


Abstract
Several recent studies show that inhibition of the hepatic transport proteins organic anion-transporting polypeptide 1B1 (OATP1B1) and 1B3 (OATP1B3) can result in clinically relevant drug-drug interactions (DDI). To avoid late-stage development drug failures due to OATP1B-mediated DDI, predictive in vitro and in silico methods should be implemented at an early stage of the drug candidate evaluation process. In the present study, we first developed a high-throughput in vitro transporter inhibition assay for the OATP1B subfamily. A total of 2000 compounds were tested as potential modulators of the uptake of the OATP1B substrate sodium fluorescein, in OATP1B1- or 1B3-transfected Chinese hamster ovary cells. At an equimolar substrate-inhibitor concentration of 10 µM, 212 and 139 molecules were identified as OATP1B1 and OATP1B3 inhibitors, respectively (minimum 50% inhibition). For 69 compounds, previously not identified as OATP1B inhibitors, concentration-dependent inhibition was also determined, yielding Ki values ranging from 0.06 to 6.5 µM. Based on these in vitro data, we subsequently developed a proteochemometrics-based in silico model, which predicted OATP1B inhibitors in the test group (20% of the dataset) with high specificity (86%) and sensitivity (78%). Moreover, several physicochemical compound properties and substructures related to OATP1B1/1B3 inhibition or inactivity were identified. Finally, model performance was prospectively verified with a set of 54 compounds not included in the original dataset. This validation indicated that 80 and 74% of the compounds were correctly classified for OATP1B1 and OATP1B3 inhibition, respectively.
Introduction
Over the past decade, increasing evidence has established the role of transport proteins in pharmacokinetic processes and drug-drug interactions. The organic anion-transporting polypeptide (OATP) 1B1 (SLCO1B1 gene) and OATP1B3 (SLCO1B3 gene) are uptake transporters predominantly expressed at the basolateral membrane of human hepatocytes. OATP1B1 and OATP1B3 exhibit broad and overlapping substrate specificities for endogenous and exogenous compounds including thyroid hormones, bile acids, repaglinide, and several statins (Hagenbuch and Gui, 2008). The importance of these hepatic transporters has been illustrated by recent studies showing that OATP1B-mediated transport can be the rate-determining step of hepatobiliary drug clearance (Fenner et al., 2012). Moreover, OATP1B inhibition or induction may be the underlying mechanism of clinically relevant drug-drug interactions (Müller and Fromm, 2011; Shitara, 2011). Therefore, the assessment of OATP1B-mediated drug-drug interactions (DDI) has become a critical aspect of early drug development, as recognized by the International Transporter Consortium (Giacomini et al., 2010). This review acknowledged OATP1B1 and OATP1B3 as two of the seven clinically most relevant transporters and provides recommendations regarding the preferred in vitro methods to be used for evaluating drug interactions mediated by these transporters. In addition to their role in hepatic drug disposition and DDI, recent findings have suggested that OATPs (in particular OATP1B3) may have an important pathophysiological role in certain pancreatic (Kounnis et al., 2011) and ovarian cancers (Svoboda et al., 2011). Therefore, OATP1B modulators could form a new class of potential therapeutic agents.
Identification (and kinetic characterization) of OATP ligands early on in the drug evaluation process has thus become a prerequisite for successful drug development. In vitro methods for assessing OATP1B-mediated hepatic drug uptake based on primary human hepatocytes are generally predictive of the in vivo situation but remain resource-intensive. Hence, several studies have successfully relied on the use of higher throughput assays with transfected human embryonic kidney (HEK)293 or Chinese hamster ovary (CHO) cells to investigate OATP1B inhibition and transporter kinetics (Baldes et al., 2006; Annaert et al., 2010; Bednarczyk, 2010; Gui et al., 2010). These studies led to the identification of new OATP1B inhibitors and/or the characterization of the inhibition potential of existing drugs (e.g., HIV protease inhibitors) (Annaert et al., 2010). By supporting the evaluation of a relative large number of compounds, these in vitro approaches also enable gaining valuable information regarding the molecular structural properties of OATP ligands. Statistical quantitative structure activity relationship (QSAR) modeling is commonly applied to computationally derive chemical features shared by compounds that are active as opposed to chemical features shared by compounds that are inactive. Similarly, as reviewed by van Westen et al. (2011b), proteochemometric modeling additionally takes into account structural protein features (this can be either the full protein or a selection of residues, usually the binding pocket) (Lapinsh et al., 2001; Prusis et al., 2006; van Westen et al., 2013).
Previously, we characterized the in vitro hepatic uptake mechanisms of sodium fluorescein and demonstrated that sodium fluorescein can be used as a probe substrate to evaluate OATP1B1/3-mediated transport (De Bruyn et al., 2011). Based on this information, we presently report the development of a high-throughput in vitro assay that was applied to evaluate the OATP1B1 and OATP1B3 inhibitory potential of a library of 2000 compounds. Next, we established an in silico proteochemometric model to predict OATP1B interaction potential of compounds and to identify chemical properties and substructures related to OATP1B1 and/or OATP1B3 inhibition. Our results demonstrate that this integrated in vitro and in silico approach leads to a particularly useful in silico algorithm for direct structure-based identification of (drug) compounds modulating OATP1B activity.
Materials and Methods
Reagents
The Spectrum Collection (2000 compounds, supplied as 10 mM Me2SO solution) was purchased from MicroSource Discovery Systems Inc. (Gaylordsville, CT). Dulbecco’s modified Eagle’s medium (DMEM) and G418 (Geneticin) were purchased from Invitrogen (Paisley, UK). l-Glutamine, penicillin-streptomycin mixture (contains 10,000 IU potassium penicillin and 10,000 IU streptomycin sulfate per ml in 0.85% saline), fetal bovine serum (FBS), trypsin EDTA, Hanks’ balanced salt solution (HBSS) and 1× phosphate buffered saline (PBS) were purchased from Lonza SPRL (Verviers, Belgium). Sodium fluorescein was obtained from UCB (Leuven, Belgium) and HEPES was purchased from MP Biochemical (Illkirch, France). Triton X-100 and sodium butyrate were purchased from Sigma-Aldrich (Schnelldorf, Germany).
In Vitro Assessment of OATP1B Inhibitory Potential
Cell Culture.
Wild-type OATP1B1- and 1B3-transfected CHO cells were cultured at passage 45 to 60, as described previously (Treiber et al., 2007). The culture medium consisted of Dulbecco’s modified Eagle’s medium containing 1 g/l d-glucose, 1 mM l-glutamine, 25 mM HEPES and 110 mg/l sodium pyruvate, supplemented with 10% fetal bovine serum, 50 μg/ml l-proline, 100 IU/ml penicillin and 100 IU/ml streptomycin. The culture medium of the transfected cell lines additionally contained 500 μg/ml G418 to maintain selection. CHO cells were grown in 75-cm2 T-flasks at 37°C in an atmosphere of 5% CO2 and 90% relative humidity and were subcultured every 3 days. For high-throughput uptake experiments, wild-type CHO cells were seeded in black 96-well cell culture plates with a transparent bottom (Greiner-Bio-One, Wemmel, Belgium) at a density of 8,000 cells/well. Culture medium was replaced every other day and uptake experiments were performed on day 4 after seeding. One day before the experiment, cells were additionally treated with 5 mM sodium butyrate to induce gene expression.
Transport Studies.
Cells were washed twice with 0.2 ml/well prewarmed uptake buffer (Hanks’ balanced salt solution with 10 mM HEPES, pH 7.4) and preincubated for 10 minutes at 37°C with 100 µl of uptake buffer. In experiments in which inhibitors were investigated, cells were preincubated with 100 µl of a double-concentrated inhibitor solution. Uptake assays were initiated by adding 100 µl of a double-concentrated sodium fluorescein solution. To determine the uptake kinetics in OATP1B1- or OATP1B3-transfected cells, sodium fluorescein was incubated at eight concentrations ranging from 1 to 80 µM. The inhibitory potential of all 2000 compounds from The Spectrum Collection library was evaluated at an equimolar substrate-inhibitor concentration of 10 µM. Uptake experiments were stopped after 3 minutes by removing the uptake solution and rinsing the cells three times with ice-cold uptake buffer. Subsequently, cells were lysed with 100 µl of 0.5% Triton X solution (in phosphate buffered saline) and placed on a plate shaker for 30 minutes at room temperature. Cell lysates were analyzed by fluorescence spectroscopy (ex 490 nm, em 524 nm) in a Tecan Infinite 200 plate reader (Tecan Benelux, Mechelen, Belgium). Uptake rates were normalized for protein content, which was measured using a BCA Protein assay kit (Pierce Chemical, Rockford, IL).
In Vitro Data Analysis.
Net uptake values were obtained by subtracting uptake in wild-type CHO cells from total uptake at 37°C in transfected cells. Cellular fluorescein uptake kinetics were determined by fitting the Michaelis–Menten equation to net uptake values:

For inhibition experiments, data were expressed as uptake of sodium fluorescein in presence of the inhibitor compared with the control cellular uptake (no inhibition).
The Inhibitory Effect Emax-model was used to describe the concentration-dependent inhibitory effect by various inhibitors in transfected CHO cells: with E representing the cellular substrate uptake, Emax the substrate uptake without inhibitor (≤100%), E0 the substrate uptake at the maximum inhibitory effect of inhibitor (≥0%), and (Emax – E0), the maximum inhibitory effect. The best fits of the above equations to the individual uptake and inhibition datasets were obtained by nonlinear regression analysis using the Solver tool in Microsoft Excel 2007.
with E representing the cellular substrate uptake, Emax the substrate uptake without inhibitor (≤100%), E0 the substrate uptake at the maximum inhibitory effect of inhibitor (≥0%), and (Emax – E0), the maximum inhibitory effect. The best fits of the above equations to the individual uptake and inhibition datasets were obtained by nonlinear regression analysis using the Solver tool in Microsoft Excel 2007.
For the estimation of Ki from IC50, assuming competitive inhibition, the Cheng–Prusoff equation was used: with IC50 representing the concentration of the inhibitor producing a 50% inhibition, S the substrate concentration, and Km the Michaelis constant of the substrate.
with IC50 representing the concentration of the inhibitor producing a 50% inhibition, S the substrate concentration, and Km the Michaelis constant of the substrate.
The data analysis tool in Microsoft Excel 2007 (P < 0.001) was used to evaluate statistical differences in the correlation between remaining sodium fluorescein uptake and the computationally predicted inactivity correlation value for all compounds in the prospective validation set.
In Silico Modeling for the Prediction of OATP1B Inhibition
Dataset.
The dataset consisted of in vitro OATP modulation data for 2000 compounds (The Spectrum Collection; MicroSource Discovery Systems, Inc.). For all test compounds, the assay activity value was expressed as a percent of the control sodium fluorescein uptake (after correction for uptake in wild-type CHO cells). When the residual activity was lower than 50%, compounds were deemed “Active” (able to inhibit the uptake of sodium fluorescein) and vice versa.
The predictivity of the in silico model was prospectively verified with an external test set of 54 compounds that were not included in The Spectrum Collection.
Molecular and Protein Descriptors.
The compounds were standardized and their ionization state was determined at pH 7.4 (identical to assay conditions). Furthermore, hydrogens were added and two-dimensional coordinates were calculated. Subsequently, compounds were converted to circular fingerprints (FCFP_6) and a number of numeric descriptors were calculated. These included molecular weight, number of hydrogen bond donors/acceptors, and polar surface area (Supplemental Table 1). Standardization and descriptor calculation steps were performed in Pipeline Pilot 8.5 (Accelrys Software Inc., San Diego, CA).
Proteins were described based on their sequence (Supplemental Table 2). Sequences of OATP1B1, OATP1B3, OATP1A2, and OATP2B1 were aligned with ClustalW (Slow alignment, Gap Open 4, Gap Extend 4; Supplemental Material, Alignment 1). The latter two OATP isoforms were included to increase the quality of the alignment. From this alignment, amino acids previously shown to be important for OATP activity (Meier-Abt et al., 2005; Mandery et al., 2011) were selected and translated to a descriptor based on Z-scales or a feature-based ProtFP descriptor (Lapinsh et al., 2002; van Westen et al., 2011a). Supplemental Figure 1 shows the similarity of the proteins as described by the Z-scales descriptors in a two-dimensional plot.
Proteochemometric Modeling.
Pipeline Pilot 8.5 (Accelrys Software Inc, San Diego, CA) was used for proteochemometric (PCM) modeling. Random forest models were trained using the R package randomForest as described previously (Liaw and Wiener, 2002). The optimal number of trees was determined to be 25, and for each tree, the optimal number of allowed descriptors per split was set at a fraction of 0.5 from the total number of descriptors. Model performance was estimated with the out-of-bag (oob) data, external validation, and prospective validation.
For all trained classification models, performance was estimated using sensitivity, specificity, the Matthews correlation coefficient (Matthews, 1975), and the fraction of correctly predicted compounds (Supplemental Table 3).
Two final models were selected, i.e., a 2-class model (distinguishing between compounds either “Active” or “Inactive”; model 8 in Supplemental Table 3), and a 4-class model (distinguishing between “Active_1B1”, “Active_1B3”, “Inactive_1B1”, or “Inactive_1B3”; model 17 in Supplemental Table 3).
Descriptor Interpretation.
To interpret the random forest models, descriptors used in the model training were converted. The numerical compound descriptors were binned into four classes based on their distribution in the dataset. The “low” bin contained the lower 20% of the frequency histogram for each descriptor, the “high” bin contained the higher 20% of the frequency histogram, and the remaining 60% was equally distributed between “medium_low” and “medium_high.” For further details per descriptor see Supplemental Table 4.
For the circular fingerprints, the “Fingerprints to properties” component from Pipeline Pilot 8.5 was used to make a selection; from a total of 16,341 chemical features present, 1308 relevant features were encoded for presence or absence. We selected the features that were most equally/unequally distributed between actives and inactives (512/512 features), supplemented with a random selection of 2% of all features (297). Thirteen features overlapped and were removed.
For the protein descriptors, each Z-scale (representing a physicochemical property) was binned similarly to the molecular descriptors. As a consequence, every amino acid obtained a score for Hydrophobicity (Z1), Size/Polarizability (Z2), Polarity (Z3), Electronegativity (Z4), and Electrophilicity (Z5). Presence or absence of each of those scores was encoded (Supplemental Table 5). The bins were selected such that each amino acid was uniquely identifiable via these binned values.
To estimate the significance of each descriptor, the model provided two importance values: Accuracy and Gini. The accuracy value was calculated by omitting one descriptor and calculating the deterioration of the model’s ability to correctly classify compounds. The Gini measure for a descriptor estimates the ability to differentiate between activity classes when the dataset is split using this descriptor. Finally, the correlation of each descriptor with each activity class allowed interpretation to see whether a descriptor is positively or negatively correlated with OATP1B activity.
Prospective Validation.
A total of 54 compounds (not present in the original compound library) were selected based on in-house availability and solubility, and computationally evaluated for OATP1B1 and OATP1B3 inhibition with the final random forest model (model 8). Using the 2-class in silico model, a total of 30 and 23 compounds were predicted to be inhibitors of OATP1B1 and OATP1B3, respectively. Activity/inactivity correlation values, reflecting the potency of the prediction, were directly obtained from the Pipeline Pilot software output. Next, we experimentally determined the OATP1B1 and OATP1B3 inhibitory potential of all 54 compounds, following the described in vitro OATP1B inhibition assay, and assessed the relationship between residual sodium fluorescein uptake and the inactivity correlation value.
Results
Uptake Kinetics of Sodium Fluorescein in OATP1B1- and OATP1B3-Transfected CHO Cells
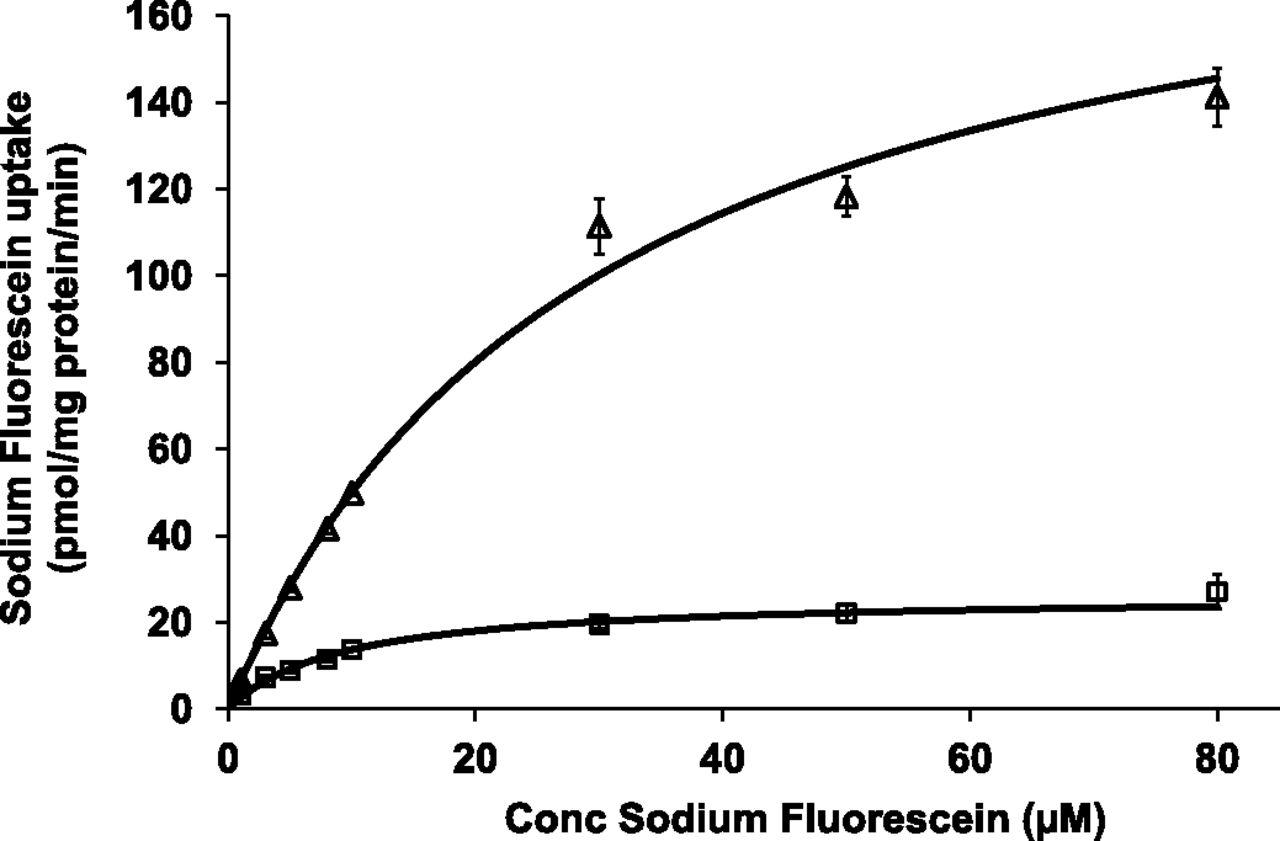
Concentration-dependent uptake of sodium fluorescein showed Michaelis–Menten type kinetics in both OATP1B1- and OATP1B3-transfected CHO cells (Fig. 1). Their respective kinetic parameters for net active uptake were 12.2 ± 4.9 μM and 33.2 ± 3.4 μM for Km and 30.1 ± 3.8 and 195.6 ± 16.6 pmol/mg protein per minute for Vmax (n = 4). This corresponds to uptake clearance values of 2.5 and 5.9 µl/mg protein per minute for OATP1B1- and OATP1B3-transfected CHO cells, respectively.

Concentration-dependent uptake of sodium fluorescein in OATP1B1- (squares) and OATP1B3- (triangles) transfected CHO cells (n = 4). CHO cells were incubated with 1–80 μM sodium fluorescein. OATP-mediated sodium fluorescein uptake was obtained by subtracting uptake in wild-type CHO cells from total uptake in OATP-transfected cells. Points represent mean (± S.D.) of triplicate determinations and lines represent best fit to experimental data according to the Michaelis–Menten equation.
Inhibition of Sodium Fluorescein Uptake by Established OATP1B Inhibitors in Transfected CHO Cells
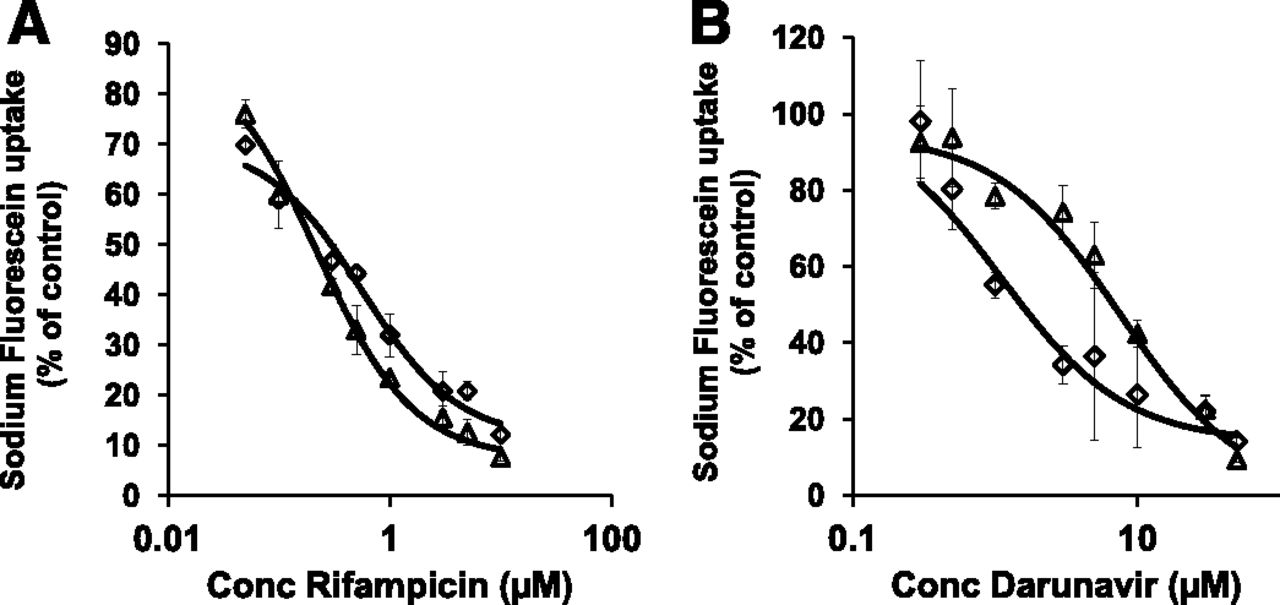
The high-throughput in vitro screening method was validated by assessing the inhibitory potential of two established OATP1B inhibitors, rifampicin and darunavir (Annaert et al., 2010). Both inhibitors decreased the uptake of sodium fluorescein in a concentration-dependent manner with Ki values of 0.30 ± 0.05 µM and 0.22 ± 0.06 µM for rifampicin and 3.6 ± 1.3 µM and 6.7 ± 2.8 µM for darunavir in OATP1B1- and OATP1B3-transfected CHO cells, respectively (Fig. 2).
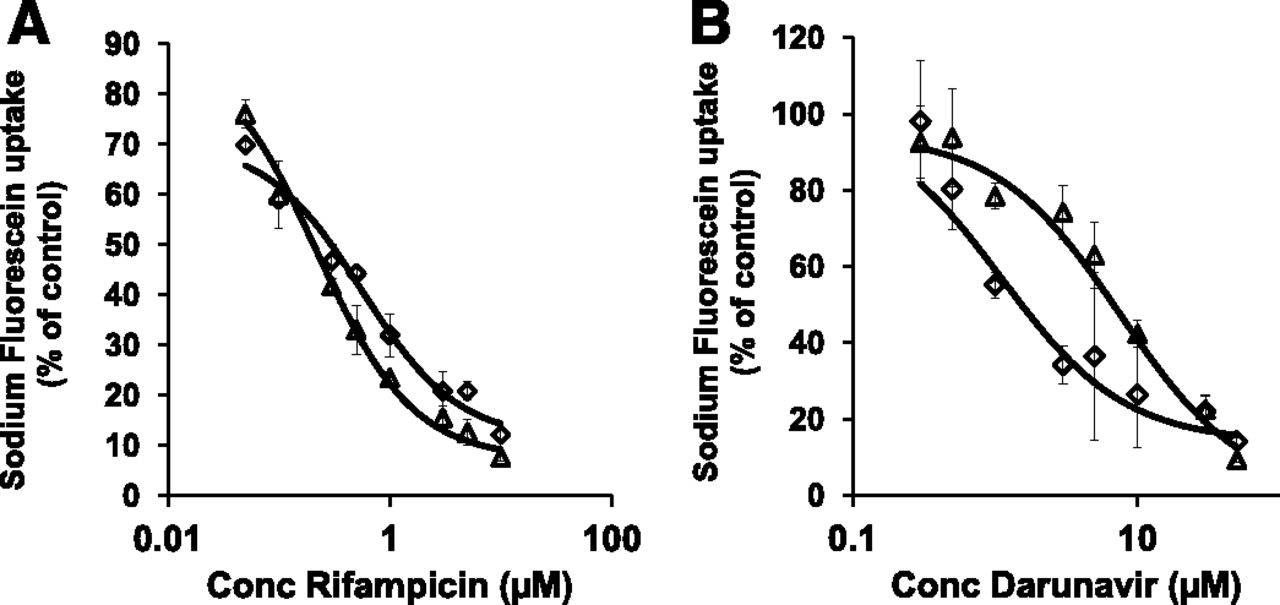
Concentration-dependent inhibition of sodium fluorescein (10 µM) uptake by the known OATP1B inhibitors rifampicin (A) and darunavir (B) in CHO cells. Diamonds represent OATP1B1-expressing CHO cells while squares represent OATP1B3-expressing CHO cells. Net uptake values were obtained by subtracting uptake in wild-type CHO cells from total uptake in transfected cells. Lines represent best fit to data as described in the method section.
High-Throughput in Vitro Screening Assay
A total of 2000 compounds were evaluated as potential inhibitors of the uptake of sodium fluorescein in OATP1B1 and OATP1B3-transfected CHO cells (Fig. 3). At an equimolar substrate-inhibitor concentration of 10 µM, 212 and 139 compounds inhibited the uptake of sodium fluorescein by at least 50% in OATP1B1 and OATP1B-transfected CHO cells, respectively (Table 1; Supplemental Material). Some of these compounds (e.g., cyclosporine, rosuvastatin, sildenafil, and paclitaxel) had been previously reported to inhibit the transport of OATP substrates, while others were demonstrated to be OATP1B1 and/or OATP1B3 inhibitors for the first time (e.g., utilin). Secondly, we measured the inhibition of sodium fluorescein uptake by 69 of the most potent inhibitors at concentrations ranging from 0.1 to 10 µM. All selected compounds inhibited the uptake of sodium fluorescein (10 µM) in a concentration-dependent manner. The corresponding Ki values are listed in Table 1.

The inhibitory effect of 2000 compounds (10 µM) on the uptake of sodium fluorescein (10 µM) in OATP1B1- (A) and OATP1B3- (B) transfected CHO cells. Relative sodium fluorescein uptake was obtained by dividing net sodium fluorescein uptake in presence of the potential inhibitor by the net uptake of sodium fluorescein in the control condition for each isoform. Compounds are ranked from potent inhibitor to potent activator of sodium fluorescein uptake.
OATP1B inhibition data for all test compounds that inhibited sodium fluorescein uptake by at least 50%
Proteochemometric Modeling
Proteochemometric Models.
The final 2-class model had an out-of-bag ROC (receiver operating characteristic) score of 0.86, a sensitivity of 0.78, a specificity of 0.86, and a percentage correctly classified compounds of 89.2% (Supplemental Fig. 2A; Supplemental Table 3). Identical models were trained per OATP leaving out the protein descriptor (and hence creating a QSAR model) to compare the performance to the state of the art as was done previously. The QSAR model reached a percentage correctly classified of 84.6% on the same set. In an external validation experiment, leaving out 30% of the compounds rather than a percentage of the bioactivity data points, similar results were seen with the PCM model reaching 84.2% and the QSAR models reaching 85.2%. Due to the increased interpretability of the PCM models we preferred PCM over QSAR.
The final 4-class model had an ROC score of 0.94 (Supplemental Fig. 2B), a sensitivity of 0.75, and a specificity of 0.85, and correctly classified 88.5% of compounds (Supplemental Fig. 2B; Supplemental Table 3). Sensitivity and specificity were calculated using the classification active or inactive (regardless of the protein isoform).
Important Chemical Properties of the 2-Class Model.
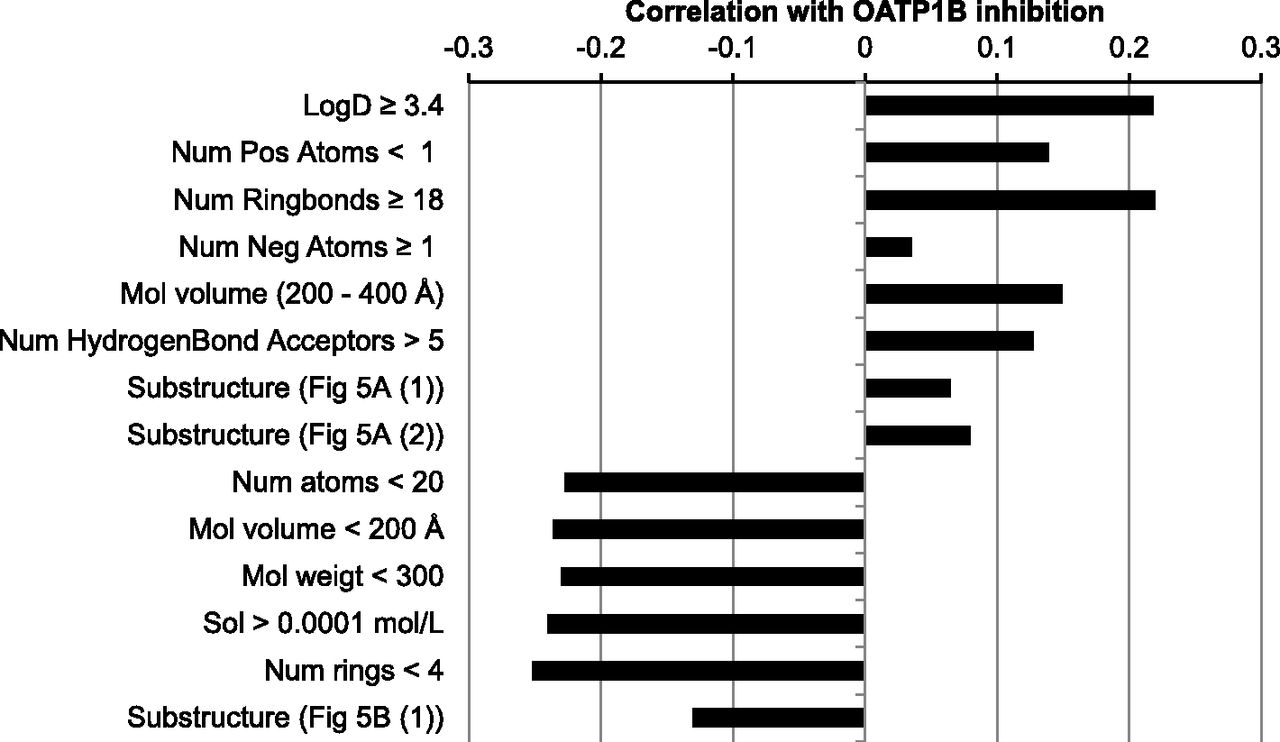
The interpretation of the 2-class model revealed several physicochemical properties and substructures that correlate with OATP inhibition (Figs. 4 and 5; Supplemental Table 6). Based on descriptor importance “gini”, the most important compound property was a LogD between 3.4 and 7.5, followed by the absence of positively charged atoms and a number of ringbonds between 18 and 32. Furthermore, the presence of an anionic group (e.g., carboxylate group), a large molecular volume (>200 Å3), and a high number of hydrogen bond acceptors (>5) are correlated with increased likelihood of OATP inhibition.
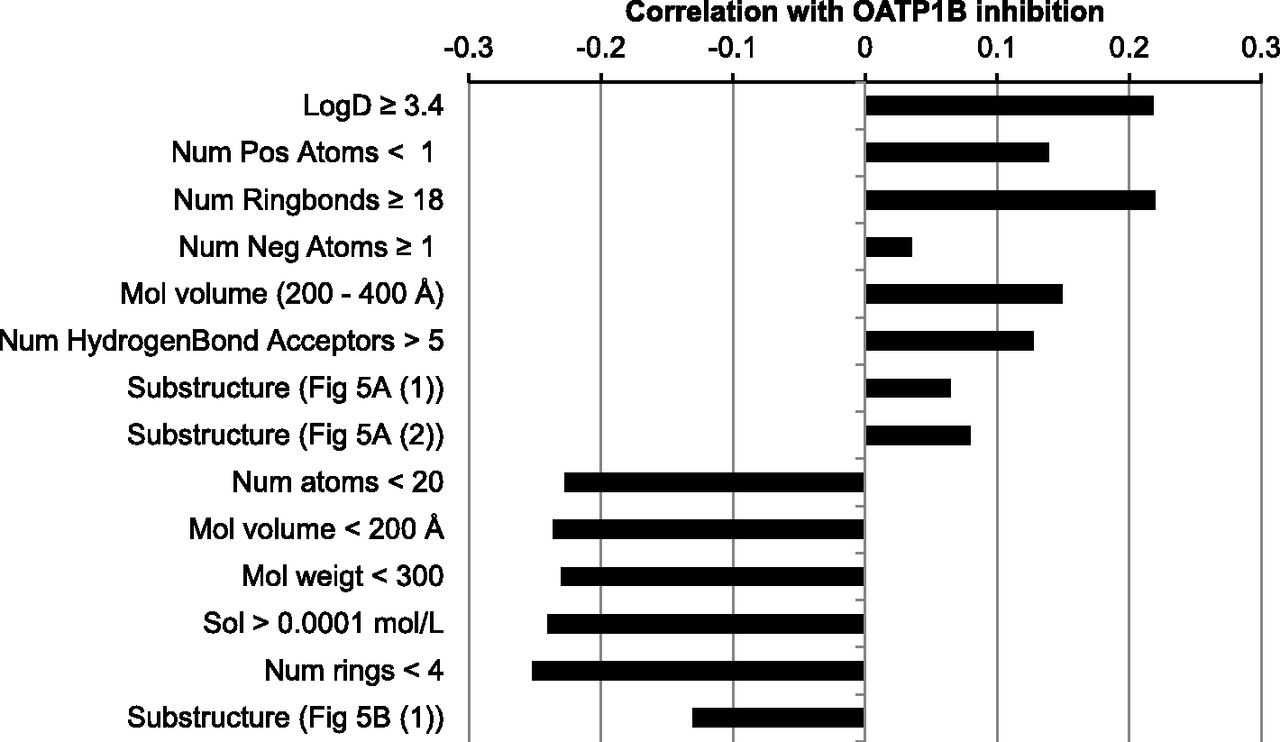
The most important compound properties that positively or negatively correlate with OATP1B inhibition.
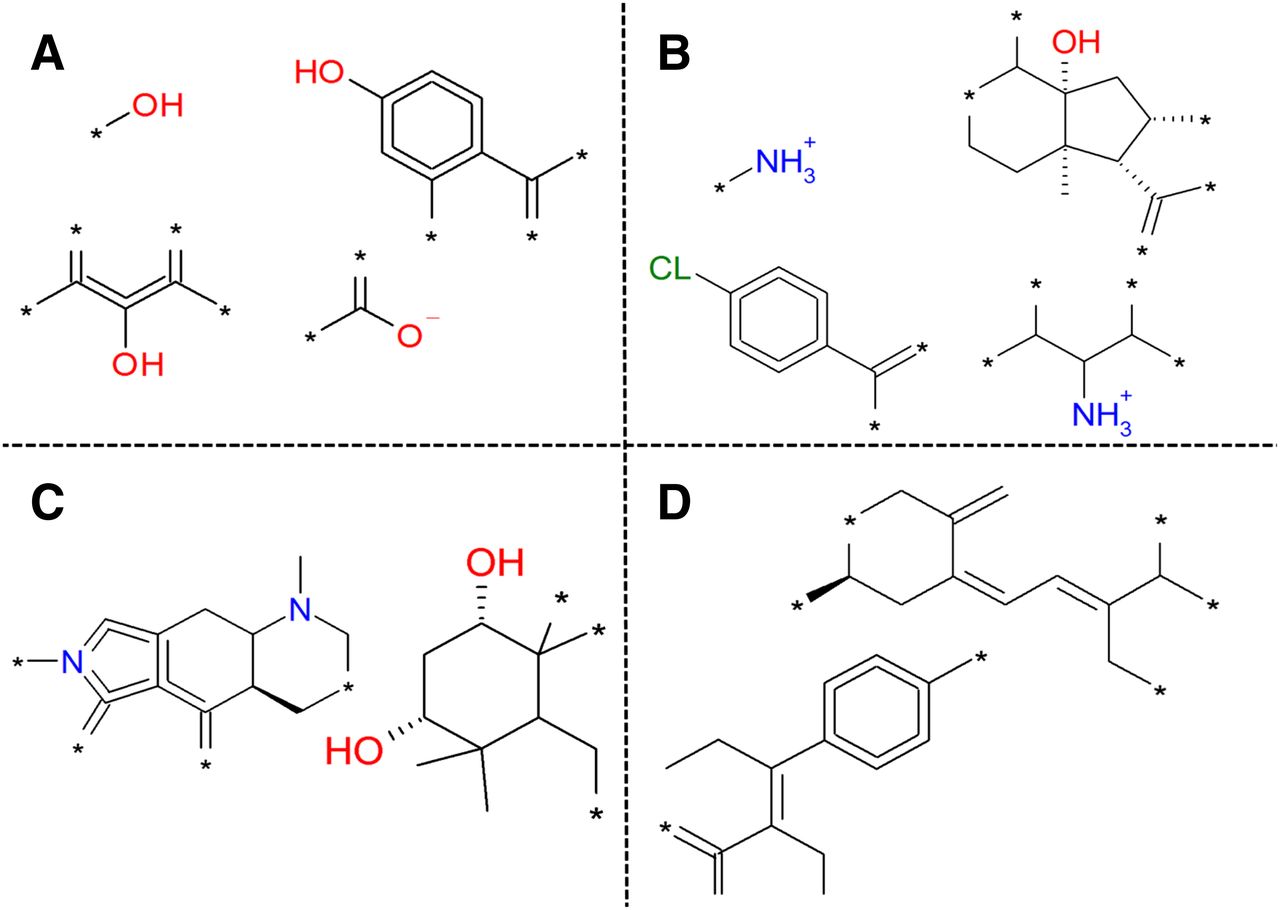
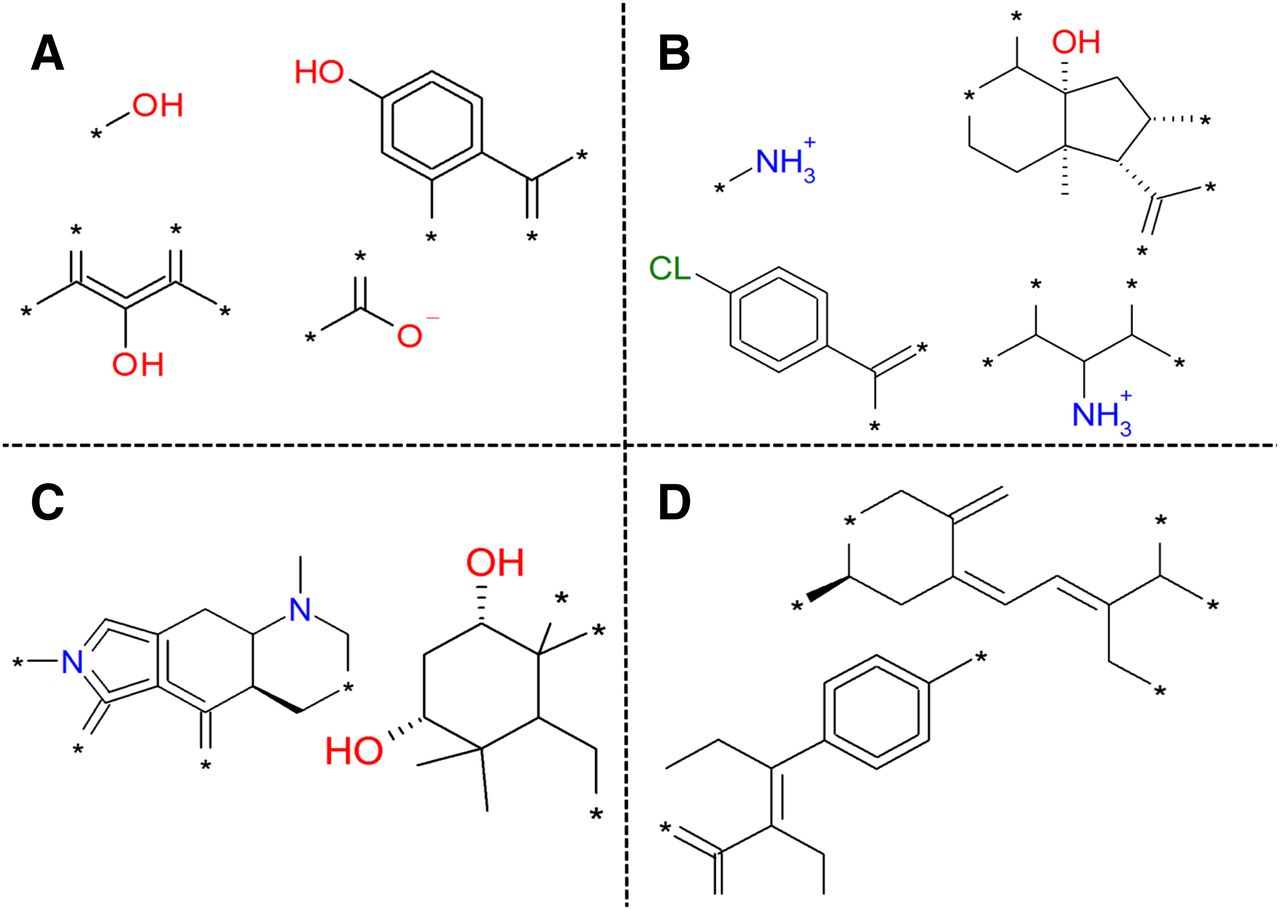
Selection of substructures that correlate with an activity class based on the 4-class model. (A) Substructures that lead to activity on both OATP1B1 and OATP1B3. (B) Substructures that lead to inactivity on both OATP1B1 and OATP1B3. (C) Substructures specific for activity on OATP1B1. (D) Substructures specific for activity on OATP1B3.
Several physiochemical properties and substructures are highly correlated with OATP1B inactivity (Figs. 4 and 5; Supplemental Table 6). The strongest negative correlations were found with substructures that are or can be positively charged at pH 7.4. Furthermore, small hydrophilic compounds with positively charged atoms and without negatively charged atoms are favored as inactives. More specifically, a total number of heavy atoms lower than 20, a molecular volume <200 Å3, a molecular weight under 300, a calculated aqueous solubility >0.1 mM, and number of rings <4 were correlated with inactivity.
Important Chemical Properties of the 4-Class Model.
In general, the 4-class model provides similar results to the 2-class model when considering the importance of chemical features for activity or inactivity on both proteins. The most important feature for activity also included a group that could be negatively charged (based on descriptor importance “gini”). Important features for inactivity were substructures that are or can be positively charged (at pH 7.4).
In addition, the 4-class model provides information regarding selectivity for either OATP1B1 or OATP1B3. A low number of aromatic bonds (<7) was found to be positively correlated with OATP1B1 activity and negatively with OATP1B3 inhibition. Conversely, a high LogD value (>7.5) and a medium low number of hydrogen bond donors (3–4) were positively correlated with OATP1B3 activity.
Finally, we did identify some substructures favoring inhibition of OATP1B1 over OATP1B3 and vice versa (Fig. 5, C and D). A complete list of the important chemical properties is available electronically as Supplemental Table 6.
Important Protein Properties.
As we constructed a PCM model on only two targets, only limited interpretation of the model with respect to the target can be performed (Supplemental Table 7). However, a follow up of the current work should include a larger group of protein targets. Previously, we have shown that incorporating a larger number of proteins (8, 14, or even >10,000 proteins) leads to improved performance and model interpretability, hence a similar approach could be followed here (van Westen et al., 2011a, 2012, 2013). A good starting point would be the ChEMBL database that lists bioactivity data for 22 OATP isoforms. Nine of these are human isoforms and include the four isoforms we used to construct our multiple sequence alignment (Gaulton et al., 2012).
Prospective Validation
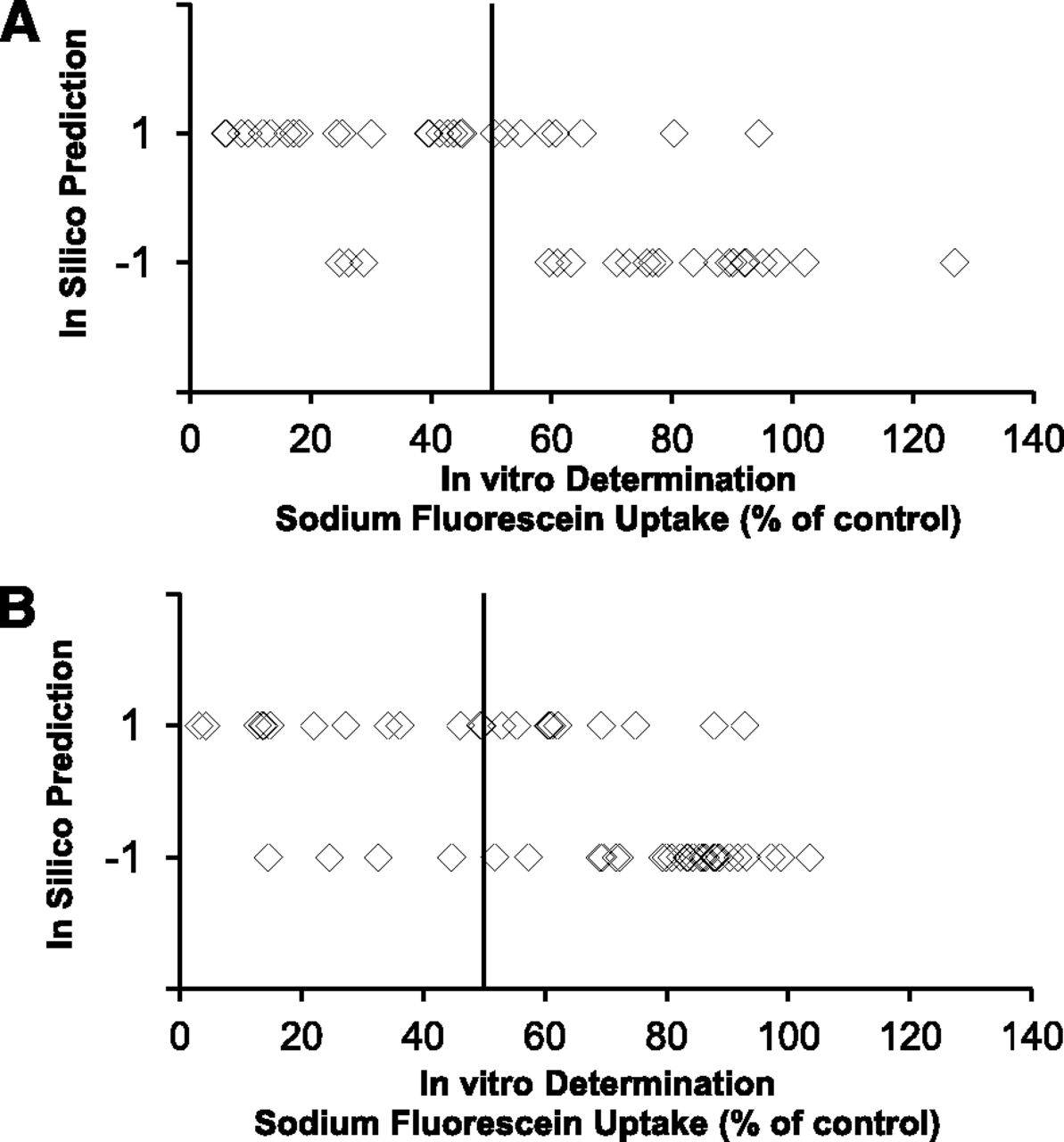
The results of the prospective validation are depicted in Table 2 and Fig. 6 and are uploaded as Supplemental Material. For OATP1B1, 22 out of 30 computationally predicted active compounds inhibited OATP1B1-mediated sodium fluorescein uptake by more than 50%; 21 out of 24 compounds predicted negative, i.e., did not inhibit OATP1B1 by more than 50% (Fig. 6A). For OATP1B3, 13 out of 23 computationally predicted active compounds inhibited OATP1B3-mediated sodium fluorescein by more than 50% and 27 out of 31 negatives did not inhibit OATP1B3 (Fig. 6B). This corresponds to a sensitivity of 0.88 and 0.77 and a specificity of 0.72 and 0.73 for OATP1B1 and OATP1B3, respectively.
Prospective validation

Prospective validation of the in silico model. A total number of 54 compounds were computationally evaluated for OATP1B1 (A) and OATP1B3 (B) inhibition with the final random forest model. Compounds that are computationally predicted to be inhibitors and noninhibitors are represented with 1 and –1, respectively. The OATP1B1 and OATP1B3 inhibitory potential of all 54 compounds was determined following the described in vitro OATP1B inhibition assay with sodium fluorescein as a substrate.
The correlations between the residual OATP1B1- and OATP1B3-mediated uptake of sodium fluorescein and the computationally generated inactivity correlation values for all 54 compounds of the prospective validation are represented in Fig. 7A (R2 = 0.45) and Fig. 7B (R2 = 0.51), respectively.

Relationship between the experimentally determined inhibitory potential (remaining sodium fluorescein uptake) and the computationally predicted inactivity correlation value for all 54 compounds evaluated in the prospective validation. (A) and (B) represent data for OATP1B1- and OATP1B3-mediated inhibition, respectively
Discussion
We previously demonstrated that sodium fluorescein can be used as a probe substrate to identify xenobiotics modulating OATP1B-mediated transport in fluorescence-based in vitro transporter assays (De Bruyn et al., 2011). In the present study, we optimized this concept into a high-throughput in vitro assay that can be implemented in an early drug development setting to assess the risk for OATP1B-mediated DDI of new drug candidates. In addition, the extensive size and structural diversity of our in vitro dataset allowed us to develop an accurate in silico proteochemometric model. In a final prospective validation, we confirmed the utility of this model to computationally predict OATP1B-mediated inhibition and to identify molecular characteristics important for OATP1B inhibition.
Fluorescence-based in vitro screening assays provide rapid and sensitive approaches for studying drug-transporter interactions (Bednarczyk, 2010; De Waart et al., 2010; Gui et al., 2010). The high-throughput assay developed here was used to assess the inhibitory potential of a very extensive and chemically diverse compound dataset. All 2000 test compounds were evaluated at an equimolar inhibitor-substrate concentration of 10 µM. In addition, compounds were only classified as inhibitors when the uptake of sodium fluorescein decreased by more than 50% of the control value. Following this approach, only relatively potent (and more likely clinically relevant) OATP1B inhibitors are identified. In this respect, the experimental conditions used in our in vitro model differ from previously reported methodologies using an excess of inhibitor. Applying a high inhibitor/substrate concentration ratio might indeed lead also to the selection of those compounds with a relatively lower OATP inhibition potency (Badolo et al., 2010; Karlgren et al., 2012a). In the present study, rifampicin and darunavir inhibited sodium fluorescein uptake by more than 80 and 60% of the control value (Fig. 2), confirming that established OATP inhibitors are indeed detected with our in vitro assay.
Different computational approaches are possible given the available data. Structure-based approaches (docking) rely on a crystal structure, but this information is not available (Rognan, 2013). Hence, we opted for a proteochemometric approach that relies on (two-dimensional) structural sequence information for a given protein. Based on in vitro OATP inhibition data, we optimized a 2-class computational model to predict OATP1B inhibition. Although Ki values were determined for 69 potent OATP1B inhibitors (Table 1), lack of accurate parameter values (such as Ki) for noninhibitors, precluded the use of these data for in silico model development. Therefore, classification data (active versus inactive) of all 2000 compounds were used for model building. In contrast to a regression model (based on Ki values), this classification approach allowed inclusion of both active and inactive compounds and therefore determination of the characteristics of compounds either inhibiting or not inhibiting OATP1B.
The predictivity of the model was first estimated based on the out-of-bag error for which the original dataset was divided in a training set (80% of the dataset) and a 20% test set. Using this approach, 89% of the compounds in the test set were correctly classified (with QSAR models reaching 84%). In an external validation experiment the PCM model classified 84% of the compounds correctly (85% for QSAR models). Arguably, the only unbiased assessment of the reliability of any in silico model is a prospective validation, i.e., on application of the in silico model on a selection of compounds unknown to the model. Therefore, we composed a validation set of 54 compounds that were not present in the original dataset. After computational prediction of OATP1B inhibition potential, experimentally obtained in vitro results revealed that 80 and 74% of the compounds were correctly classified for OATP1B1 and OATP1B3, respectively (Fig. 6; Table 2). This prospective validation confirms the reliability and applicability of the computational model. Further, there appears to be a reasonable correlation (R2 = 0.45–0.51) between the residual values for OATP1B-mediated uptake of sodium fluorescein and the computationally generated correlation values with inactivity for all 54 compounds. This suggests that this correlation value could be reliably used for in silico estimation of the residual sodium fluorescein uptake in the presence of a given putative inhibitor with known structure and physicochemical properties.
The in silico model was further applied to 51 compounds that were not present in the original dataset and for which an OATP1B1 inhibition value was published by Karlgren et al. (2012a). Our model correctly classified 26 of the 35 OATP1B1 inhibitors (74%) and 14 of the 16 noninhibitors (88%). The relatively lower prediction for inhibitors confirmed the hypothesis that an excess of inhibitor compared with the substrate concentration is reflected in a higher number of compounds classified as (weakly) active that are not detected by our in silico model. The moderate sensitivity but high specificity of our in silico model toward published inhibition data results in a limited number of false positive results, and thus the model favors compounds with a potent OATP1B inhibitory potency. This could be an important advantage when searching for the most potent OATP1B inhibitors among a (very) large set of test compounds. However, to further improve the predictive value of our model, the dataset supporting this model can be expanded further. The ChEMBL database, a large public database of bioactivity data, also contains OATP bioactivity data for nine human OATP proteins (including the four isoforms we used to generate our multiple sequence alignment). Because inclusion of more protein targets has previously been shown to lead to better prospective qualities (van Westen et al., 2012), we encourage a follow-up study to create a second generation OATP PCM model (Gaulton et al., 2012; Kramer et al., 2012).
The 2-class model was further expanded to a 4-class model for the descriptor interpretation. However, predictive information regarding compound properties extracted from both models was essentially comparable. The 4-class model additionally offered the advantage of pinpointing compound and protein properties important for inhibition of only one of the OATP1B isoforms.
The key descriptors important for an increase in OATP1B inhibition potency were the increase in lipophilicity and polar surface, the presence of anionic atoms or several hydrogen bond acceptors, and the absence of cationic atoms. These findings are consistent with previous results from Chang et al. (2005), who constructed a pharmacophore model for OATP1B1 and concluded that lipophilicity and hydrogen bond-acceptor strength are important properties for OATP1B1 inhibition. Our results also confirm the conclusions drawn by Badolo et al. (2010), who determined that lipophilicity and the number of hydrogen bond acceptors (next to polarity and pKa) are the key properties for inhibiting the OATP1B1 probe substrate estradiol-17β-glucuronide in human hepatocytes. The key descriptors identified with our computational model are consistent with the data published by Karlgren et al. (2012b), who concluded that lipophilicity and polar surface area are key molecular features for inhibition of several OATP1B isoforms. More recently, Soars et al. (2012) developed an OATP1B1-HEK inhibition assay and also concluded that the prime molecular descriptors for the prediction of OATP1B1 inhibition are maximal hydrogen bonding strength followed by lipophilicity.
Besides these previously reported key descriptors, we identified several new compound properties (e.g., high number of ring bonds) and substructures important for OATP1B inhibition (Fig. 5; Supplemental Table 6). Moreover, defined ranges for these physicochemical compound properties were specified rather than just a qualitative interpretation. To our knowledge, physicochemical compound properties along with compound substructures important for the OATP1B isoform selectivity are reported here for the first time (Fig. 5). Finally, due to the nature of our proteochemometric models, we were also able to identify residues in both OATP1B1 and OATP1B3 that are important for OATP1B1 inhibition.
In conclusion, we developed an in vitro OATP1B inhibition assay that can immediately be implemented in drug discovery settings for enhanced throughput evaluation of the OATP1B inhibition potency of large numbers of drug candidates. For a total of 69 compounds we generated for the first time Ki values regarding their inhibition potential toward OATP1B1 and or OATP1B3. Based on our extensive amount of in vitro data we developed a proteochemometric model that accurately predicts OATP1B1 and OATP1B3 inhibition for a given new chemical entity based on its structure. We conclude that a high lipophilicity and polar surface, the presence of anionic atoms or several hydrogen bond acceptors, and the absence of cationic atoms are key features for OATP1B inhibition. In addition, our proteochemometric approach allowed identification of residues in both OATP1B1 and OATP1B3 that are important for OATP1B1 inhibition.
The in silico models developed in the present study are included in the Supplemental Data, Supplemental Methods, and Supplemental Fig. 3 and can be used within Pipeline Pilot 8.5 (Accelrys Software Inc) to predict the ability of any compound to inhibit OATP1B1 or OATP1B3.
Authorship Contributions
Participated in research design: De Bruyn, van Westen, Augustijns, Annaert.
Conducted experiments: De Bruyn, van Westen.
Contributed new reagents or analytic tools: Stieger, de Witte, IJzerman, Augustijns.
Performed data analysis: De Bruyn, van Westen, Annaert.
Wrote or contributed to the writing of the manuscript: De Bruyn, van Westen, IJzerman, Stieger, de Witte, Augustijns, Annaert.
Footnotes
- Received December 9, 2012.
- Accepted April 8, 2013.
This study was supported by grants from “Fonds voor Wetenschappelijk Onderzoek,” Flanders [G.0662.09N] and “Onderzoeksfonds” of the KU Leuven, Belgium.
T.D.B. received a Ph.D. scholarship [093306] from the Agency for Innovation by Science and Technology, Flanders; G.V.W. received funding from Tibotec BVBA.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- CHO
- Chinese hamster ovary
- DDI
- drug-drug interactions
- geneticin
- (2R,3S,4R,5R,6S)-5-amino-6-[(1R,2S,3S,4R,6S)-4,6-diamino-3-[(2R,3R,4R,5R)-3,5-dihydroxy-5-methyl-4-methylaminooxan-2-yl]oxy-2-hydroxycyclohexyl]oxy-2-(1-hydroxyethyl)oxane-3,4-diol
- OATP/Oatp
- organic anion transporting polypeptide (human/rat)
- PBS
- phosphate buffered saline
- PCM
- proteochemometric
- QSAR
- quantitative structure activity relationship
- SLC
- solute-linked carrier
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}