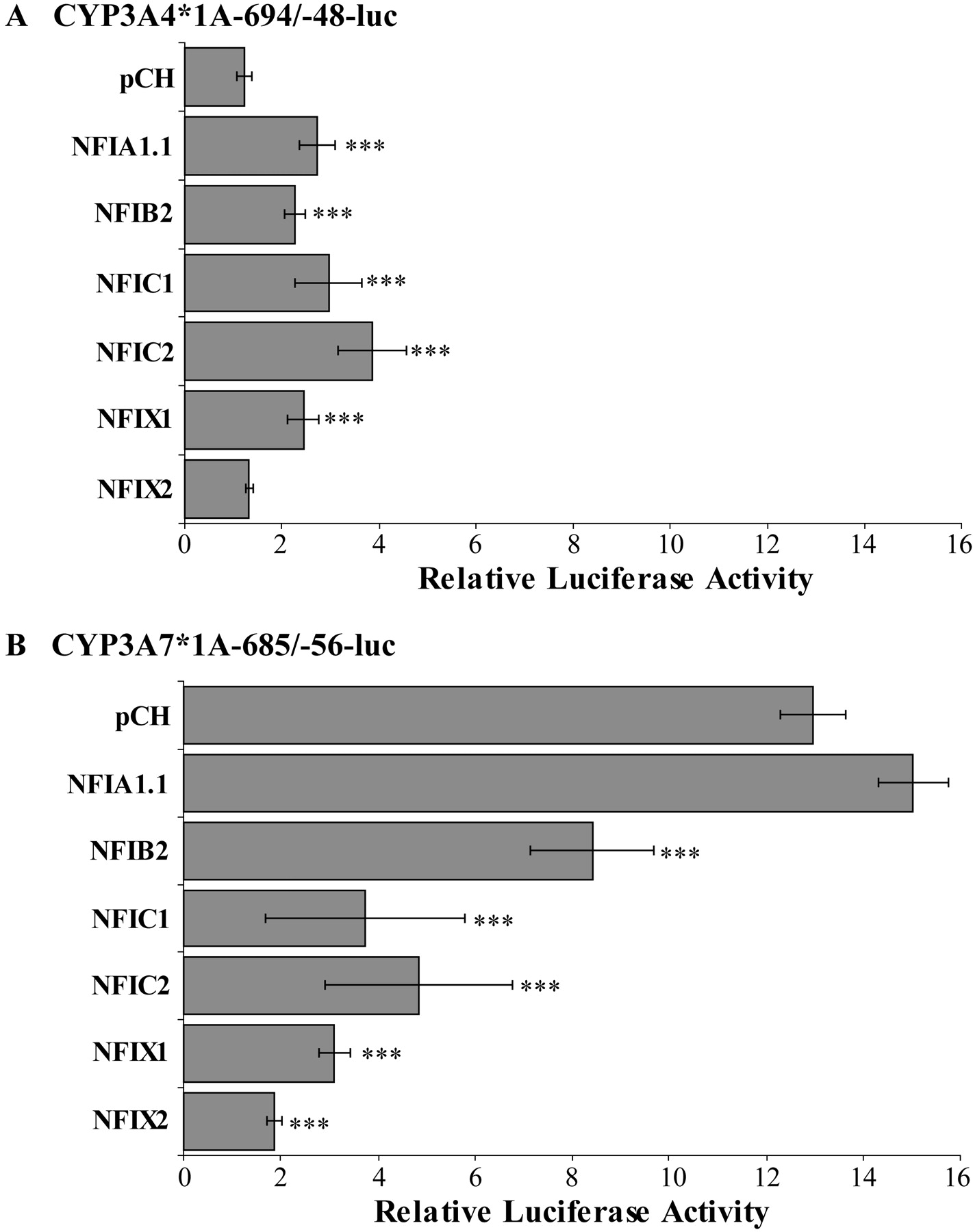
Regulation of CYP3A4*1A and CYP3A7*1A proximal promoters by NFI isoforms. Transient transfections of HepG2 cells were performed as described to determine whether NFI isoforms differentially regulate the CYP3A4*1A (A) and CYP3A7*1A (B) proximal promoters. Firefly luciferase activity was normalized to R. reniformis luciferase activity from the cotransfected control vector and bars represent luciferase activity relative to pGL3-basic cotransfected with the respective NFI isoform. Data shown are average ± S.D. of at least two independent transfections with four replicates in each transfection. Statistically significant differences (one-way analysis of variance, with Tukey's post hoc analysis, SPSS 17.0; SPSS Inc., Chicago, IL) from the relative luciferase activity from cells cotransfected with pCH are represented with ***, p < 0.001.
Although the proximal promoters of CYP3A4 and CYP3A7 are greater than 90% identical, the two promoters differ by a single base pair (A/C) within the sequences shown to be bound by the NFI and HNF3γ transcription factors in EMSAs (Figs. 1 and 2). To determine whether the differential regulation of the CYP3A4*1A and CYP3A7*1A proximal promoters by isoforms of the NFI family is dependent upon this sequence difference within the NFI/HNF3γ binding site, site-directed mutagenesis was performed to exchange the variant base between the two promoters (CYP3A7 m-232 A→C or CYP3A4 m-233 C→A). Mutation of position −233 (C→A) did not affect basal activity of the CYP3A4*1A−694/−48 proximal promoter (Fig. 7A); in contrast, a significant decrease in basal activity of the CYP3A7*1A−685/−56 promoter was observed upon mutation of position −232 (A→C) (Fig. 7B). Mutation of position −233 (C→A) of the CYP3A4*1A luciferase reporter construct blocked the NFIA1.1-mediated activation of luciferase activity in transfected HepG2 cells relative to that observed with the wild-type CYP3A4*1A promoter (Fig. 7A versus 6A) but had no effect on activation by the other NFI isoforms. Expression of NFIA1.1 up-regulates luciferase activity in HepG2 cells cotransfected with CYP3A7*1A m-232 (Fig. 7B) in a statistically significant manner, similar to results observed in transfections with the CYP3A4*1A proximal promoter (Fig. 6A) and in contrast to results observed with the wild-type CYP3A7*1A promoter (Fig. 6B). In contrast to the wild-type CYP3A7*1A proximal promoter, expression of NFIB2 and NFIC1 modestly increased promoter activity of CYP3A7*1A m-232, although the increase was not statistically significant (Fig. 7B). Mutation of position −232 (A→C) of the CYP3A7*1A luciferase promoter-reporter construct blocked down-regulation of luciferase activity in HepG2 cells cotransfected with NFIC2 and NFIX1 in contrast to results observed with the CYP3A7*1A wild-type proximal promoter (Fig. 7B versus 6B). Mutation of position −232 (A→C) of CYP3A7*1A had no effect on the ability of expression of NFIX2 to repress luciferase activity in cotransfected HepG2 cells (Fig. 7B versus 6B). These data suggest that isoforms of NFI regulate the CYP3A4 and CYP3A7 proximal promoters via multiple mechanisms.

Mutation of the HNF3/NFI binding sites in CYP3A4*1A and CYP3A7*1A alters regulation by NFI isoforms. Transient transfections of HepG2 cells were performed with the CYP3A4*1A m-233 (C→A) and CYP3A7*1A m-232 (A→C) variant promoters to determine the role of the HNF3/NFI binding site in differential regulation by isoforms of NFI. Firefly luciferase activity was normalized to R. reniformis luciferase activity from the cotransfected control vector and bars represent luciferase activity relative to pGL3-basic cotransfected with the respective NFI isoform. Data shown are average ± S.D. of at least two independent transfections with four replicates in each transfection. Statistically significant differences (one-way analysis of variance, with Tukey's post hoc analysis, SPSS 17.0; SPSS Inc., Chicago, IL) from the relative luciferase activity from cells cotransfected with pCH are represented as follows: ***, p < 0.001; **, p < 0.01; and *, p < 0.05.
Site-directed mutagenesis of the NFI/HNF3γ binding sites of the CYP3A4*1A and CYP3A7*1A proximal promoters suggests that although at least some of the activity of NFI transcription factor family members on these promoters is mediated by direct interactions with DNA of this region, additional direct or indirect interactions with promoter DNA and/or transcription factors nearby are partially responsible for differential regulation. The CYP3A7*1C allele is characterized by a region between −291 and −232 base pairs that has been replaced with the corresponding sequences of the CYP3A4*1A gene promoter, resulting in seven single base-pair changes within this region of the CYP3A7 proximal promoter, including an A→C at base −232 within the NF1/HNF3γ binding site. The variation occurring in the CYP3A7*1C promoter results in increased activation by pregnane X receptor and constitutive active receptor in vitro and aberrant postnatal expression of CYP3A7 in vivo, suggesting that this region may be important for developmental regulation (Burk et al., 2002). Transient transfection studies with a luciferase reporter construct containing −685 to −56 of the CYP3A7*1C promoter were performed to determine whether variation within the CYP3A7*1C allele in addition to −232 (Fig. 7B) mediates differential regulation of the CYP3A7 proximal promoter by members of the NFI transcription factor family (Fig. 8 versus Fig. 7B). Expression of NFIA1.1 and NFIB2 significantly increased luciferase activity from HepG2 cells cotransfected with the CYP3A7*1C promoter construct, whereas NFIC1 and NFIC2 modestly increased luciferase activity under the control of the CYP3A7*1C promoter. Expression of NFIX1 and NFIX2 had no effect on luciferase activity when cotransfected with the CYP3A7*1C luciferase construct. These data suggest that additional protein-protein or protein-DNA interactions that may be required occur through elements outside of those lying within the region defined by the CYP3A7*1C allele.

Regulation of CYP3A7*1C proximal promoter by NFI isoforms. Transient transfections of HepG2 cells were performed with the CYP3A7*1C variant promoter to determine the role of the HNF3/NFI binding site in differential regulation by isoforms of NFI. Firefly luciferase activity was normalized to R. reniformis luciferase activity from the cotransfected control vector and bars represent luciferase activity relative to pGL3-basic. Data shown are average ± S.D. of three independent transfections with four replicates in each transfection. Statistically significant differences from the relative luciferase activity from cells cotransfected with pCH are represented as follows: ***, p < 0.001 and **, p < 0.01.
Discussion
The ontogeny of the enzymes of the CYP3A family has been well characterized; however, the mechanisms responsible for changes in expression of CYP3A4 and CYP3A7 remain undetermined. We present data here from in vitro binding assays that demonstrate developmentally distinct transcription factor complexes from human liver that are capable of binding to the CYP3A4 and CYP3A7 proximal promoters. Distinct differences in mobility of the protein-DNA complexes formed using prenatal or postnatal nuclear proteins were observed to bind to sequences from the −243 to −220 region of the CYP3A4*1A and CYP3A7*1A promoters (Fig. 1). However, both prenatal and postnatal protein-DNA complexes contained members of the NFI transcription factor family as determined by both competitive binding assays and immunodepletion with an antibody that recognizes all NFI isoforms (Figs. 1 and 2). In mammals, the NFI transcription factor family is composed of isoforms that are differentially expressed during development (Chaudhry et al., 1997). Isoforms of the NFI transcription factor family are expressed from four genes with multiple splice variants from each gene possessing differing abilities to activate or repress transcription (Gründer et al., 2003). Further results from immunoblotting (Fig. 3A) confirm that isoforms of NFI are likely to be developmentally regulated in human liver. Furthermore, quantitative RT-PCR demonstrates developmental changes in expression of NFIC isoforms (Fig. 4B). However, alternative splicing of the NFI isoforms may further contribute to developmental changes in NFI expression and function in human liver.
Although developmental differences were observed in the mobility of protein-DNA complexes, there was only one complex that differed between assays with the CYP3A4*1A or CYP3A7*1A sequences. Competitive binding assays and immunodepletion demonstrate that HNF3γ from prenatal liver nuclear extracts is capable of interacting with the −243 to −219 element from the CYP3A7*1A proximal promoter but not the CYP3A4*1A promoter. Similar to the NFI transcription factor family, there are three members of the HNF3 (FOXA) transcription factor family that are important beginning very early in liver development and that are required for maintenance of homeostasis. Furthermore, the observation that HNF3γ does not interact with the CYP3A4 promoter element is consistent with a previous report in which overexpression of HNF3γ had no effect on the CYP3A4*1A proximal promoter in transient transfections of HepG2 or HeLa cells (Rodríguez-Antona et al., 2003).
In addition to the existence of developmentally distinct NFI complexes in human liver, results from reporter assays indicate that members of the NFI transcription factor family differentially regulate the CYP3A4 and CYP3A7 proximal promoters via multiple mechanisms that are promoter- and isoform-specific. All NFI isoforms, with the exception of NFIA1.1, were capable of reducing the promoter activity of a CYP3A7*1A reporter construct. Mutation of the A at −232 of the CYP3A7*1A HNF3/NFI binding site to a C, which occurs in the CYP3A4*1A element and is bound by NFI only, reduced the basal promoter activity of CYP3A7*1A. In addition, NFIB2 induced luciferase activity from the CYP3A7*1A m-232 promoter when cotransfected into HepG2 cells, whereas NFIC and NFIX1 isoforms were no longer able to inhibit promoter activity. These data suggest that inhibition of the CYP3A7*1A promoter activity by the NFIB2, NFIC1, NFIC2, and NFIX1 isoforms is probably due to NFI transcription factors displacing HNF3γ from the CYP3A7*1A proximal promoter. The results of the transactivation studies on the CYP3A7*1A proximal promoter along with the developmental expression patterns of the NFIC isoforms are consistent with the hypothesis that developmental changes in NFIC may play a role in the suppression of CYP3A7 expression in postnatal tissue.
Although regulation of the CYP3A7*1A proximal promoter by NFIB2, NFIC1, NFIC2, and NFIX1 seems to rely on competitive binding to promoter sequences, studies of the regulation of the CYP3A4*1A promoter suggest a more complex mechanism that is dependent upon cooperative interactions with other transcription factors. Addition of a HNF3γ binding site by the mutation of nucleotide −233 (C→A) increased basal activity modestly (approximately 2-fold) but had no effect on the ability of NFIB2, NFIC1, NFIC2, or NFIX1 to trans-activate the CYP3A4*1A proximal promoter. These data are consistent with those presented previously (Rodríguez-Antona et al., 2003) and suggest that DNA-HNF3γ interactions alone are not sufficient for either activation of the CYP3A7*1A promoter or to reverse repression of the CYP3A4*1A proximal promoter. A comparative study of the two proximal regulatory regions identified a binding site for the YY1 transcription factor in the CYP3A4*1A, but not CYP3A7*1A, promoter (Saito et al., 2001). YY1 is a possible candidate transcription factor in the differential regulation of CYP3A4/CYP3A7 because of its unique ability to either activate or repress transcription in a cell- and promoter-specific manner. The role of interactions among HNF3γ, NFI proteins, and YY1 on the differential regulation of the CYP3A7 and CYP3A4 promoters remains to be determined.
Regulation of CYP3A4*1A and CYP3A7*1A promoter activity by NFIA1.1 occurs by an alternative mechanism. NFIA1.1 does not affect the wild-type CYP3A7*1A promoter activity but activates both the CYP3A4*1A promoter and CYP3A7*1A m-232 variant promoter. Transient transfections with the CYP3A4*1A m-233 variant promoter and CYP3A7*1C proximal promoter provide additional evidence to suggest that NFIA1.1 does not bind to the CYP3A7*1A HNF3/NFI element and thus is not able to disrupt HNF3γ binding to the CYP3A7*1A promoter.
Similar to the other NFI isoforms, NFIX2 demonstrates differential regulation of the CYP3A4*1A and CYP3A7*1A proximal promoters. However, in contrast to the other NFI isoforms, regulation of the CYP3A7*1A proximal promoter by NFIX2 does not occur as a result of interactions with the sequences between −242 and −219 because mutation of this site does not affect the ability of NFIX2 to repress luciferase activity in transient cotransfections. In addition, cotransfection of NFIX2 had no effect on the CYP3A4*1A wild-type, CYP3A4*1A m-233 variant, or CYP3A7*1C proximal promoters. The absence of an effect on the CYP3A7*1C promoter suggests that NFIX2 interacts with sequences just upstream of the HNF3/NFI site at −242/−219, which includes the proximal pregnane X receptor response element or an additional HNF3 site (Bombail et al., 2004), both of which are affected by the CYP3A7*1C polymorphism.
The binding of HNF3 and NFI proteins to the CYP3A7 gene promoter is intriguing because developmental regulation of α-fetoprotein has been shown to involve both HNF3 and NFI transcription factors (Bois-Joyeux and Danan, 1994; Crowe et al., 1999; Huang et al., 2002). Both NFI and HNF3 transcription factors possess chromatin remodeling activity, which facilitates the binding of other transcription factors such as C/EBP and the glucocorticoid receptor (Cirillo et al., 2002; Hebbar and Archer, 2003, 2007). The ability of HNF3 transcription factors to reposition nucleosomes near the α-fetoprotein gene has been suggested to be one component of the mechanisms underlying developmental gene expression (Crowe et al., 1999). The interactions of HNF3 and NFI with sequences from the CYP3A7 gene promoter make a mechanism of developmental regulation via chromatin remodeling plausible. However, the in vitro binding and reporter assays used in this study do not address the role of chromatin structure in protein-DNA or protein-protein interactions at the CYP3A locus.
In conclusion, the data presented indicate that variant isoforms of NFI are capable of differentially regulating the CYP3A4*1A and CYP3A7*1A promoters; however, in vitro binding data indicate that NFI-containing complexes found in prenatal as well as postnatal liver interact with elements from both promoters without distinction. In contrast, HNF3γ from prenatal liver is capable of interacting with only the CYP3A7*1A proximal promoter and may play a role in prenatal expression of this enzyme. However, data from transient transfection assays suggest that additional determinants play a role in regulating developmental expression of the human CYP3A enzymes. These determinants may include protein-protein interactions between transcription factors binding to proximal and distal promoter elements or chromatin remodeling that modulates access of transcription factors to their cognate DNA response elements as well as direct protein-protein interactions with other transcription factors binding to the CYP3A locus. In addition, NFI regulation of the CYP3A7*1C promoter suggests an alternative mechanism for postnatal expression of CYP3A7 in individuals carrying this allele. Further studies are required to determine the developmental expression of NFI splice variants, the role of HNF3γ and YY1 in the regulation of CYP3A7 and CYP3A4 expression, and the role of chromatin structure on the interaction of transcription factors with the CYP3A locus.
Footnotes
-
This work was supported by the Katherine Berry Richardson Foundation [Grant 01.4223].
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.109.055699
-
ABBREVIATIONS:
- Received February 17, 2009.
- Accepted August 25, 2009.
- © 2009 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}