


Abstract
Compared with rodents and many other animal species, the human cytochrome P450 (P450) Cyp2c gene cluster varies significantly in the multiplicity of functional genes and in the substrate specificity of its enzymes. As a consequence, the use of wild-type animal models to predict the role of human CYP2C enzymes in drug metabolism and drug-drug interactions is limited. Within the human CYP2C cluster CYP2C9 is of particular importance, because it is one of the most abundant P450 enzymes in human liver, and it is involved in the metabolism of a wide variety of important drugs and environmental chemicals. To investigate the in vivo functions of cytochrome P450 Cyp2c genes and to establish a model for studying the functions of CYP2C9 in vivo, we have generated a mouse model with a deletion of the murine Cyp2c gene cluster and a corresponding humanized model expressing CYP2C9 specifically in the liver. Despite the high number of functional genes in the mouse Cyp2c cluster and the reported roles of some of these proteins in different biological processes, mice deleted for Cyp2c genes were viable and fertile but showed certain phenotypic alterations in the liver. The expression of CYP2C9 in the liver also resulted in viable animals active in the metabolism and disposition of a number of CYP2C9 substrates. These mouse lines provide a powerful tool for studying the role of Cyp2c genes and of CYP2C9 in particular in drug disposition and as a factor in drug-drug interaction.
Introduction
The heme-containing cytochrome P450 (P450) enzymes play a central role in the oxidative metabolism of a vast range of small molecule substrates, such as endogenous compounds, environmental chemicals, and drugs. Although 57 P450 enzymes have been described in humans (Nelson et al., 2004), only a small number account for the majority of P450-mediated metabolism of clinically used drugs. In this regard, it has been estimated that ∼95% of drug metabolism is mediated by either CYP3A4/5, CYP2D6, CYP2C9/2C19, or CYP1A1/1A2 (Guengerich, 2008). To overcome the differences that exist in the substrate specificity, regulation of expression, and multiplicity of P450s between species, significant efforts have been made in recent years to humanize mice for some of these key metabolic enzymes, the aim being to provide animal models that better predict human pathways of drug metabolism. In this regard, humanized mouse models for CYP3A4 (Cheung et al., 2006; van Herwaarden et al., 2007; Hasegawa et al., 2011), CYP2D6 (Corchero et al., 2001; Scheer et al., 2012), CYP2C19 (Löfgren et al., 2008), and CYP1A1/1A2 (Cheung et al., 2005; Jiang et al., 2005; Dragin et al., 2007) have been created using a variety of different approaches, either with or without deletion of the corresponding mouse genes. In addition, fertile and viable knockout models of the mouse Cyp3a (van Herwaarden et al., 2007; Hasegawa et al., 2011), Cyp2d (Scheer et al., 2012), and Cyp1a (Dragin et al., 2007) gene clusters have been described, which are associated with only minor overt phenotypic changes compared with WT controls.
The mouse and the human CYP2C cluster differ significantly in their genomic organization, with 15 functional genes described in mice compared with only 4 genes in human (Nelson et al., 2004). Because of these differences in multiplicity and also in sequence variation, it is not possible to define orthologous genes between both species. Fourteen of the functional genes within the mouse Cyp2c cluster are located in close proximity within approximately 1.2 Mb of mouse chromosome 19, whereas Cyp2c44, albeit on the same chromosome, is separated from the other genes by approximately 4 Mb (Nelson et al., 2004). The four functional genes within the human CYP2C cluster are CYP2C8, CYP2C9, CYP2C18, and CYP2C19. Together they account for approximately 25% of all P450-mediated hepatic drug metabolism (Williams et al., 2004; Guengerich, 2008). Among the human CYP2C enzymes, CYP2C9 is of key importance, because, apart from CYP3A4, it is the largest contributor to the total human liver microsomal P450 content. CYP2C9 is involved in the disposition of some major classes of therapeutic drugs, including anticoagulants and nonsteroidal anti-inflammatory agents, such as warfarin or diclofenac, respectively. It has been estimated to be responsible for the metabolic clearance of up to 15% of all drugs that undergo phase I metabolism and has been associated with a number of drug-drug interactions. CYP2C9 is also polymorphic in the human population, with a number of null or low-activity alleles, and such polymorphisms are directly linked to the adverse effects of, for example, warfarin (Stubbins et al., 1996; Rettie and Jones, 2005).
Despite their important role in drug metabolism and the significant species differences in Cyp2c genes, there are only a few published reports on humanized mouse models. A mouse humanized for CYP2C18/19 that expressed catalytically active CYP2C19 in the liver and intestine has recently been reported (Löfgren et al., 2008). However, neither a Cyp2c knockout nor a CYP2C9 humanized model has been described. The complex genetic organization of the mouse Cyp2c cluster, which is significantly larger and contains many more functional genes than any other Cyp gene cluster, could explain why such models have not been generated. Furthermore, CYP2C enzymes are expressed in the vascular epithelium, and it has been speculated that enzymes such as CYP2C9 play important roles in angiogenesis and vascular development (Michaelis et al., 2008; Webler et al., 2008). Therefore, the deletion of the Cyp2c cluster could potentially have deleterious effects both during and after embryonic development.
Here we describe the generation of a Cyp2c cluster knockout (Cyp2c KO) mouse line, in which 14 of the 15 functional genes of this gene family have been deleted. Furthermore, we used a sophisticated Cre recombinase-mediated insertion strategy to replace the mouse Cyp2c cluster with a liver-specific CYP2C9 expression cassette. Both the Cyp2c KO and the CYP2C9 humanized (hCYP2C9) mouse line were viable and fertile but showed certain phenotypic alterations in the liver. The initial characterization of these mouse lines is described, as well as the changes in activity toward selected CYP2C9 probe substrates.
Materials and Methods
Animal Husbandry.
Mice were kept as described previously (Scheer et al., 2008).
DNA Constructs and Cloning.
For targeting the Cyp2c55 gene locus, a basic vector containing a neomycin expression cassette, a loxP and f3 site, was constructed in pBluescript. For the neomycin cassette, the translational start ATG and the corresponding promoter are separated from an ATG-deficient neomycin (5prime]ΔNeo) in frame by the loxP site, such that additional amino acids encoded by the loxP site are fused to the N terminus of neomycin. This constellation gives rise to a functional protein resulting in G418 resistance upon expression (Hasegawa et al., 2011). A 5.4-kb genomic sequence immediately upstream from the translational start site of the mouse Cyp2c55 gene and a 3.0-kb fragment comprising exons 2 and 3 of Cyp2c55, both used as targeting arms for homologous recombination, were obtained by Red/ET recombineering (Zhang et al., 1998) and subcloned into the basic targeting vector as depicted in Fig. 1C.

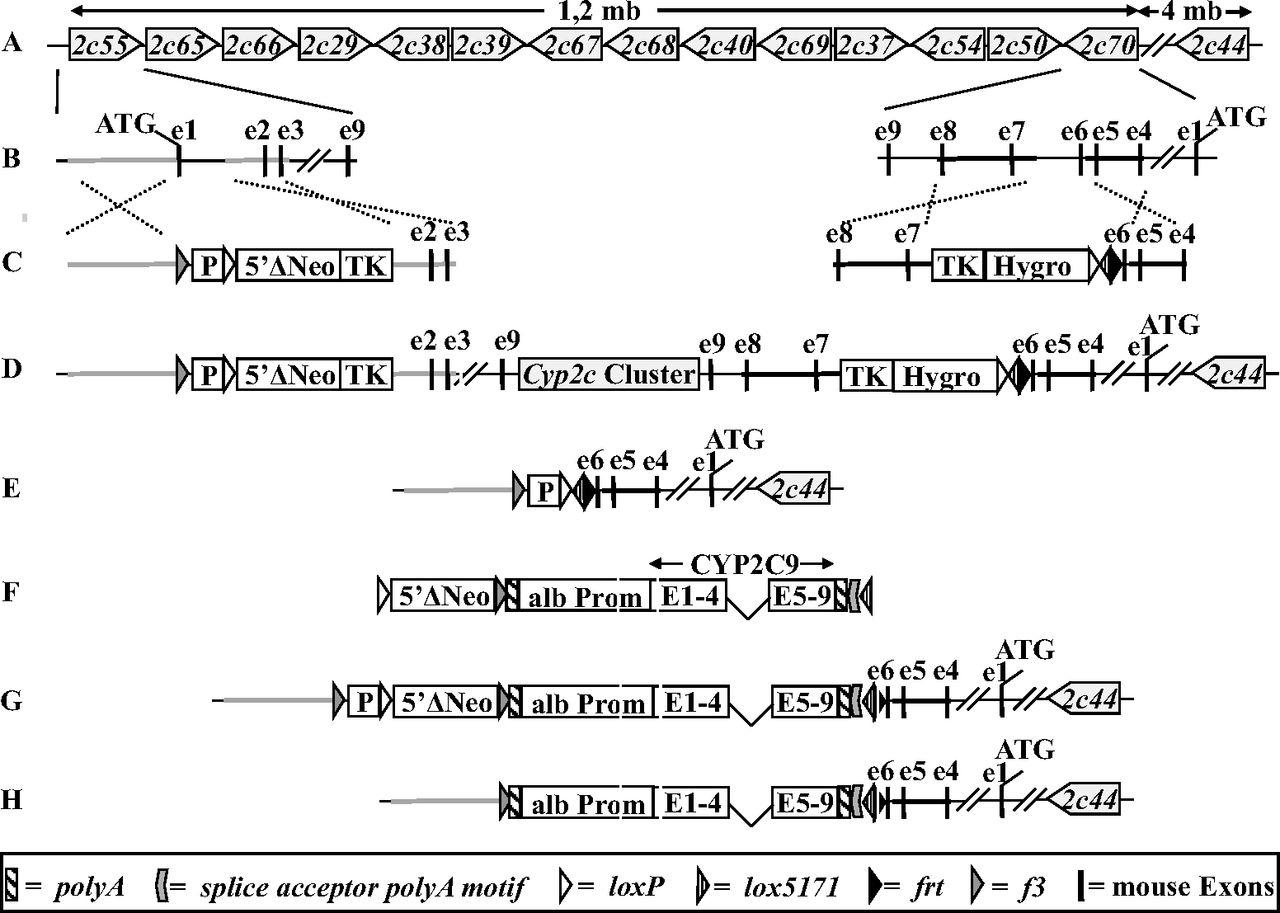
Strategy for generating Cyp2c KO and hCYP2C9 mice. A, schematic representation of the chromosomal organization and orientation of functional genes within the mouse Cyp2c cluster. B, exon/intron structure of Cyp2c55 and Cyp2c70. Exons are represented as black bars, and the ATGs mark the translational start sites of both genes. The positions of the targeting arms for homologous recombination are highlighted in gray (Cyp2c55) and black (Cyp2c70), respectively. C, vectors used for targeting of Cyp2c55 (left) and Cyp2c70 (right) by homologous recombination. loxP, lox5171, frt, and f3 sites are represented as white, stripped, black, and gray triangles, respectively. D, genomic organization of the Cyp2c cluster in double-targeted ES cells after insertion of the targeting vectors. E, deletion of the mouse Cyp2c cluster after Cre-mediated recombination at the loxP sites. F, CYP2C9 expression cassette used for Cre-mediated insertion via the loxP and lox5171 sites. G, mouse Cyp2c locus after Cre-mediated insertion of the CYP2C9 expression cassette. H, mouse Cyp2c locus in the hCYP2C9 model after Flp-mediated deletion of the neomycin expression cassette. For the sake of clarity sequences are not drawn to scale. Hyg, hygromycin expression cassette; TK, thymidine kinase expression cassette; alb Prom, mouse albumin enhancer/promoter element; P, promoter that drives the expression of neomycin; 5prime]ΔNeo, ATG-deficient neomycin.
For targeting the Cyp2c70 gene locus, a basic vector containing thymidine kinase and hygromycin expression cassettes, a loxP, lox5171, and frt site, was constructed in pBluescript. A 5.5-kb genomic sequence comprising exons 7 and 8 of the mouse Cyp2c70 gene and a 3.1-kb fragment comprising exons 4 and 5 of Cyp2c70, both used as targeting arms for homologous recombination, were obtained by Red/ET recombineering and subcloned into the basic targeting vector as depicted in Fig. 1C.
Generation and Molecular Characterization of Targeted Embryonic Stem Cells.
Culture and targeted mutagenesis of embryonic stem (ES) cells were performed as described previously (Hogan et al., 1994). Details on the generation and molecular characterization of ES cell clones targeted at Cyp2c55 and Cyp2c70 gene loci are described in the Supplemental Materials and Methods.
Generation and Molecular Characterization of Cyp2c Knockout and CYP2C9 Humanized Mice.
For the generation of Cyp2c KO and hCYP2C9 mice, ES cell clones with a deletion of the Cyp2c cluster and an insertion of the human CYP2C9 expression cassette, respectively, were expanded, injected into BALBc blastocysts and transferred into foster mothers as described previously (Hogan et al., 1994). Litters from these foster mothers were inspected visually, and chimerism was determined by hair color. Highly chimeric animals were used for breeding with either C57BL/6 WT mice (Cyp2c KO) or with an efficient flipase (Flpe) deleter strain carrying a transgene that expresses Flpe in the germ line (hCYP2C9). The latter approach led to a deletion of the neomycin expression cassette in the offspring (Fig. 1H). The Flpe-deleter strains was generated in-house on a C57BL/6 genetic background. A detailed description on the molecular characterization of the Cyp2c KO and hCYP2C9 mice is given in the Supplemental Materials and Methods.
Animal Experimentation.
All animal procedures were performed under a United Kingdom Home Office license, and all animal studies were approved by the Ethical Review Committee, University of Dundee. Homozygous mice for each transgenic line were used for experimental studies. C57BL/6 animals of the same age purchased from Harlan Olac (Bicester, Oxon, UK) were used as WT controls. All housing conditions were as described previously (Scheer et al., 2012).
Terminal Procedures.
Mice were killed by exposure to a rising concentration of CO2, and blood was collected by cardiac puncture into lithium/heparin-coated tubes for plasma preparation. Details on the procedures of tissue preparation, immunoblot analysis of Cyp2c and CYP2C9 protein expression, quantitative reverse transcriptase-polymerase chain reaction, and in vitro and in vivo determination of CYP2C-dependent activities are described under the Supplemental Materials and Methods.
Results
Generation of CYP2C9 Humanized and Cyp2c Cluster Knockout Mice.
The strategy of generating hCYP2C9 and Cyp2c KO mice is illustrated in Fig. 1. The mouse Cyp2c cluster with the exception of Cyp2c44, which is located 4 Mb away, was flanked with Cre recombinase recognition (loxP) sites using two consecutive rounds of targeting in mouse ES cells (Fig. 1, A–D). Subsequent Cre-mediated recombination between the loxP sites resulted in a deletion of all exons and introns of Cyp2c55, Cyp2c65, Cyp2c66, Cyp2c29, Cyp2c38, Cyp2c39, Cyp2c67, Cyp2c68, Cyp2c40, Cyp2c69, Cyp2c37, Cyp2c54, and Cyp2c50, as well as a deletion of exons 7 to 9 of Cyp2c70 (Fig. 1E). The Cyp2c deleted ES cells were used to derive Cyp2c KO mice.
hCYP2C9 mice were generated from the Cyp2c-deleted ES cells described above by further Cre-mediated insertion of an expression cassette in which human CYP2C9 is under control of the liver-specific mouse albumin promoter (Fig. 1F). Cre-mediated insertion of this expression cassette was achieved by recombination via the loxP site that resulted from deletion of the mouse Cyp2c cluster and a heterospecific lox5171 site (Lee and Saito, 1998), which was coinserted with the Cyp2c70 targeting vector (Fig. 1G). A neomycin complementation approach was used to achieve high stringency for the selection of ES cell clones with a correct Cre-mediated insertion (Hasegawa et al., 2011). To terminate any potential transcription from the Cyp2c55 or Cyp2c70 promoters, a polyA motif and a splice acceptor polyA motif were included upstream of the albumin promoter and downstream of the CYP2C9 expression cassette, respectively. Transgenic mice from correctly targeted ES cells were generated subsequently, and by further crosses with a mouse line expressing the Flp-recombinase in the germ line the neomycin expression cassette was deleted via recombination at the Flp recombinase recognition (frt) sites (Fig. 1H). Homozygous hCYP2C9 and Cyp2c KO mice were obtained by breeding.
Phenotypic Characterization of CYP2C9 Humanized and Cyp2c Cluster Knockout Mice.
Homozygous humanized and knockout mice appeared normal, could not be distinguished from WT animals, and had normal body weights, liver weights, and fertility (data not shown). To further characterize the hCYP2C9 and Cyp2c KO mice, plasma samples were analyzed for albumin, alkaline phosphatase, alanine amino transferase, aspartate aminotransferase, direct and total bilirubin, high- and low-density lipoproteins, triglycerides, and cholesterol (n = 3 mice/line). The only significant phenotypic change in the hCYP2C9 mice was an ∼0.5-fold decrease in alkaline phosphatase activity. The Cyp2c KO mice exhibited a similar change and also a small but significant decrease (∼0.7-fold) in high-density lipoprotein and cholesterol (Supplemental Fig. 1). Alanine amino transferase and aspartate aminotransferase activities were also increased in both genetically modified mouse lines, but the variability between individual mice were high and the changes were not statistically significant. To assess whether the increased alanine amino transferase and aspartate aminotransferase activities may indicate hepatotoxicity, we carried out a hematoxylin and eosin analysis on livers of hCYP2C9 and Cyp2c KO mice. Compared with WT controls, this analysis indeed showed an increased infiltration by lymphocytes and neutrophils and unidentified “ovoid” cells in the portal areas of samples from both the Cyp2cKO and hCYP2C9 mice (Supplemental Fig. 2). The degree of this pathology was variable between samples.
Human CYP2C9 and Mouse Cyp2c Expression in Cyp2c KO and CYP2C9 Humanized Mice.
The expression of human CYP2C9 mRNA in the liver of WT, Cyp2c KO, and hCYP2C9 mice was determined by TaqMan analysis. This confirmed the expression of CYP2C9 in the hCYP2C9 model (Fig. 2A). The average Ct value of 21.8 (n = 3 mice) for CYP2C9 mRNA in the hCYP2C9 mice was comparable to many hepatic mouse cytochrome P450 genes. The CYP2C9 mRNA level in other organs was negligible (data not shown). For example, the average Ct value for CYP2C9 in the duodenum was 29.0, i.e., at the limit of detection. The loss of hepatic mouse Cyp2c mRNA expression in the Cyp2c KO and hCYP2C9 models was confirmed through the finding that the mRNAs for the three distant mouse Cyp2c genes Cyp2c55, Cyp2c39, and Cyp2c70 could be readily detected in WT animals but not in homozygous Cyp2c KO and hCYP2C9 mice (Fig. 2B). No significant changes in the expression of the remaining mouse Cyp2c gene Cyp2c44 were measured in either the Cyp2c KO or hCYP2C9 animals (Fig. 2C).
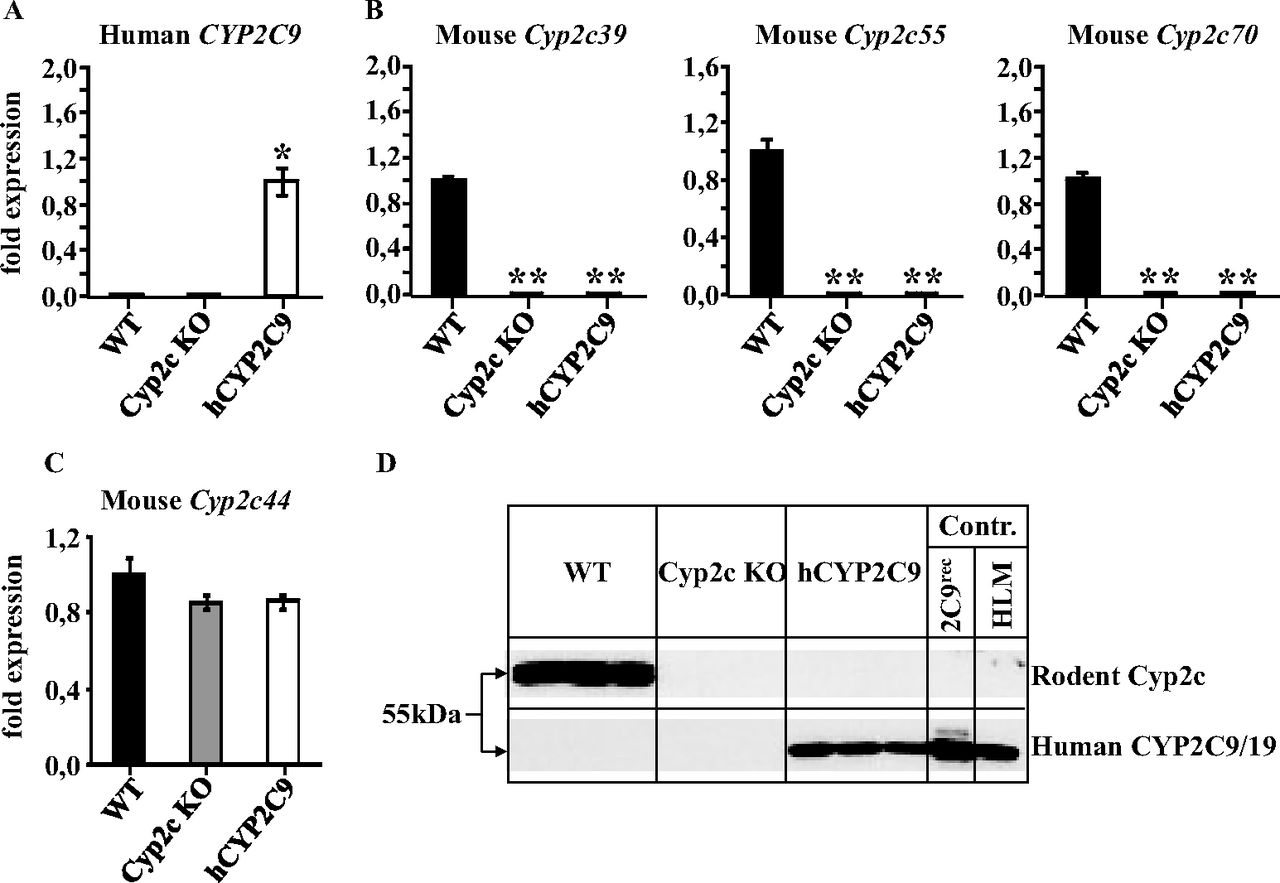
Analysis of Cyp2c and CYP2C9 expression in WT, Cyp2c KO, and hCYP2C9 mice. Relative expression levels of human CYP2C9 (A), mouse Cyp2c39, Cyp2c55, and Cyp2c70 (B), and mouse Cyp2c44 mRNA (C) in the liver of WT, Cyp2c KO, and hCYP2C9 mice. Expression levels in hCYP2C9 mice (in case of CYP2C9 mRNA) and in WT animals (in case of Cyp2c39, Cyp2c55, Cyp2c70, and Cyp2c44 mRNA) were arbitrarily set as 1. Data are expressed as means ± S.D. (n = 3 mice/genotype). Transgenic mice were compared with WT animals using a Student's t test (two-sided). Statistically different from control: *, p < 0.05; **, p < 0.01. D, mouse Cyp2c (upper lane) and human CYP2C9 (lower lane) protein expression in liver microsomes from WT, Cyp2c KO, and hCYP2C9 mice shown by Western blot analysis using a mouse-specific monoclonal anti-Cyp2c or a human-specific CYP2C9/19 antibody, respectively. The positive controls were recombinant CYP2C9 (2C9rec) and human liver microsomes (HLM), respectively.
To analyze the expression of CYP2C proteins in the livers of WT, Cyp2c KO, and hCYP2D9 mice, microsomes from these three mouse lines were studied by Western blotting. With use of a mouse Cyp2c-specific antibody, immunoreactive Cyp2c proteins were detected in WT but not in Cyp2c KO or hCYP2C9 mice, confirming the loss of mouse Cyp2c protein expression in these transgenic mouse lines (Fig. 2D). With use of a human-specific CYP2C9/19 antibody, CYP2C9 was only detected in liver microsomes from hCYP2C9 mice (Fig. 2D), the level of expression being comparable to that detected in human liver microsomes.
Cytochrome P450-Mediated Catalytic Activity in WT, Cyp2c KO, and hCYP2C9 Mice.
In Cyp2c KO mice, tolbutamide methylhydroxylase activity, a prototypical CYP2C substrate, was markedly decreased (91%) compared with WT controls, suggesting that murine Cyp2c proteins play a major role in the metabolism of this compound in mice (Fig. 3A). Tolbutamide methylhydroxylase activity in microsomes from the hCYP2C9 mice was significantly higher than that in mice null for the Cyp2c proteins (4.5-fold), demonstrating that the CYP2C9 protein is catalytically active. The tolbutamide methylhydroxylase activity of human liver microsomes was in between that of WT and hCYP2C9 mice.

In vitro metabolism of CYP2C9 probe substrates by liver microsomes from WT, Cyp2c KO, and hCYP2C9 mice and humans. A, tolbutamide methylhydroxylation. B, diclofenac 4prime]-hydroxylation. Data are expressed as means ± S.D. (n = 3 for all mouse lines). Activities in samples from Cyp2c KO mice were compared with that from WT and hCYP2C9 mice using a Student's t test (two-sided) Statistically different from these mouse lines: *, p < 0.05; **, p < 0.01. HLM, human liver microsomes.
Diclofenac 4-hydroxylation in hCYP2C9 mice was comparable to that in human liver microsomes and was significantly higher than for WT and Cyp2c KO animals (Fig. 3B). The ∼2-fold higher activity in hCYP2C9 compared with Cyp2c KO mice further confirmed the functionality of the CYP2C9 protein in the hCYP2C9 mouse line. Of interest, diclofenac 4-hydroxylase activity was similar in WT and Cyp2c KO mice, indicating that diclofenac is not a major substrate of the deleted mouse Cyp2c enzymes.
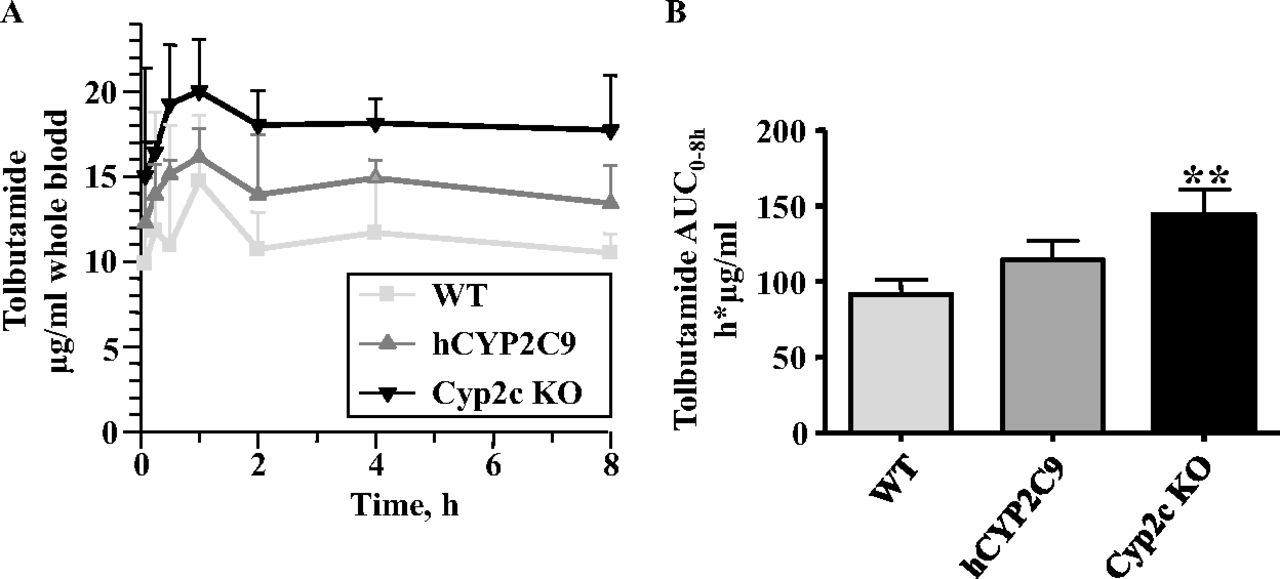
In vivo pharmacokinetics of tolbutamide in WT, Cyp2c KO, and CYP2C9 humanized mice. The pharmacokinetics of tolbutamide was determined in WT, Cyp2c KO, and hCYP2C9 mice. Over a time period of 8 h, the highest tolbutamide levels were observed in Cyp2c KO animals, followed by hCYP2C9 and WT mice (n = 3 mice/line) (Fig. 4A). Furthermore, compared with WT controls, the tolbutamide area under the concentration versus time curve (AUC) was significantly increased by ∼1.6-fold in Cyp2c KO mice (Fig. 4B). hCYP2C9 mice showed an ∼1.25-fold higher AUC than WT animals, but this change was not statistically significant. Overall, the pharmacokinetics of tolbutamide in these three mouse lines was in agreement with the in vitro results and confirmed the contribution of the mouse Cyp2c enzymes and human CYP2C9 in the metabolism of this compound in vivo. It should be noted that signs of toxicity were observed in all three mouse lines at the tested dose level (5 mg/kg i.p.), so that it was decided to terminate the study at 8 h after administration of tolbutamide. Conclusions from the in vivo study therefore need to be drawn with caution.

Pharmacokinetics of tolbutamide in WT, Cyp2c KO, and hCYP2C9 mice. A, concentration versus time profiles. B, areas under the concentration versus time (0–8 h) curves (AUC0–8h). Data are expressed as means ± S.D. (n = 3 mice/genotype). AUCs for Cyp2c KO and hCYP2C9 mice were compared with those for WT controls using a Student's t test (two-sided). Statistically different from control: **, p < 0.01.
Inhibition of Tolbutamide Hydroxylation in WT, Cyp2c KO, and hCYP2C9 Mice.
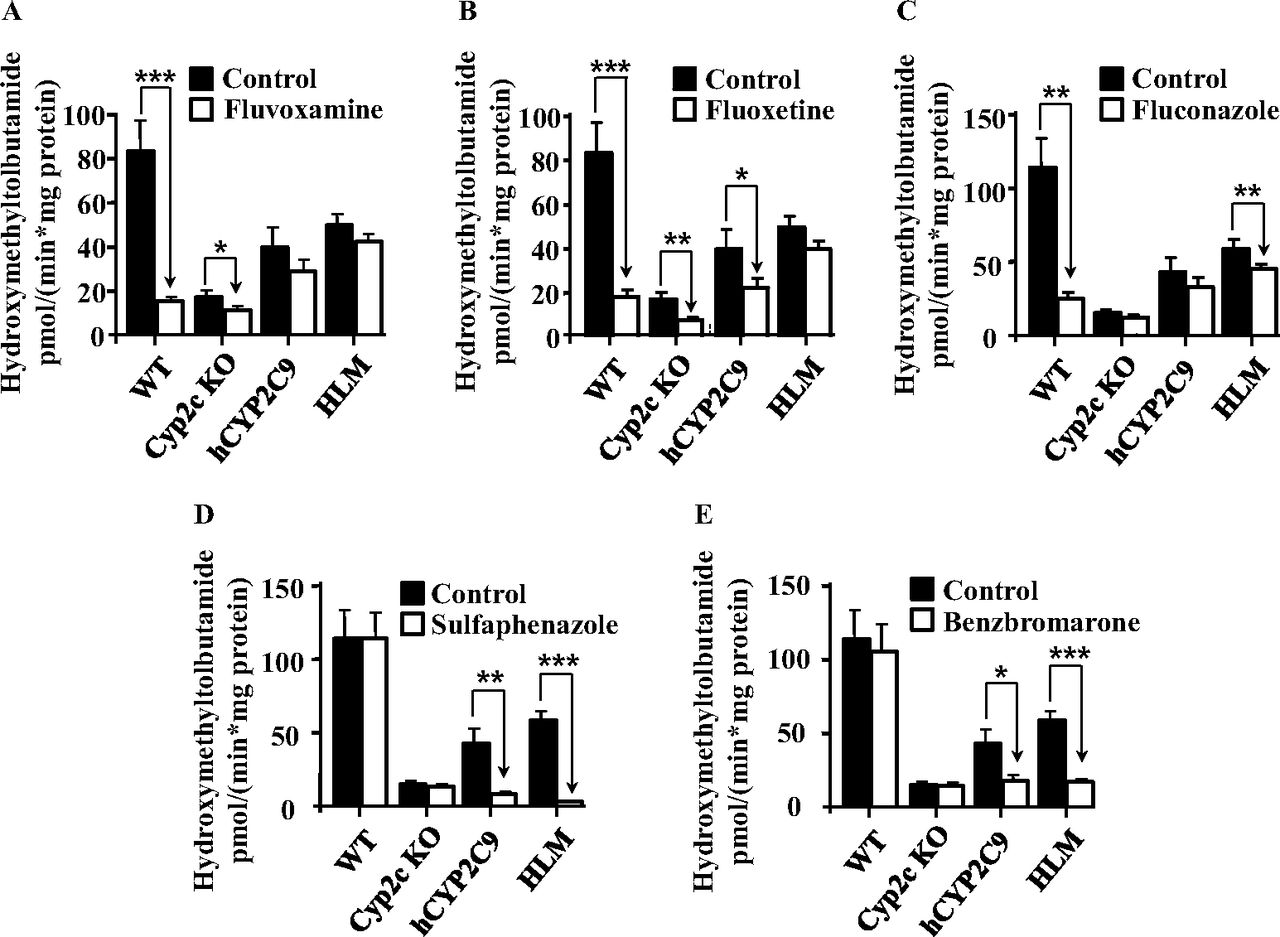
The effect of different CYP2C inhibitors on the hydroxylation of tolbutamide was determined using liver microsomes from WT, Cyp2c KO, and hCYP2C9 mice, and the results were compared with the inhibition observed in human liver microsomes. At the concentrations used, fluvoxamine, fluoxetine, and fluconazole strongly inhibited tolbutamide methylhydroxylase activity in microsomes from WT mice by >75%, but with 14 to 40% inhibition the effect in samples from hCYP2C9 mice or human liver was much smaller (Fig. 5, A–C). Of interest, some minor inhibition in liver microsomes from Cyp2c KO mice was also observed with fluvoxamine and fluoxetine, indicating that other enzymes might be involved. In contrast, sulfaphenazole and benzbromarone strongly inhibited the hydroxylation of tolbutamide in hepatic microsomal samples from hCYP2C9 mice and humans by 60 to 90%, but had no effect on this reaction in microsomes from WT or Cyp2c KO mice (Fig. 5, D–E). In summary, the activity of the inhibitors in samples of the hCYP2C9 mice was in good agreement with those from human liver microsomes.

Inhibition of vitro metabolism of tolbutamide in liver microsomes from WT, Cyp2c KO, and hCYP2C9 mice and humans. Tolbutamide methylhydroxylation either without inhibitor (squlf]) or during coincubation with 2 μM fluvoxamine (A), 15 μM fluoxetine (B), 5 μM fluconazole (C), 50 μM sulfaphenazole (D), and 0.25 μM benzbromarone (E) (squlo]). Tolbutamide concentration was 100 μM, microsomal protein concentration was 0.5 mg/ml, and incubation time was 20 min in all experiments. Data are expressed as means ± S.D. (n = 3 for all mouse lines). Activities of inhibitor-treated samples were compared with that from the corresponding control group using a Student's t test (two-sided). Statistically different from control: *, p < 0.05; **, p < 0.01; ***, p < 0.001. HLM, pooled human liver microsomes.
Compensatory Changes in the Expression of Other Drug-Metabolizing Enzymes in Cyp2c KO and CYP2C9 Humanized Mice.
The deletion of cytochrome P450 genes in the mouse can result in compensatory changes in other pathways of drug metabolism (van Waterschoot et al., 2008) We therefore compared the mRNA levels of a selected number of genes coding for key mouse drug-metabolizing enzymes by quantitative real-time polymerase chain reaction analysis (n = 3 mice/line). Compared with the WT controls, no significant changes in any of the hepatic cytochrome P450 genes analyzed (Cyp3a11, Cyp3a13, Cyp2d9, Cyp2d22, and Cyp2d26) were observed in hCYP2C9 or Cyp2c KO mice (Fig. 6). However, moderate but statistically significant decreases in the hepatic mRNA expression levels of Ugt1a6 (0.7- and 0.6-fold), Ugt2b5 (0.6- and 0.7-fold), and Ugt2b34 (0.6- and 0.6-fold) were found in hCYP2C9 and Cyp2c KO mice, respectively. No significant changes in the expression of any of these genes were observed in the intestine of the two transgenic mouse lines (data not shown).

Hepatic mRNA expression levels of selected genes coding for drug-metabolizing enzymes in WT, Cyp2c KO, and hCYP2C9 mice. A, relative expression levels of mouse Cyp3a11, Cyp3a13, Cyp2d9, Cyp2d22, Cyp2d26, Ugt1a6, Ugt2b5, and Ugt2b34 mRNA in the liver of WT, Cyp2c KO, and hCYP2C9 mice. Expression levels in WT animals were arbitrarily set as 1. Data are expressed as means ± S.D. (n = 3 mice/genotype). Transgenic mice were compared with WT animals using a Student's t test (two-sided). Statistically different from control: *, p < 0.05; **, p < 0.01.
Discussion
CYP2C9 plays a major role in the metabolic clearance of many important classes of therapeutic drugs, such as nonsteroidal anti-inflammatory oral anticoagulant, and oral hypoglycemic agents. Furthermore, it is one of the most abundant cytochrome P450 enzymes expressed in human liver, and pharmacological inhibition or pharmacogenetic variability of CYP2C9 can be associated with severe adverse drug reactions (Rettie and Jones, 2005).
In this report we have generated a model that can be used to study the in vivo consequences of CYP2C9-mediated drug metabolism and drug-drug interactions. The value of traditional preclinical animal models for such studies is compromised, because of the significant species differences in the multiplicity and substrate specificity of CYP2C enzymes (Nelson et al., 2004; Baillie and Rettie, 2011). Although humanized and knockout mouse models for other major drug-metabolizing enzymes have been developed recently (Corchero et al., 2001; Cheung et al., 2005, 2006; Jiang et al., 2005; Dragin et al., 2007; van Herwaarden et al., 2007; Löfgren et al., 2008; Hasegawa et al., 2011; Scheer et al., 2012), this is the first report of a humanized CYP2C9 or a Cyp2c knockout mouse model. To create these mice, the significant technical challenge and biological risk of deleting the very large mouse Cyp2c cluster needed to be overcome.
To this end, a sophisticated combination of homologous recombination, Cre-mediated deletion and Cre-mediated targeted insertion was applied. Both the hCYP2C9 and the Cyp2c KO mice were viable and fertile and did not display any visual physiological abnormalities, suggesting that the deleted mouse Cyp2c genes have no vitally important role during development. This observation was further sustained by the fact that compared with WT controls no changes in body and liver weights and most clinical chemistry parameters were detected in the transgenic animals. However, ∼50% decreased alkaline phosphatase activities were observed in hCYP2C9 and Cyp2c KO mice, as well as small, but statistically significant, decreases in high-density lipoproteins and cholesterol in the Cyp2c KO model. These changes are presumably a consequence of the deletion of the mouse Cyp2c genes. For example, changes in alkaline phosphatase activities can be associated with the levels of different vitamins, such as vitamin B6, B12, C, or D (Faiz et al., 2007; Aasheim et al., 2008), and it is known that cytochrome P450 enzymes can be involved in vitamin metabolism (Guengerich, 2003). On the other hand, we cannot rule out the possibility that the observed differences are caused by subtle variations in the genetic background of the mouse strains used in the experiments. Sequence polymorphisms in the alkaline phosphatase 2 gene have been found between different mouse strains, which are associated with differences in alkaline phosphatase levels (Foreman et al., 2005). However, this explanation appears unlikely because all mouse lines used in our studies were on a consistent C57BL/6 genetic background. In addition to the changes described above, the average alanine amino transferase and aspartate aminotransferase activities were higher in both genetically modified mouse lines than in WT animals. This result is in agreement with the liver pathology observed by hematoxylin and eosin analysis in both transgenic models. The fact that the changes observed in the hCYP2C9 mice were also visible in the Cyp2c KO animals indicates that these effects reflect roles of the mouse Cyp2c enzymes in endogenous processes and that they are not a consequence of the expression of human CYP2C9. The Cyp2c KO model should facilitate studies to assess these roles.
Human CYP2C9 was expressed in the liver of the hCYP2C9 mice at levels similar to those found in human liver. Furthermore, the enzyme was active in the hydroxylation of the two established CYP2C9 probe substrates tolbutamide and diclofenac. Tolbutamide methylhydroxylase activity was higher in WT mice than in hCYP2C9 mice, suggesting that tolbutamide is a preferred substrate of the mouse Cyp2c enzymes. In contrast, diclofenac 4-hydroxylation was higher in the hCYP2C9 samples. The fact that the deletion of the mouse Cyp2c cluster did not result in a reduction in diclofenac 4-hydroxylation demonstrates that either the murine Cyp2c enzymes are not active toward this substrate or that there is a compensatory increase in other P450s active in its metabolism. This result might be explained by previous observations in rats, indicating that the metabolic activation of diclofenac through oxidation is catalyzed not only by Cyp2c but also by Cyp2b and Cyp3a enzymes (Tang et al., 1999).
One potential use of the hCYP2C9 model is for CYP2C9 inhibition studies, for example, to predict drug-drug interactions in humans. To this end, we tested the effect of a number of CYP2C inhibitors on the hydroxylation of tolbutamide in liver microsomes from WT, Cyp2c KO, and hCYP2C9 mice and compared the results with those obtained from human liver microsomes. Whereas a clear difference was observed between samples from WT mice and humans, the hCYP2C9 model reliably predicted the inhibition of activity seen in human liver microsomes.
In a previous report, the deletion of the mouse Cyp3a gene cluster was associated with considerable compensatory changes in other gene families involved in drug metabolism and disposition (van Waterschoot et al., 2008, 2009). For the limited number of genes analyzed in the present study, only some small reductions in the expression of different Ugt genes were measured in the Cyp2c KO mice, which were not reversed in the hCYP2C9 model. This is in contrast to the Cyp3a knockout model for which the increased expression of, for example, Cyp2c55 was reversed when a transgene expressing CYP3A4 was expressed in the liver (van Waterschoot et al., 2009). The effect of the deletion of the mouse Cyp2c cluster on the expression of other genes not analyzed in the present work needs to be assessed by further studies.
Cytochrome P450 humanized mouse models provide a powerful approach to overcome the species differences in drug metabolism (Gonzalez and Yu, 2006). In the new model, CYP2C9 is expressed off the albumin promoter, which implies that it should be expressed throughout the liver acinus, whereas Cyp2c proteins are expressed in centrilobular hepatocytes. We have attempted to perform immunohistochemical analysis to compare Cyp2c/CYP2C9 protein expression in hCYP2C9 and WT mice, but to date these experiments have been unsuccessful. Furthermore, because of use of the albumin promoter, the expression of CYP2C9 is not inducible by exogenous chemicals as has been shown to be in humans (Ferguson et al., 2002; Gerbal-Chaloin et al., 2002; Chen et al., 2004; Al-Dosari et al., 2006). Finally, in humans, CYP2C9 is expressed in the intestine (Paine et al., 2006), which is not the case in the hCYP2C9 model and the observed liver pathology may complicate the use of the model for certain applications. These points should be taken into account when data using this model are interpreted. Despite these limitations, the novel hCYP2C9 and Cyp2c KO mice provide valuable models for evaluating the role of this enzyme in the metabolism and disposition of drugs and foreign compounds; for example, to assess the role of CYP2C9 in systemic drug clearance, for CYP2C9 inhibition studies, or, relating to regulatory guidance, for the identification and safety assessment of CYP2C9 metabolites (Powley et al., 2009). Furthermore, it was recently shown that the mouse Cyp2c enzymes play an important role in the metabolism of substrates that are otherwise metabolized by CYP3A4 in humans. For example, the mouse Cyp2c enzymes significantly contributed to the in vivo metabolism of midazolam, a highly specific CYP3A4 substrate in humans (van Waterschoot et al., 2008). Therefore, the combination of the hCYP2C9 model with one of the previously described CYP3A4 humanized mouse lines (van Herwaarden et al., 2007; Hasegawa et al., 2011) might help to overcome the background effects of mouse Cyp2c enzymes on the metabolism of CYP3A4 substrates.
Authorship Contributions
Participated in research design: Scheer, Kapelyukh, and Wolf.
Conducted experiments: Kapelyukh, Chatham, Rode, and Buechel, and Chatham.
Performed data analysis: Scheer, Kapelyukh, and Wolf.
Wrote or contributed to the writing of the manuscript: Scheer, Kapelyukh, and Wolf.
Acknowledgments
We thank Vincent Beuger, Lisa Antoni, Sylvia Krueger, and Anja Mueller (TaconicArtemis, Cologne, Germany), Barbara Elcombe, Marie Bowers, Lori Woods, Corinne Haines, Nick Birse, and Sol Gibson (CXR Biosciences, Dundee, UK), and Colin Henderson (University of Dundee, Dundee, UK) for technical assistance.
Footnotes
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.This work was supported in part by ITI Life Sciences, Scotland.
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
ABBREVIATIONS:
- P450
- cytochrome P450
- WT
- wild-type
- Cyp2c KO
- Cyp2c knockout
- hCYP2C9
- CYP2C9 humanized
- ES
- embryonic stem
- Flpe
- flipase
- AUC
- concentration versus time curve.

- Received May 25, 2012.
- Accepted August 23, 2012.
- Copyright © 2012 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}