


Abstract
Organic anion transporters play an essential role in the distribution and excretion of numerous endogenous metabolic products and exogenous organic anions, including a host of widely prescribed drugs. The expression and activity of these transporters is influenced by several conditions, including transcriptional regulation, gender-dependent regulation, and genetic variation. In addition, the interaction of these transporters with several drugs and endogenous substrates has been well documented and may play a significant role in drug disposition and development of various disease states, such as nephrotoxicity and familial idiopathic hypouricemia. Members of this family of transporters have been localized mainly to the renal epithelia of various species. Much of the early research in this field has focused on their role in renal drug transport, yet increasing research on this family of transporters has localized them to various other epithelial tissues, including liver, brain, and placenta. Thus, an understanding of the role of these transporters in drug interaction and disposition in the kidney and other tissues may help in the determination of individual drug response, susceptibility to drug toxicity, and chemical carcinogenesis. This review seeks to summarize current knowledge of the molecular function and substrate profile of cloned organic anion transporters and to discuss recent progress in the understanding of the impact of interindividual variability, transcriptional regulation, and tissue distribution on individual drug response.
I. Introduction
Over the past decade, drug transporters have increasingly gained importance, bringing this group of proteins into the forefront of pharmacology research, after almost a century of trailing behind systematic investigations on drug-metabolizing enzymes, such as cytochrome P450 (Marshall, 1997; Pirmohamed and Park, 2001; Zanger et al., 2008). Extensive physiological and molecular cloning studies on various human transporter systems and their effects on patient disposition to clinically essential drugs have been on a steep ascent (Lopez-Nieto et al., 1997; Sekine et al., 1997; Sweet et al., 2001; Enomoto et al., 2002a; Nies et al., 2011). Among these transporters, organic anion transporters (OATs1) of the amphiphilic solute transporter family 22A (SLC22A), a subset of the major facilitator superfamily, have been the focus of intense scientific and medical interest because of their roles as exchangers in the elimination of many therapeutically relevant drugs and hormone conjugates (VanWert et al., 2010; Burckhardt and Burckhardt, 2011).
OATs act in secretion of drugs and their metabolites between blood and intracellular fluid in a protein-mediated process that frequently involves direct or indirect expenditure of energy (Pritchard and Miller, 1993; Simonson et al., 1994). All OATs are expressed in the two major excretory organs of the body (i.e., kidney and/or liver). Furthermore, individual family members (discussed in section II) are also expressed in different barrier epithelia, including the placenta and choroid plexus (Koepsell, 1998; Cha et al., 2000; Sweet et al., 2002). A composite of sequence analyses of the various OAT members exposes a family of proteins composed of 541 to 568 amino acid residues and 12 membrane-spanning α-helices (Koepsell, 1998; Sekine et al., 2000). The segments connecting these helices are relatively short with the exception of two large interconnecting loops between helices 1 and 2 and helices 6 and 7, where several glycosylated sites and multiple protein kinase C (PKC) phosphorylation sites of functional significance have been identified (Robertson and Rankin, 2006; Zhou and You, 2007).
OATs are characterized by two notable features. First, they display broad substrate specificity and interact with a large number of compounds that are capable of handling not only small, amphiphilic organic anions of varied chemical structures but also uncharged molecules and even some organic cations (Pritchard and Miller, 1993; Sweet and Pritchard, 1999; Van Aubel et al., 2000). Several studies investigating the structural requirements for OAT substrates suggest that substrate recognition is based on diverse physicochemical properties, such as hydrophobicity, the presence of one or more hydrogen-bonding sites, and an ionized or partial negative charge distribution (Ullrich, 1997; Burckhardt and Wolff, 2000). A typical OAT substrate is a type I organic anion with a relatively low molecular mass (up to 400–500 Da), such as para-aminohippurate (PAH) (Sekine et al., 2006). Thus, given this broad substrate specificity and the fact that drugs and their metabolites are often ionized in the blood, it is quite expected that these transporters interact with many clinically relevant and commonly prescribed anionic drugs, including penicillin, cephalosporin, diuretics, nonsteroidal anti-inflammatory drugs (NSAIDs), antiviral agents, and methotrexate. Furthermore, owing to their localization to the epithelial boundary of cells, these transporters may play a significant role in the absorption, distribution and elimination of clinically relevant drugs (Sweet et al., 2001; Eraly et al., 2004; Shugarts and Benet, 2009). In addition, these features allow OATs to mediate drug-drug interactions during competition of two or more agents for the same transporter and mediate cellular toxicity as a result of transporting cytotoxic drugs (Koepsell and Endou, 2004; Rizwan and Burckhardt, 2007). These factors imply that OATs have a fundamental role in drug interaction and response.
The second remarkable feature of this family of genes is their tendency to occur in pairs (as immediate chromosomal neighbors) in the genome. This has been observed for various members of this family, including OAT1 and -3, as well as OAT4 and urate transporter 1 (URAT1) (Eraly et al., 2003b). Furthermore, these pairs are also phylogenetically very closely related, suggesting that they are also one-another's closest relations (Eraly et al., 2004). One standing hypothesis for this observation implicates an evolutionary mechanism that allows sharing of regulatory elements between pair members. Such pairing exists to assist the coregulation of pair members through the use of shared regulatory elements (Pennacchio and Rubin, 2001; Eraly et al., 2003b). This and the distribution of OATs in multiple tissue compartments further support the “remote sensing” theory, which suggests that coregulation may further allow remote chemosensation among different tissue compartments, body fluids, and environments (Kaler et al., 2007; Ahn and Nigam, 2009). Although much of early research in the field of OA transport system is focused on the role of OATs in renal drug transport, the remote-sensing hypothesis might provide a means of exploiting the coregulatory capability among different tissues for improving drug design and delivery. This suggests the need for a more comprehensive understanding of the role of these proteins in drug disposition, not only in the kidney, but also in other tissues.
Recent functional and molecular analyses of OATs have improved our knowledge of this group of transporter proteins and their fundamental role in clinically significant drug interactions and adverse drug reactions. However, with this knowledge has come the realization that a complete characterization of this group of proteins is required for the development of patient and condition-specific drugs.
This review summarizes the current state of knowledge of molecular and functional characteristics of multispecific transport family of OATs, with a particular focus on tissue specific expression, as well as the impact of transcriptional, environmental, and hormonal regulation. Furthermore, we summarize currently available data on the genetic variability of OAT members and discuss their potential functional and pathophysiological impact, as well as how these findings may be further explored for drug development and the strategy of a more individualized method of therapy.
II. Localization, Substrate Specificity and Molecular Pharmacology of Organic Anion Transporter Family Members
A. Organic Anion Transporter 1 (SLC22A6)
In 1997, OAT1 was the first organic anion transporter to be cloned from rat kidney and was further functionally characterized using the Xenopus laevis oocyte expression system (Sekine et al., 1997; Sweet et al., 1997). Subsequent cloning attempts have led to the identification of the human ortholog, hOAT1, containing two functional splice variants, as well as orthologs from monkey, pig, rabbit, and Caenorhabditis elegans (Lopez-Nieto et al., 1997; George et al., 1999; Bahn et al., 2002; Hagos et al., 2002). The gene for hOAT1 (i.e., SLC22A6) is located on chromosome 11q12.3, very near the OAT3 gene, SLC22A8 (Eraly et al., 2003b) (Table 1). As will be discussed later, the close pairing of these two genes suggests a common ancestor and may suggest an underlying mechanism for coregulation. The mammalian OAT1s are composed of 545 to 551 amino acids. In particular, mouse and rat Oat1s consist of 546 and 551 amino acids, respectively (Hosoyamada et al., 1999; Race et al., 1999; Sweet and Pritchard, 1999). As mentioned above, in humans, there exist a longer splice variant that is composed of 563 amino acids (OAT1-1), a more predominantly expressed shorter isoform that is composed of 550 amino acids (OAT1-2), and two shorter, nonfunctional splice variants known as OAT1-3 and OAT1-4 (Race et al., 1999; Bahn et al., 2000). Studies of the secondary structure of this protein have predicted twelve putative transmembrane helices consisting of N and C termini located at the cytosolic side of the plasma membrane, a large extracellular loop between transmembrane helices (TM) 1 and 2, and a large intracellular loop between TM6 and TM7. This predicted topology has been further verified in the hOAT1 protein. Furthermore, the large extracellular loop between TM1 and TM2 carries several glycosylation sites and the C terminus, whereas the intracellular loop between TM6 and TM7 carry several consensus sequences for phosphorylation by protein kinases. The glycosylation of OAT1, including hOAT1, has been shown to be important for the correct shuttling of newly synthesized transporters to the cell membrane; however, the role of the identified phosphorylation sites is unclear and remains to be elucidated (Koepsell and Endou, 2004; Hong et al., 2007).
Gene loci and tissue distribution of hOATs
Early Northern blot analyses have revealed that OAT1/Oat1 transcripts are strongly expressed in human kidney as well as kidneys of monkeys, dogs, sheep, pigs, rabbits, rats, and mice (Lopez-Nieto et al., 1997; Sweet et al., 1997; Lu et al., 1999; Bahn et al., 2002; Hagos et al., 2002; Tahara et al., 2005b; Wood et al., 2005; Bleasby et al., 2006). Immunohistological studies have revealed the expression of hOAT1 and rat and mouse Oat1 at the basolateral membrane of proximal tubule kidney cells (Sekine et al., 1997; Hosoyamada et al., 1999). Low levels of OAT1/Oat1 mRNA were further detected in various other tissues and organs, including brain, liver, stomach, pancreas, salivary glands, urinary bladder, skeletal muscle, mammary glands, and some blood cells, such as leukocytes and neutrophils (Bleasby et al., 2006). In particular, studies on OAT1 expression in the human brain using microarray technology have revealed some mRNA expression levels in the temporal cortex, hypothalamus, hippocampus, and cerebellum (Bleasby et al., 2006). Immunohistological studies of the brain identified OAT1 staining on both the apical and basolateral sides of the cell membranes of human choroid plexus cells; however, the polarized localization of this transporter in human remains to be investigated (Alebouyeh et al., 2003). In rats, Oat1 is localized to the apical membrane of the choroid plexus cells, in addition to the neurons and axons in the cortex cerebri and hippocampus in the mouse (Bahn et al., 2005; Bleasby et al., 2006). Thus far, OAT1/Oat1 expression levels have been shown to be generally much higher in the kidneys than in any other organ, such that it can be identified as the transporter specific to the kidneys (Lopez-Nieto et al., 1997; Lu et al., 1999; Bleasby et al., 2006).
Along with OAT3, which will be discussed in section II.C, OAT1 is the transporter responsible for the first step of renal organic anion secretion, which involves the uptake of organic anions from the blood into the proximal tubule cells in exchange for intracellular α-ketoglutarate (Wright and Dantzler, 2004). As a result, OAT1s from different species interact with a wide selection of endogenous and exogenous organic anions of various chemical structures. The prototypical substrate of the OAT family of transporters (i.e., PAH) was first used for the basic characterization of OAT1. Various expression systems revealed a wide range of Km values for hOAT1, between 3.1 and 112.7 μM (Pritchard and Miller, 1993; Hosoyamada et al., 1999; Lu et al., 1999; Hagos et al., 2002). Furthermore, PAH uptake by hOAT1 was increased by an outwardly directed concentration gradient of α-ketoglutarate, which is in agreement with the theory that OAT1 is an organic anion/dicarboxylate exchanger and that the relative intracellular and extracellular concentration of α-ketoglutarate provides the driving force of the uptake of organic anions (Burckhardt and Burckhardt, 2003) (Fig. 1). In addition to PAH and α-ketoglutarate, OAT1/Oat1 has been shown to transport a large variety of endogenous compounds, including the local hormones prostaglandin E2 and prostaglandin F2α, the second messengers cAMP and cGMP, the purine breakdown product urate, the purine metabolites xanthine and hypoxanthine, the hormones corticosterone and dehydroepiandrosterone sulfate (DHEAS), although the transport of estrone sulfate (ES) is ambiguous and not yet confirmed (Sekine et al., 1997; Sugawara et al., 2005; Aslamkhan et al., 2006; Ueo et al., 2007; Sato et al., 2008). Furthermore, OAT1s have been shown to transport several anionic neurotransmitter metabolites, including the acidic metabolites of the neurotransmitters norepinephrine, dopamine, 5-hydroxytryptamine, and of cerebral tryptophan, suggesting a dual role of OAT1 in the removal of these metabolites from brain in addition to kidney (Sekine et al., 1997; Lu et al., 1999; Béery et al., 2003; Hasegawa et al., 2003; Sugawara et al., 2005; Kaler et al., 2007).
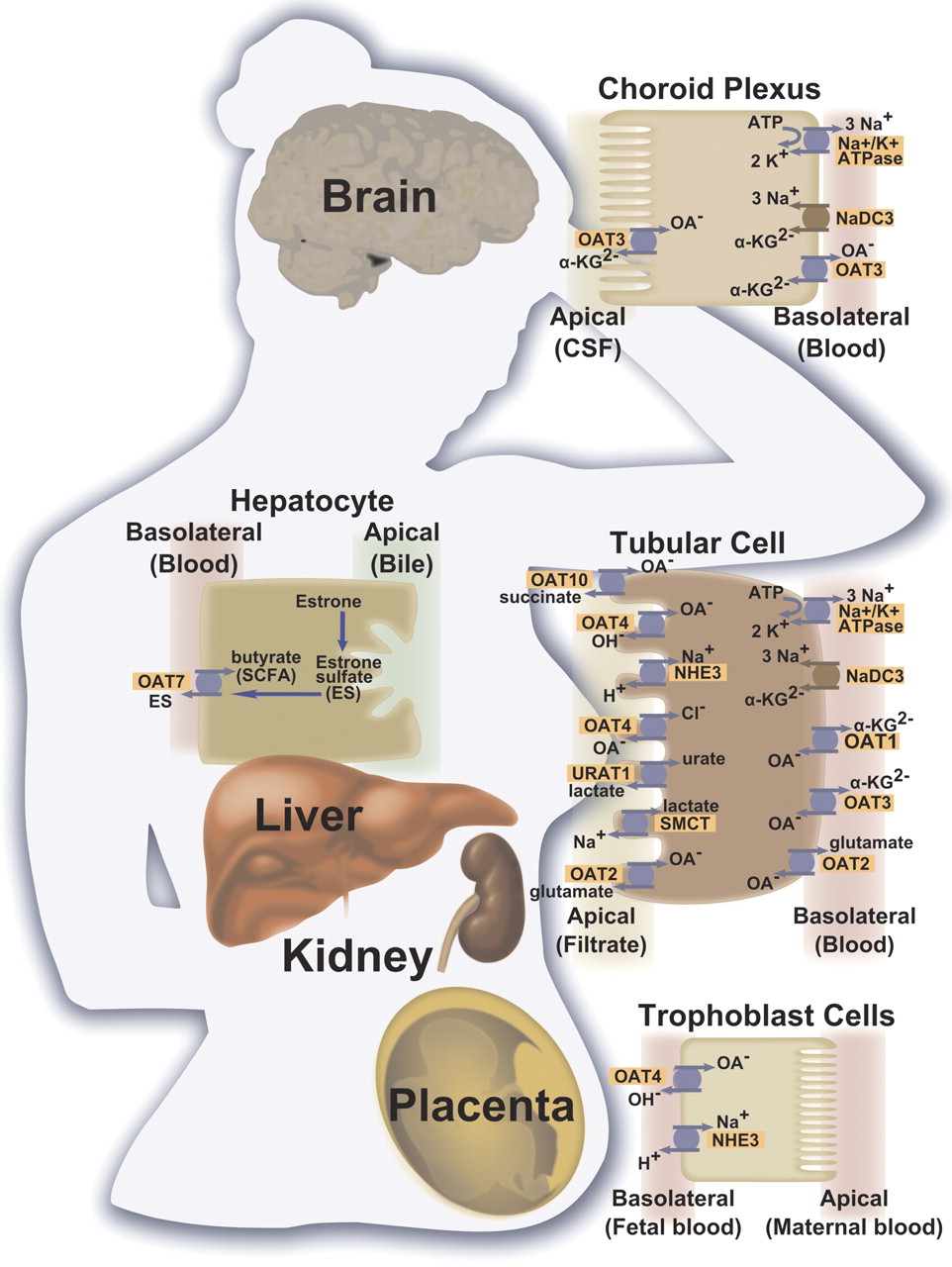
Tissue localization of selected human organic anion transporter proteins and their mechanism of action. Transporters located on the membrane domains of kidney proximal tubules, hepatocytes, choroid plexus epithelial cells, and placenta trophoblast cells are presented. OA−, organic anion; α-KG, α-ketoglutarate; SMCT, sodium-coupled monocarboxylate transporter I (SLC5A8); NHE3, sodium-hydrogen antiporter 3 (SLC9A3); NADC3, sodium-dependent dicarboxylate transporter 3 (SLC13A3); SCFA, short-chain fatty acid.
Eraly et al. (2006) generated a mouse knockout (KO) model of Oat1. The mice developed normally and were fertile. Furthermore, the kidneys were histologically unchanged, displaying normal glomerular filtration rate in addition to normal salt and water excretion. However, clearance of PAH and the excretion of 3-hydroxybutyrate, 3-hydroxyisobutyrate, 3-hydroxypropionate, benzoic acid, 4-hydroxyphenylpyruvate, 4-hydroxyphenyllactate, 4-hydroxyphenylacetate, and N-acetylaspartate were decreased, suggesting that these compounds are endogenous substrates of Oat1. This study showed that OAT1 contributes to the renal excretion of several additional endogenous substrates, but that normal function and survival of the mice is not dependent on the transport activity of this protein, most likely as a result of compensation by closely related family members, such as OAT3 (Eraly et al., 2006). Furthermore, several recent studies have reported a significant loss of urinary renal excretion of PAH in Oat1 KO mice, suggesting that the role of Oat3 in the transport of PAH in vivo is relatively minor (Eraly et al., 2006; VanWert et al., 2007; Vallon et al., 2008a). Yet analysis at the mRNA level has shown normal renal PAH secretion in Oat3 KO mice regardless of significant reduction of Oat1 expression, which may be due to their close genetic link. These observations suggest that the lack of OAT1 is not fully compensated by OAT3 for all substrates and may suggest the presence of alternative transporters, which compensate for the loss of Oat1 or Oat3 (Vallon et al., 2008a).
In addition to endogenous compounds, OAT1/Oat1 has been shown to interact with a large number of frequently prescribed therapeutic agents, including antihypertensives, statins, antibiotics, antivirals, histamine receptor 2 (HR2) antagonists, NSAIDs, and many more. A list of these compounds and their kinetic data is given in Table 2. Additional details regarding some of these compounds are provided below.
Summary of selected drugs interacting with OATs 1–4 and URAT1 in humans
1. Antihypertensives.
This group of compounds includes angiotensin-converting enzyme (ACE) inhibitors, and angiotensin II receptor blockers (ARBs). Some examples of ACE inhibitor transport by hOAT1 are shown in Table 2; to date, however, a systematic study has not been carried out on the transport of this group of compounds in any species, and thus the role of OAT1 in the excretion of this group of chemicals remains to be elucidated (Ueo et al., 2007; Yamada et al., 2007; Sato et al., 2008; Yuan et al., 2009).
2. Diuretics.
Loop and thiazide diuretics act to inhibit salt transporters in the tubule lumen in the thick ascending limb of the loop of Henle and in the distal convoluted tubule, respectively. As a result, it is believed that OAT1s are involved in their secretion in the proximal tubules and in aiding them reach their downstream target. This is supported by the high to intermediate potency of inhibition of OAT1s by these compounds (Uwai et al., 2000; Hasannejad et al., 2004; Hagos et al., 2007a). In general, OAT1 seems to have a higher affinity for most of the thiazide diuretics. Although loop diuretics have displayed greater interaction with OAT3 (Hasannejad et al., 2004), studies by Eraly et al. (2006) have indicated that Oat1 KO mice display a significant reduction in the tubular secretion of the loop diuretic furosemide, indicated by a rightward shift in a natriuresis dose-response curve compared with wild-type mice. Similar findings have been reported in mouse KO models by Vallon et al. (2008b) with respect to the thiazide diuretic bendroflmethiazide. It is possible that other diuretics are transported by OAT1 as well; however, direct experimental evidence proving this is still lacking.
3. Antibiotics.
Many different antibiotics, including penicillins, cephalosporins, tetracyclines, quinolones, aminoglycosides, and macrolides, have been tested as possible substrates of OAT1 (Hosoyamada et al., 1999; Jariyawat et al., 1999; Lu et al., 1999; Babu et al., 2002a; Takeda et al., 2002a; Ueo et al., 2005). For most compounds, interaction with and inhibition of OAT1 function have been observed, and corresponding kinetic values have been summarized in Table 2. However, tetracycline uptake into mouse proximal tubule cells transfected with hOAT1 was very similar to uptake into nontransfected cells, suggesting that OAT1 is not a great contributor to renal tetracycline excretion (Babu et al., 2002a). Similar conflicting results make it difficult to determine the extent of excretion of benzylpenicillin by hOAT1 (Hosoyamada et al., 1999; Lu et al., 1999).
4. Antiviral Agents.
OAT1/Oat1s are involved in the uptake of most antivirals from the blood into the proximal tubule cells, including the acyclic nucleoside analogs, which do not bear a negative charge (Lu et al., 1999; Wada et al., 2000; Takeda et al., 2002c; Hasegawa et al., 2003; Bleasby et al., 2005; Moss et al., 2011). Of interest is the ability of OAT1 to mediate the nephrotoxic potential of antiviral drugs, such as adefovir and cidofovir, in cellular expression systems, which can be attenuated through coadministration of probenecid or NSAIDs, an intended drug-drug interaction (Ho et al., 2000; Mulato et al., 2000).
5. Histamine Receptor 2 Antagonists.
As shown in Table 2, both cimetidine and ranitidine are translocated by OAT1 but not famotidine (Khamdang et al., 2004; Motohashi et al., 2004; Nagata et al., 2004). Furthermore, transport of cimetidine has been shown to display pH dependence, such that the uncharged form of cimetidine is the substrate preferentially transported by OAT1, whereas the charged form is most likely translocated by renal organic cation transporter 2 (OCT2) and by OAT3 (Tahara et al., 2005a).
6. Nonsteroidal Anti-Inflammatory Drugs.
Various nonsteroidal anti-inflammatory drugs were tested on OAT1 and were found to have moderate to high affinity for hOAT1. However, it should be noted that most NSAIDs display high plasma protein binding and little renal excretion, such that the free plasma concentrations may be much smaller than the IC50 values measured in vitro and an interaction between most of the compounds reported in Table 2 may not occur in vivo (Mulato et al., 2000; Khamdang et al., 2002; Nozaki et al., 2007).
7. Statins.
As can be seen in Table 2, fluvastatin, pravastatin, and simvastatin all inhibited OAT1; however, neither atorvastatin nor rosuvastatin display any interaction with this transporter. Nevertheless, the free plasma concentrations of fluvastatin, pravastatin, and simvastatin are much lower than the measured IC50 values, indicating that OATs most likely do not contribute greatly to the renal excretion of statins (Khamdang et al., 2004; Takeda et al., 2004; Windass et al., 2007).
As a result of its broad substrate specificity, it might be expected that OAT1 is a common site of drug-drug interactions when two or more drugs compete with each other for renal proximal tubular secretion. A classic example of drug-drug interaction involves the uricosuric drug probenecid and penicillin, which results in a decreased renal excretion of penicillin in patients. Although it is now believed that OAT3 is the main transporter responsible for the excretion of penicillin, the same principle applies to the renal excretion of various cephalosporins, which are primarily believed to be transported by OAT1 (Tsuji et al., 1990; Brown, 1993). A similar example of such a drug-drug interaction is that of probenecid and the diuretic furosemide, although OAT3 has a higher affinity for furosemide, it is also very likely that probenecid interacts with other loop and thiazide diuretics that are more specific substrates of OAT1 and decreases their renal excretion. Finally, as mentioned above, probenecid may prevent kidney damage of antivirals by reducing renal accumulation; therefore, cytotoxicity of these compounds (Brown, 1993; Cihlar et al., 1999; Uwai et al., 2000).
Another family of OAT1 substrates involved in drug-drug interactions is the NSAIDs, which act to potently inhibit OAT1. Such an interaction may explain the clinical observation that coadministration of ketoprofen with the chemotherapeutic drug methotrexate (MTX) resulted in life-threatening suppression of hematopoiesis as a consequence of MTX accumulation (Thyss et al., 1986). Although OAT3 has a higher affinity for the transport of MTX and, at low doses of MTX, might be the major site of interaction, OAT1 becomes an additional site of interaction in high dose therapy with MTX, resulting in severe toxicity (Khamdang et al., 2002; Takeda et al., 2002b; Nozaki et al., 2004). However, ketoprofen shows high protein binding, and it is not yet understood why these side effects are observed as a result of ketoprofen interaction.
Finally, recent findings indicate involvement of this transporter in reinforcing the protective role of mesna or dimesna, chemicals coadministered with chemotherapeutics such as cyclophosphemide or cisplatin to reduce their toxic side effects (Cutler et al., 2011). Hence, drug-drug interactions at this transporter may lead to a significant reduction in the efficacy of these chemoprotectants.
B. Organic Anion Transporter 2 (SLC22A7)
In 1994, an unknown liver protein with sequence motifs resembling those of the major facilitator superfamily was identified in rat liver and was named novel liver-specific transporter (NLT), although it was not yet functionally characterized (Simonson et al., 1994). Later, in 1998, in light of findings on OAT1, NLT was recloned from rat liver and expressed in X. laevis oocytes for functional characterizations (Sekine et al., 1998). These studies indicated that this protein was capable of mediating transport of organic anions. Furthermore, sequence comparisons indicated 42% sequence identity with previously characterized rat OAT1. The OAT-like functions of NLT suggested that it might be a member of the OAT family of proteins. NLT was established as the second member of the OAT protein family and was subsequently named Oat2 (Sekine et al., 1998). Since then, human and mouse orthologs have also been cloned and functionally characterized (Sun et al., 2001; Kobayashi et al., 2002). The gene for the human OAT2, known as SLC22A7, is located on chromosome 6p21.2-1 and is not paired with any other gene from the SLC22 family. OAT2/Oat2 protein isoforms consist of 535-546 amino acids arranged in a membrane topology that is identical to that of OAT1/Oat1 and other OATs (Simonson et al., 1994; Sun et al., 2001; Kobayashi et al., 2002). Two N-glycosylation sites and two putative phosphorylation sites for protein kinase A and C have been identified in rat Oat2 proteins and three potential N-glycosylation and six putative protein kinase C phosphorylation sites have been identified in rat Oat2 proteins, although the functional significance of these sites is not yet known (Kobayashi et al., 2002; Ljubojević et al., 2007; Chen et al., 2009).
Northern blot analyses have identified liver expression and, to a considerably lesser extent compared with OAT1 and OAT3, kidney expression of OAT2/Oat2 mRNA in mice, rats, and humans, as well as choroid plexus expression in mice and rats (Sekine et al., 1998; Sun et al., 2001; Kojima et al., 2002; Augustine et al., 2005) (Table 1). Immunohistological studies of hOAT2 have localized it to the basolateral membrane of the proximal tubules in the kidney, whereas the mouse and rat Oat2 protein have been localized to the apical membrane of the late proximal tubules (segment S3) or even the collecting duct of the kidneys (Enomoto et al., 2002b; Kobayashi et al., 2002; Kojima et al., 2002; Hilgendorf et al., 2007). Surprisingly, although it is commonly assumed that OAT2/Oat2 is localized to the sinusoidal membrane of hepatocytes, the subcellular localization of this protein in the liver, which is the main site of its expression, has only been demonstrated in rat but not in humans (Simonson et al., 1994).
Furthermore, the driving force influencing the transport function of OAT2/Oat2 is still in dispute, especially after the findings regarding the dissimilar subcellular localization of this protein in different species. At first, hOAT2 was believed to transport the dicarboxylate α-ketoglutarate, yet its PAH transport functions were not inhibited by this substrate, as was observed for hOAT1 (Sekine et al., 1998; Sun et al., 2001; Kobayashi et al., 2005). Similarly conflicting results exist for the rOAT2 with regard to its interaction with α-ketoglutarate (Sekine et al., 1998; Morita et al., 2001). Furthermore, the dicarboxylate glutarate was also found to be transported by hOAT2 expressed in oocytes, but again, it did not trans-stimulate ES uptake, as observed for other OAT proteins (Kobayashi et al., 2005). In addition, studies by Kobayashi et al. (2005) in X. laevis oocytes found that the uptake of radioactively labeled PAH via hOAT2, as well as rat and mouse Oat2, were not affected when extracellular sodium was replaced with lithium, choline, or mannitol, suggesting that OAT2/Oat2 is a sodium-independent transporter. Furthermore, Kobayashi et al. (2005) proposed that the 4-carbon dicarboxylates, succinate and fumarate, are transported by OAT2 and are also capable of trans-stimulating the uptake of estrone-sulfate, which was recently, however, questioned by Rizwan and Burckhardt (2007). Fork et al. (2011) studied the substrate specificity of this transporter again to clarify the confusion over its precise mechanism of transport. Their studies involved the use of human embryonic kidney (HEK) 293 cells, which may be a more reliable expression system than X. laevis oocytes. The authors showed that OAT2 functions as a high-affinity glutamate efflux carrier, such that even in the absence of extracellular substrates to be exchanged, it releases glutamate from the cell. Hence, the physiological function of OAT2 was determined to be to release glutamate from cells (Fig. 1). Furthermore, in the article by Fork et al. (2011), OAT2 was shown to transport orotic acid with high specificity (Fork et al., 2011). Because orotic acid transport proteins have only been identified in bacteria (Defoor et al., 2007), OAT2 is one of few organic anion transporters that has been shown to carry out this function in mammals. As discussed by the authors, orotic acid is not retained in the cells, but rather, efficiently eliminated by the kidney; thus, it may seem that hepatic uptake of orotic acid by OAT2 is not imperative (Fork et al., 2011). However, OAT2 may play an important role in orotic aciduria, a disease state associated with excessive urinary excretion of orotic acid, by facilitating the transport of large stores of orotic acid from hepatocytes. This further introduces the possibility of targeting OAT2 for therapeutic intervention (Brosnan and Brosnan, 2007; Fork et al., 2011).
Furthermore, OAT2/Oat2 plays a role in the transport of various other endogenous and exogenous substrates (Table 2), although compared with OAT1 and OAT3, only a limited number of therapeutic drugs have been tested on this transporter thus far. With respect to endogenous compounds, as mentioned above, α-ketoglutarate may be transported by OAT2, and fumarate and succinate are believed to be counteranions for the exchange of ES by this transporter. Furthermore, hOAT2 was reported to transport the second messengers cAMP and cGMP as well as adenine, adenosine, cytidine, guanidine, guanosine, inosine, thymine, and thymidine, but not cytosine (Sun et al., 2001; Cropp et al., 2008). In addition, prostaglandin E2 is transported by hOAT2 and to a lesser extent by rOAT2. As well, both human and rat OAT2/Oat2 transport prostaglandin F2α but with lower affinity compared with prostaglandin E2 (Morita et al., 2001; Enomoto et al., 2002b; Kimura et al., 2002). Sulfated steroid hormones, such as DHEAS and estrone sulfate, are also taken up by hOAT2 but not by rOAT2. It must be noted, however, that results regarding DHEAS are still unclear, because it was found to be transported by hOAT2 in one study and not in another (Sun et al., 2001; Kobayashi et al., 2005).
OAT2-mediated transport of therapeutic compounds belonging to various drug classes have been investigated, including antibiotics, diuretics, antivirals, NSAIDs, HR2 antagonists, and antineoplastic drugs. A list of these compounds and their kinetic data is presented in Table 2, and a brief summary of these drug classes is given below. Furthermore, no known OAT2-related drug-drug interactions have yet been reported.
1. Diuretics.
Both loop and thiazide diuretics have been tested for interaction with OAT2. Compared with OAT1, the affinities for most diuretics are lower. hOAT2 displayed intermediate (bumetanide, ethacrynate) to low (furosemide, hydrochlorothiazide, chlorothiazide) affinity for selected diuretics (Hasannejad et al., 2004; Kobayashi et al., 2005). Bumetanide was shown to be transported with high affinity in one study (Kobayashi et al., 2005) but not in another (Hasannejad et al., 2004). In addition, rOAT2 was inhibited by bumetanide and mOAT2 showed a high affinity for bumetanide transport (Sekine et al., 1998; Kobayashi et al., 2005).
2. Antibiotics.
OAT2 interacted with cephalosporins, tetracyclines, and the macrolide erythromycin, although cephalosporins inhibited this transporter with really low affinities, especially compared with OAT1 transport (Khamdang et al., 2002; Kobayashi et al., 2005). In addition, erythromycin was transported with good affinity by hOAT2 but not rOAT2, and tetracycline was transported with low affinity by hOAT2 (Table 2) (Morita et al., 2001; Babu et al., 2002a; Kobayashi et al., 2005).
3. Antiviral Agents.
As shown in Table 2, except for transporting zidovudine (AZT), hOAT2 does not display any major interaction with other antiviral drugs (Takeda et al., 2002c). In addition, rOAT2 was found to mediate the transport of AZT with an affinity similar to that of hOAT2 (Morita et al., 2001).
4. Histamine Receptor 2 Antagonists.
Cimetidine and ranitidine are translocated by hOAT2, but conflicting data exist with regard to cimetidine transport and inhibition in rats (Morita et al., 2001; Khamdang et al., 2004; Tahara et al., 2005a).
5. Nonsteroidal Anti-Inflammatory Drugs.
Again, the IC50 values of OAT2 for NSAIDs are considerably higher than that of OAT1. The affinity of hOAT2 ranges from intermediate (diclofenac, indomethacin, mefenamate, and piroxicam) to low or very low (ibuprofen, ketoprofen, naproxen, sulindac, and phenacetin). In general, hOAT2 displays a very low affinity for hydrophilic NSAIDs (Khamdang et al., 2002).
6. Antineoplastic Drugs.
hOAT2 displayed very high affinity for transport of 5-fluorouracil, MTX, and paclitaxel (Taxol) (Kobayashi et al., 2005). Some conflicting data exist for rOAT2 with relation to MTX transport (Sekine et al., 1998; Sun et al., 2001; Kimura et al., 2007).
Overall, despite initial characterization of OAT2, further studies on its function and cellular expression have yielded highly variable results. Especially with respect to substrate specificity and species- or sex-dependent expression patterns (discussed in chapter III), there is considerable uncertainty with respect to the specific role of this protein in organic anion transport.
C. Organic Anion Transporter 3 (SLC22A8)
Kusuhara et al. (1999) isolated a third member of the OAT family by isolating a rat brain cDNA library through homology screening with an Oat1-based probe. Since then, orthologs from various other species have been cloned, including human, monkey, pig, rabbit, and mouse (Brady et al., 1999; Cha et al., 2001; Zhang et al., 2004; Hagos et al., 2005; Tahara et al., 2005b). The gene for the human OAT3 protein is designated SLC22A8 and is located on chromosome 11q12.3, where it is paired with SLC22A6, the gene coding for OAT1 (Cha et al., 2001; Eraly et al., 2003b) (Fig. 2). The mammalian OAT3/Oat3 proteins consist of 536 to 542 amino acids, again with a secondary structure identical to that of other members of the OAT family. The large intracellular loop between helices 6 and 7 contains potential phosphorylation sites for its regulation by protein kinases (Kusuhara et al., 1999; Cha et al., 2001). PKC activation in rat by Takeda et al. (2000) caused an inhibition of rOAT3, believed to be a result of internalization.

A phylogenetic tree of all known human SLC22 family members, including all human organic anion transporters discussed in this review. This tree depicts the pairing of specific members of the OAT family, which may provide evidence for the remote sensing and signaling hypothesis. [Reprinted from Nies AT, Koepsell H, Damme K, and Schwab M (2011) Organic cation transporters (OCTs, MATEs), in vitro and in vivo evidence for the importance in drug therapy. Handb Exp Pharmacol (201):105–167. Copyright © 2011 Springer. Used with permission.]
Northern blot analyses localized OAT3/Oat3 mRNA to the kidneys in humans, mice, monkeys, dogs, pigs, and rats (Brady et al., 1999; Kusuhara et al., 1999; Race et al., 1999; Cha et al., 2001; Hagos et al., 2005; Tahara et al., 2005b; Bleasby et al., 2006). Although OAT3 mRNA expression was highest in the kidneys of all species, it was also detected in blood-brain barrier (BBB) endothelium, choroid plexus, skeletal muscle, liver, and adrenal gland (Cha et al., 2001; Alebouyeh et al., 2003; Kikuchi et al., 2003; Kobayashi et al., 2004; Asif et al., 2005). In addition, hOAT3 mRNA levels in the kidney exceeded those described for OAT1 (Motohashi et al., 2002). Immunohistochemical analyses revealed that in the kidneys, OAT3/Oat3 is located on the basolateral membrane of the proximal tubular cells in humans, mice, and rats (Kojima et al., 2002; Motohashi et al., 2002; Bahn et al., 2005). In rats, OAT3 was additionally found in other segments of the nephron, including the thick ascending limb of the loop of Henle, the distal convoluted tubule, and collecting ducts (Kojima et al., 2002; Bahn et al., 2005). Although the basolateral localization of OAT3/Oat3 fits with its role in the uptake of organic anions from blood into proximal tubule cells, its function in deeper nephron segments is not yet clear. In the choroid plexus, hOAT3 staining was found on both the apical and basolateral sides of cell membranes of epithelial cells; however, more studies are needed to confirm its exact polarized localization (Alebouyeh et al., 2003). At the BBB, rOat3 has been localized to the abluminal membrane of endothelial cells (Kikuchi et al., 2003; Mori et al., 2003) (Table 1).
Analogous to OAT1, OAT3/Oat3 is believed to operate as an organic anion/α-ketoglutarate exchanger (Fig. 1). Hence, the lists of substrates transported by these two transporters are overlapping, though not identical. In general, OAT3 has been shown to handle bulkier and more lipophilic organic anions compared with OAT1 (Bakhiya et al., 2003; Sweet et al., 2003). Surprisingly, the transport of α-ketoglutarate was not proven experimentally, but inhibition of OAT3 by this substrate was shown in human, monkey, and rabbit (Bakhiya et al., 2003; Zhang et al., 2004; Tahara et al., 2005b). A list of these substrates is shown in Table 2 and is further summarized below.
Among the endogenous substrates, estrone sulfate is the prototypical test substrate for OAT3/Oat3 because it is transported with very high affinity (Km values ranging between 2.2 and 21.2 μM). As will be discussed shortly, the ability of OAT3 to transport ES and taurocholate distinguishes it from OAT1 (Cha et al., 2001; Takeda et al., 2001; Erdman et al., 2006). Furthermore, hOAT3 has been found to transport cAMP, cortisol, prostaglandins E2 and F2α, DHEAS, estradiol-17β-glucuronide, taurocholate, and urate (Cha et al., 2001; Kimura et al., 2002; Kobayashi et al., 2004; Asif et al., 2005; Nilwarangkoon et al., 2007; Chen et al., 2008). A large number of neurotransmitters and their metabolites were tested with human, rat, and mouse transporters. In general, the tests showed that the OAT3/Oat3 located at the BBB and choroid plexus was likely involved in the efflux of acidic metabolites from brain tissue (Kusuhara et al., 1999; Alebouyeh et al., 2003; Mori et al., 2003). Furthermore, studies by Vallon et al. (2008a) demonstrated lowered blood pressure in Oat3 KO mice, suggesting that OAT3 specifically transports endogenous regulators of blood pressure. In particular, thymidine was shown to be transported in vitro and resulted in a reduction in blood pressure in vivo.
Oat3 KO [Oat3(−/−)] mice have been generated by Sweet et al. (2002) to determine the specific function of this protein in kidney and choroid plexus. In these mice, no Oat3 mRNA expression was detected in the kidney, liver, or choroid plexus, yet they appeared normal and fertile with no gross morphological tissue abnormalities. This may be due to the fact that other OATs, such as OAT1, provide backup transport function in the absence of OAT3. However, a particular phenotype involved a substantial decrease in the transport of organic anions, including renal excretion of diuretics, benzylpenicillin, quinolones, methotrexate, taurocholate, and estrone sulfate, suggesting that these organic anions are largely transported by OAT3, whereas other OATs, such as OAT1 and OAT4, may play a larger role in the transport of other identified organic anions (Sweet et al., 2002; VanWert et al., 2007, 2008; Vallon et al., 2008b; VanWert and Sweet, 2008). Furthermore, approximately 75% reduction was observed in the accumulation of fluorescein in the choroid plexus of KO mice, whereas capillary accumulation was unchanged (Sweet et al., 2002). These observations suggest that the effects of Oat3 loss may be restricted to the apical entry step and that Oat3 plays a significant role in controlling the systemic detoxification and distribution of organic anions within the cerebrospinal fluid. In addition, no differences were observed in the hepatic activities of wild-type compared with KO mice, suggesting that Oat3 does not play a key role in hepatic OA uptake (Sweet et al., 2002).
In addition to endogenous compounds, a myriad of exogenous compounds and clinical substances have been tested with human OAT3. A list of these compounds and their kinetic data is provided in Table 2. They include diuretics, antibiotics, antivirals, histamine receptor 2 antagonists, and NSAIDs.
1. Diuretics.
A comprehensive database is available on the interaction of hOAT3 with carboanhydrase inhibitors, thiazide diuretics, and loop diuretics. Bumetanide, ethacrynate, and furosemide (loop diuretics), were found to display the highest inhibitory potency toward hOAT3, whereas most thiazide diuretics, as well as torasemide (a loop diuretic) displayed intermediate affinity. Acetazolamide (carboanhydrase inhibitor) and hydrochlorothiazide (thiazide diuretic) displayed low affinity (Kusuhara et al., 1999; Hasannejad et al., 2004; Chu et al., 2007; Hagos et al., 2007a; Vallon et al., 2008b).
2. Antibiotics.
Because of its ability to transport benzylpenicillin with a greater affinity than OAT1, OAT3 is believed to be the transporter mainly involved in renal excretion of penicillin in all species, including humans, rats, mice, and monkeys. Benzylpenicillin has been shown to be transported in mice and rats, Km values ranging from 40 to 85.1 μM, respectively (Kusuhara et al., 1999; Hasegawa et al., 2002; Ohtsuki et al., 2004). In vivo studies in KO mouse models showed a 2-fold increase in the half-life of penicillin G and a significant reduction in its volume of distribution, indicating that Oat3 plays a significant role in the elimination and efficacy of this compound (VanWert et al., 2007). Other antibiotics, including tetracycline, also displayed high-affinity transport by OAT3; however, inhibition studies could not confirm whether this specific transporter plays a role in the excretion of these compounds (Cha et al., 2001; Babu et al., 2002a; Takeda et al., 2002a; VanWert et al., 2007).
3. Antiviral Agents.
Conflicting data are available on the transport of different antivirals by OAT3. OAT3 is believed to contribute equally to the renal excretion of AZT, whereas adefovir and ganciclovir have not been shown to be transported by OAT3, and valacyclovir has been shown to be only weakly transported (Takeda et al., 2002c; Kobayashi et al., 2004; Uwai et al., 2007). As a result, it is believed that OAT1/Oat1 plays a greater role in the renal transport of antivirals compared with OAT3/Oat3.
4. Histamine Receptor 2 Antagonists.
hOAT3 transported cimetidine, famotidine, and ranitidine (Khamdang et al., 2004; Motohashi et al., 2004; Nagata et al., 2004; Tahara et al., 2005a; Erdman et al., 2006). Although it was shown that hOAT1 can also transport cimetidine, it does so with a lower affinity, whereas it has been shown to have very low affinity for ranitidine transport and no affinity for famotidine transport (Tahara et al., 2005a). These observations taken together suggest that OAT3 has generally a greater preference for HR2 antagonists than hOAT1.
5. Nonsteroidal Anti-Inflammatory Drugs.
hOAT3 has been shown to interact with various NSAIDs with high (diclofenac, ibuprofen, indomethacin, ketoprofen), intermediate (etodolac, phenacetin, salicylate), and low to very low (acetylsalicylate) affinity (Cha et al., 2001; Khamdang et al., 2002; Uwai et al., 2004; Nozaki et al., 2007). Compared with the affinity of other transporters, including hOAT1, hOAT2, and hOAT4, hOAT3 has considerably smaller IC50 values, suggesting that NSAIDs have comparatively the highest affinity for inhibition of this transporter.
A noteworthy drug-drug interaction at the site of hOAT3 involves MTX and β-lactam antibiotics. hOAT3 has been reported to carry out the high-affinity renal uptake of MTX (Balis et al., 1998; Takeda et al., 2002). MTX is mainly excreted through the urine in its unchanged form, and simultaneous use of acidic drugs, especially β-lactam antibiotics, such as penicillin G, has been shown to result in severe bone marrow suppression as a result of competitive inhibition for the uptake of MTX at the site of transport (Cha et al., 2001; Takeda et al., 2002). This interaction is clinically significant in the case of multi-drug treatment, especially in the treatment of patients with decreased renal function or when large dose antibiotics are administered.
D. Organic Anion Transporter 4 (SLC22A11)
Cha et al. (2000) cloned the human OAT4 from a kidney library. This transporter is unique among previously identified OATs in that it is human-specific; i.e., there are no known orthologs in other species. It must be noted that a year later, Sun et al. (2001) cloned another human gene that was also named OAT4; however, it was soon determined that this gene was not identical in function or localization to the one initially identified as OAT4, and it was later renamed OAT7 (gene SLC22A9) (Shin et al., 2007). The gene for OAT4 has been designated SLC22A11 and is located on chromosome 11q13.1, paired with SLC22A12, the gene encoding URAT1 (Cha et al., 2000; Enomoto et al., 2002b; Eraly et al., 2003b). OAT4 protein consists of 550 amino acids and the secondary structure characteristic of all OATs (i.e., five potential N-glycosylation and nine putative protein kinase C phosphorylation sites) (Cha et al., 2000). The glycosylation was found to be required for targeting OAT4 to the membrane and influencing its affinity toward estrone sulfate (Zhou et al., 2004a,b, 2005). Furthermore, as will be discussed in section III.B, OAT4 was found to interact with the scaffolding proteins PDZK1 and NHERF1 at its PDZ (postsynaptic density 95/disc-large/zona occludens) binding motif found at the C terminus, which has been shown to play a role in protein-protein interaction (Hung and Sheng, 2002). This interaction has been shown to increase the surface expression of OAT4 and is believed to play an important role in the correct membrane targeting and maintenance of this transporter in renal cells (Kato et al., 2004; Miyazaki et al., 2005).
Northern blot analyses have located OAT4 mRNA transcripts in kidneys, placenta, and adrenal glands (Cha et al., 2000; Asif et al., 2005; Bleasby et al., 2006). Immunohistochemical studies further detected this protein in the apical membrane of the proximal tubule cells (Ekaratanawong et al., 2004). This finding suggests that OAT4 is either involved in the absorption of organic anions from the primary filtrate into the proximal tubule cells or in the secretion of organic anions that have been collected in the cell by OAT1 and OAT3 back into the urine. With respect to the placenta, OAT4 has been localized to the basal (fetal) side of the syncytiotrophoblast, where it allows the transport of potentially toxic compounds from the fetus back into the mother and the transfer of sulfated C19-steroid precursors, which is required for placental estrogen synthesis, from the mother into the fetus (Ugele et al., 2003).
The prototypical substrate used in the study of OAT4 is the conjugated hormone estrone sulfate, because it is transported with very high affinity (Km, 1.01–21.7 μM) (Cha et al., 2000; Ekaratanawong et al., 2004; Hagos et al., 2007b). OAT4 carries out the transport of organic anion substrates in a sodium-independent manner. Furthermore, trans-stimulation of OAT4 has been reported to take place by dicarboxylates, suggesting that it is a bidirectional exchanger (Cha et al., 2000; Zhou et al., 2005). However, further functional assessments of OAT4 indicated that it transports uric acid and PAH unidirectionally in exchange for chloride and/or hydroxide ions (Hagos et al., 2007b). Consequently, it became evident that OAT4 has various mechanisms of transport. During secretion (efflux), organic anions, including PAH, are exchanged against extracellular chloride, whereas, during absorption (influx), the uptake of estrone sulfate or urate is coupled to the release of dicarboxylates. As well, OAT4 may be coupled to the Na+/H+ exchanger 3 (NHE3 on brush-border membrane) to carry out organic anion/hydroxyl ion exchange (Ekaratanawong et al., 2004; Hagos et al., 2007b) (Fig. 1). Hence, not only does this transporter display diverse modes of action, it is also capable of interacting with both organic and inorganic anions. Examples of these compounds and their kinetic data are provided in Table 2, and more details regarding various families of xenobiotics are given below.
1. Diuretics.
hOAT4 interacted with bumetanide, but with a much slower uptake rate than hOAT3. In addition, it showed very low to no affinity for thiazide diuretics and very variable affinity for loop diuretics, including intermediate affinity for ethacrynate, furosemide, and torasemide (Hasannejad et al., 2004; Hagos et al., 2007a).
2. Antibiotics.
To date, interaction of hOAT4 has conclusively been shown only for tetracycline, such that it has the highest affinity for this compound among other OATs (i.e., hOAT1, hOAT2, and hOAT3) (Babu et al., 2002a). Other tetracyclines, including oxytetracycline and minocycline, did not show an interaction with hOAT4. Most cephalosporins also showed very low affinity for transport by hOAT4 (Takeda et al., 2002a). In addition, benzylpenicillin was shown to inhibit hOAT4, but the IC50 value has not been reported (Cha et al., 2000). Hence, OAT4 does not seem to display very high levels of interaction with this group of xenobiotics.
3. Nonsteroidal Anti-Inflammatory Drugs.
In general, NSAIDs display lower affinity for the inhibition of hOAT4-mediated transport compared with hOAT1 and hOAT3 but higher than those reported for hOAT2-mediated transport. Most NSAIDs have been shown to interact with hOAT4 with only intermediate affinity, including diclofenac, indomethacin, ketoprofen, naproxen, and mefenamate (Khamdang et al., 2002). This suggests that NSAIDs act to inhibit OAT4, even if they are not transported by it to a great extent (Rizwan and Burckhardt, 2007).
E. Organic Anion Transporter 5 (SLC22A19)
An additional member of the OAT family was identified through in silico analysis of the Ensembl mouse genome database by Youngblood and Sweet (2004) and was designated Oat5. The following year, its rat ortholog was discovered by Anzai et al. (2005) through a homology search of an expressed sequence tag database using a human OAT4 sequence. Rat Oat5 was found to have only 55% amino acid identity to a human gene product that had been discovered by Sun et al. (2001) and named hOAT5 in 2001 (Table 1). As a result, human and rodent forms have been named differently, with the human form keeping the systematic name SLC22A10, whereas the rodent protein was designated SLC22A19. To date, SLC22A10 has not been functionally characterized and there is much debate around whether these two proteins are in fact orthologs. Hence, further discussion in this section will be referring to the rodent transporters, because more functional studies have been carried out in these species.
Mouse and rat Oat5 proteins consist of 551 amino acids, 12 putative transmembrane helices, four N-glycosylation, and five potential PKC phosphorylation sites, with no known regulatory roles. Mouse OAT5 gene, Slc22a19, was localized to mouse chromosome 19 near the genes encoding Oat1 and Oat3 (Youngblood and Sweet, 2004; Anzai et al., 2005).
Northern blot analyses demonstrated that in both adult mice and rats Oat5 is expressed exclusively in the kidneys, with a potential gender difference that will be discussed later in this review (Anzai et al., 2005; Kwak et al., 2005; Breljak et al., 2010a). Furthermore, immunohistochemical studies have localized Oat5 to the brush-border (apical) membrane of the late segments (S2–S3) of the proximal tubule (Koepsell and Endou, 2004).
An interesting finding with respect to the transport function and mechanism of Oat5 is the interaction of this transporter with succinate, a 4-carbon dicarboxylate (Youngblood and Sweet, 2004; Anzai et al., 2005). All previously identified transporters (i.e., OAT1–4) were shown to function as C5 dicarboxylate exchangers, which suggests that Oat5 operates in the same way as the classic renal OAT system, but with a different preferred counterion (i.e., succinate). In addition, this finding confirms that Oat5 mainly functions at the apical membrane for re-absorption of organic anions, because the classic renal OATs mediating the basolateral transport of organic anions use the five-carbon dicarboxylate (i.e., α-ketoglutarate) as a counteranion. Yet, studies on mOAT5 by Youngblood and Sweet (2004) found that mOAT5-mediated transport was neither cis-inhibited nor trans-stimulated by dicarboxylates glutarate or succinate, which Anzai et al. (2005) attribute to the use of low concentrations of dicarboxylates (Youngblood and Sweet, 2004; Anzai et al., 2005). Nevertheless, these conflicting results suggest the need for more studies on these systems to elucidate the specific mode of action of this transporter. Because of a lack of more comprehensive studies, only a small number of endogenous substrates or clinical drugs have been studied to date. Among the endogenous substrates, sulfate conjugates of steroids, estrone sulfate and DHEAS, as well as ochratoxin A, were transported with high affinity by both mOat5 (Km: ES, 2.2 μM; DHEAS, 3.8 μM; ochratoxin A, 2.0 μM) and rOat5 (Km: ES, 18.9 μM; DHEAS, 2.3 μM; ochratoxin A, 0.34 μM) (Youngblood and Sweet, 2004; Anzai et al., 2005; Kwak et al., 2005). The prototypical endogenous substrate traditionally transported by other OATs (i.e., PAH) was not transported in either mOat5- or rOat5-expressing X. laevis oocytes (Youngblood and Sweet, 2004; Anzai et al., 2005). With respect to clinically relevant substrates, rOAT5 was found to be inhibited by the diuretic furosemide, the antibiotic benzylpenicillin, and the NSAIDs, diclofenac, ibuprofen, and salicylate. Salicylate was also found to inhibit mOAT5. No known drug-drug interactions at this transporter have been reported (Youngblood and Sweet, 2004; Anzai et al., 2005).
F. Organic Anion Transporter 7 (SLC22A9)
OAT7 was discovered by Shin et al. (2007) through a comprehensive search of the human genome database for sequences showing homology to human OAT genes using the BLAST program. The nucleotide sequence of one predicted gene of interest matched an expressed sequence tag from human liver and was further used to screen a human liver cDNA library, leading to the cloning of the gene encoding hOAT7 (SLC22A9). The hOAT7 gene was mapped to chromosome 11q13.1 and is not paired to other known SLC22 family members (Jacobsson et al., 2007; Shin et al., 2007). In fact, this transporter displays only 35 to 46% sequence identities compared with sequences from hOAT1–4 (Fig. 2). In addition, orthologs of OAT7 have not been identified in other species to date, although predictional sequence analysis studies reveal a high sequence homology with the mouse Slc22a19 gene, also known as organic anion transporter 5 (Sun et al., 2001). As shown in Fig. 2, UST6 (SLC22A25) and Oat5 (SLC22A19) are two members of the SLC22 family that display some of the higher degrees of sequence homology compared with hOAT7.
Northern blot analyses identified OAT7 exclusively in the liver, because the strongest OAT7 mRNA expression was initially only detected in human liver (adult and fetal) and only after prolonged exposure in additional tissues (Shin et al., 2007). Furthermore, immunohistochemical investigations of the cellular and subcellular distribution of hOAT7 protein in intact human liver tissues suggest that OAT7 is consistently distributed across liver acini, specifically localized to the sinusoidal membrane of hepatocytes (Fig. 1) (Shin et al., 2007).
hOAT7 is not only unique for being the first liver-specific functional OAT member to be discovered in humans but also displays very specific and dissimilar substrate interaction capabilities. Several well known and previously mentioned OAT substrates and inhibitors, such as probenecid, PAH, glutarate, taurocholic acid, and bumetanide failed to inhibit OAT7, suggesting a lack of interaction (Shin et al., 2007). Furthermore, glutathione and glucuronide that had been shown to interact with other more exceptional members of the OAT family, such as OAT10, also failed to display any inhibitory effects on OAT7. Instead, monocarboxylates such as butyrate, trans-stimulated OAT7, which indicates unique substrate-exchange properties of this liver-specific transporter (Shin et al., 2007). In addition, OAT7 was found to preferentially interact with sulfate conjugates of xenobiotics and steroid hormones. Although other OATs have also been reported to mediate the transport of sulfate conjugates (i.e., OAT3 and OAT4), they also mediate the transport of a myriad of other organic anions and xenobiotics, whereas OAT7 seems to display a very narrow substrate recognition capability (Shin et al., 2007). Consequently, Shin et al. (2007) believe that OAT7 might play a significant role in mediating the transport of sulfated steroid hormone metabolites, such as estrone sulfate, from hepatocytes into the bloodstream in exchange for circulating short-chain fatty acids, such as butyrate, that are not used by colonocytes for metabolic fuel, a physiological role that had not previously been assigned to any other protein.
To date, only a very limited number of clinically relevant drugs have been tested, including salicylate, indomethacin, and benzylpenicillin, all of which were incapable of inhibiting the activity of this transporter (Shin et al., 2007).
G. Organic Anion Transporter 10 (SLC22A13)
OAT10 was first identified in 1998 as an OCT, based on its structural similarity to previously identified OCTs from the SLC22 family. The protein was initially named organic cation transporter-like 3 (ORCTL3), but remained an orphan transporter as further functional analyses ceased (Nishiwaki et al., 1998). Bahn et al. (2008) expressed hORCTL3 in X. laevis oocytes and found an uptake of organic anions, such as nicotinate, PAH, and urate, instead of the expected organic cations. Furthermore, several protein sequence alignments of all the known OCTs and OATs, including the nonclassified orphan transporters from the SLC22 family, indicated the presence of highly conserved amino acids. Subsequently, based on these amino acid comparisons and substrate studies, hORCTL3 was classified as an organic anion transporter and renamed hOAT10 (Bahn et al., 2008). The gene encoding hOAT10 is located on chromosome 3p21.3, paired with ORCTL4, and codes for 551 amino acids with a 44% homology to hOAT1 (Nishiwaki et al., 1998). Phylogenetic analyses revealed that hOAT10 constitutes its own branch and demonstrates only weak homologies to other members of the SLC22 family (Fig. 2). Furthermore, mouse and rat orthologs have been identified through BLAST searches, although they have not yet been functionally characterized (Bahn et al., 2008).
Initial Northern blot studies on ORCTL3 suggested that it is ubiquitously expressed with a preference for kidneys, small intestine, and colon, such that it was believed to be a polyspecific organic cation transporter in various tissues (Nishiwaki et al., 1998). Later, however, through mRNA expression studies, Bahn et al. (2008) found that only the OAT10 splice variants, which did not contain exons 3 and/or 4, were ubiquitously expressed and that the full-length OAT10 protein was mainly expressed in the kidneys and to a much lesser extent in brain, heart, small intestine, and colon (Bahn et al., 2008). Western blot analyses further localized the OAT10 protein to the brush-border membrane in the kidneys and not the basolateral membranes, suggesting that it is localized to the apical membrane of tubule cells (Bahn et al., 2008).
OAT10 transport of urate was found to be cis-inhibited and trans-stimulated by nicotinate. In addition, similar to Oat5, hOAT10 uses the 4-carbon dicarboxylate, succinate as a driving force to transport urate into the cell (Bahn et al., 2008). This counteranion specificity further confirms that, unlike the classic OATs that carry out basolateral transport of anions, OAT10 mainly functions at the apical membrane (Fig. 1). Furthermore, OAT10-mediated transport of PAH was found to be greatly increased by a low extracellular pH, suggesting that another mode of transport by this protein might be an exchange against hydroxyl ions (Bahn et al., 2008). The physiological substrate of OAT10 is believed to be nicotinate because of its high-affinity transport (Km, 22 μM). In fact, OAT10 may play a physiologically significant role in the uptake of nicotinate from the diet in the intestine and reabsorption from the proximal tubules in the kidneys to avoid loss in the urine (Bahn et al., 2008).
In addition to the above-mentioned endogenous substrates, extracellular lactate has been found to cis-inhibit OAT10-mediated urate uptake, whereas intracellular lactate has been shown to trans-stimulate this uptake, suggesting a further role of OAT10 in urate-lactate exchange; however, the affinity of OAT10 for lactate has not yet been established. Furthermore, GSH has been shown to trans-stimulate urate uptake (Bahn et al., 2008). Previous studies in mice have proposed the involvement of the organic anion transporting polypeptide 1 (Slco1a1) and multi-drug resistance-related proteins 2 and 4 (Abcc2 and Abcc4), in the luminal secretion of GSH. These findings further suggest that OAT10 may be yet another transporter involved in the luminal GSH cycle in the kidney (Frey et al., 2007).
Among the clinically relevant compounds that have been tested (diuretics, probenecid, and immunosuppressive agents), all reduced nicotinate uptake substantially, indicating a potentially important role in the reduction of nicotinate and urate in vivo (Bahn et al., 2008). These compounds included diuretics (i.e., furosemide and hydrochlorothiazide) that were shown to inhibit OAT10. However, it cannot be verified whether hydrochlorothiazide is, in fact, an OAT10 substrate, because it was not capable of trans-stimulating urate uptake as was shown for the other substrates, including the suggested physiological substrate, nicotinate. In addition, the immunosuppressive agent cyclosporine A was shown to cis-inhibit OAT10-mediated nicotinate uptake and trans-stimulate urate uptake (Bahn et al., 2008). This observation may provide a possible molecular mechanism underlying the development of cyclosporine A-induced hyperuricemia, because secretion of cyclosporine A leads to an increase in urate reabsorption.
H. Urate Transporter 1 (SLC22A12)
URAT1 was originally detected in mice and cloned as renal specific transporter (Rst) due to its predominant renal expression (Mori et al., 1997). It was found to consist of 553 amino acids that were arranged in 12 transmembrane helices. However, no specific cellular function was attributed to this protein (Mori et al., 1997). In 2002, the human ortholog of Rst was identified by screening the human genome database for genes related to OAT genes and in silico cloning (Enomoto et al., 2002a). Initial functional analyses on this ortholog indicated that it is involved in urate reabsorption, because urate exists primarily as a weak acid at physiological pH. This finding classified Rst as an OAT, and it was then renamed URAT1/Urat1 (Enomoto et al., 2002a). The hURAT1 (SLC22A12) gene is located on chromosome 11q13.1 and is paired with the gene encoding OAT4, showing a 42% sequence identity to this protein (Fig. 2) (Enomoto et al., 2002a; Eraly et al., 2003b).
URAT1/Urat1 has been found to be mainly localized to the kidneys (Mori et al., 1997). Real-time polymerase chain reaction and Western blot analyses have further located hURAT1 in vascular smooth muscle cells and to some extent in testis (Nishimura and Naito, 2005; Price et al., 2006). mURAT1 protein and mRNA were additionally detected in blood-brain barrier and choroid plexus (Imaoka et al., 2004). However, it remains unclear whether this protein is expressed to a notable extent in these nonrenal tissues. Further immunolocalization studies in the kidney placed this protein in the apical membrane of the epithelium of proximal tubules (Hosoyamada et al., 2004). This apical localization is believed to be maintained through the interaction of URAT1 with putative scaffolding protein PDZK1. The C-terminal domain of URAT1 has been shown to contain a PDZ domain, which, as previously mentioned, is known to participate in protein-protein interactions (Hung and Sheng, 2002). Interaction of URAT1 with PDZ domains of PDZK1 is believed to regulate the function of URAT1 by increasing transport activity and Vmax of transport without affecting this protein's affinity for urate (Anzai et al., 2004). Furthermore, this interaction is believed to be further modulated through other regulatory factors that are colocalized with PDZK1, including protein kinase A-anchoring protein and NHERF. However, the exact mechanism of PDZK1's modulation of URAT1 transport function has not yet been elucidated (Gisler et al., 2003; Anzai et al., 2004).
As the name suggests, the prototypical substrate for URAT1 is urate, the final product of purine metabolism in humans and higher primates (Sica and Schoolwerth, 2000). Other endogenous compounds transported by this protein include bromide, iodide, lactate, nicotinate, orotate, and DHEAS (Enomoto et al., 2002a; Hosoyamada et al., 2004). Similar to OAT4, URAT1 acts as an anion exchanger. It is believed to take up urate from the kidney filtrate and transport it into the cell in exchange for lactate, which is released into the filtrate (Enomoto et al., 2002a). Lactate is then transported back into the cell through a sodium-coupled lactate transporter (SMCT1, SLC5A8), which implies that urate is absorbed through a tertiary active transport pathway, URAT1 acting as the tertiary system that is driven by the lactate gradient (Fig. 1) (Rizwan and Burckhardt, 2007). Estrone sulfate does not interact with human or mouse URAT1 (Enomoto et al., 2002a; Hosoyamada et al., 2004). As well, interaction with α-ketoglutarate and succinate were demonstrated only at very high concentrations (Enomoto et al., 2002a).
Rst KO mice have been established by Eraly et al. (2008); Hosoyamada et al. (2010) in an attempt to study the extent of contribution of this transporter to renal excretion of urate. These mice appeared physiologically normal but displayed increased concentration ratio of urinary urate-creatinine, suggesting the involvement of this transporter in renal reabsorption of urate. Yet, the urate reabsorption ability of KO mice was not completely diminished and reabsorption was largely preserved. These observations indicate the presence of an overlapping or independent mechanism that might contribute to the reabsorptive transport of urate. Because humans with genetic defects in URAT1 seem to display significantly impaired urate reabsorption, manifested as hypouricemia, these observations may depend partly on species and ethnic differences (Eraly et al., 2008). Many xenobiotics have yet to be tested for transport with URAT1, but a summary of some classes that have been tested and their kinetic data are given in Table 2 and briefly discussed below.
1. Antihypertensives.
ARBs, specifically, losartan, pratosartan, and telmisartan interacted with hURAT1 with very high affinity (Iwanaga et al., 2007). In addition, other ARBs, including candesartan, olmesartan, losartan, and pratosartan, trans-stimulated the uptake of urate, indicating that ARBs may serve as counteranions for urate uptake (Iwanaga et al., 2007).
2. Diuretics.
Furosemide and, to some extent, torasemide inhibited hURAT1; however, other diuretics did not show a large degree of interaction with this transporter (Enomoto et al., 2002a; Hagos et al., 2007a).
3. Antibiotics.
Benzylpenicillin is the only antibiotic that has been tested for transport by URAT1 and has shown an inhibition in Rst-expressing HEK 293 cells (Hosoyamada et al., 2004; Imaoka et al., 2004).
4. Nonsteroidal Anti-Inflammatory Drugs.
Salicylate, phenylbutazone, sulfinpyrazone, and indomethacin have been shown to interact with hURAT1 (Enomoto et al., 2002a).
In brief, the information provided in this section is summarized in Table 1 and Fig. 1, which elucidate, among other aspects, the broad tissue distribution of OATs, especially the occurrence of the same OAT isoforms in different tissues.
A recent theory suggests that this broad tissue distribution may allow for members of the OAT family to participate in “remote sensing” between different epithelial tissue compartments and environments. This theory, known as the “remote sensing and signaling hypothesis,” suggests that signaling molecules and metabolites excreted through one OAT isoform in one tissue may be taken up by another OAT isoform in a different tissue, allowing a broader mode of substrate transport between different organ systems (Ahn and Nigam, 2009). This hypothesis is still under investigation. If true, it may aid in drug design to facilitate drug administration, such that simply by altering the primary route of administration, a drug might bypass the blood-brain barrier or first-pass effect and reach its intended target directly. An example of this is a newly identified olfactory-specific OAT, known as Oat6 (thus far identified only in the mouse), that shares 40 to 60% sequence homology with mOat1 and mOat3 (Monte et al., 2004). mOat6 has shown a high affinity for the transport of small volatile compounds that have been found to accumulate in the plasma of Oat1 KO mice (Ahn and Nigam, 2009). Hence, such substrates that are normally eliminated into the urine through an Oat1-mediated pathway may also be transported through an Oat6-mediated pathway in the olfactory mucosa. Thus, an intranasal mode of drug administration may allow scientists to avoid routes of transport that may influence drug pharmacokinetics and instead allow rapid absorption and increased bioavailability.
III. Regulation Mechanisms of Organic Anion Transporters
A. Sex Hormone Differences
Dating back to Hippocrates, discrepancies between the male and female members of a species in disposition and toxicity of drugs and endogenous compounds have been described. In describing symptoms of gout, a condition believed to arise from deficiencies in probenecid-sensitive urate reabsorption in the kidney, Hippocrates wrote, “A woman does not take the gout unless her menses has stopped,” suggesting a sex-dependent increased susceptibility of men to development of gout (Enomoto and Endou, 2005). Today, the availability of molecular probes for individual OATs has provided considerable evidence for the existence of remarkable gender differences in the mRNA and/or protein expression of Oat1, Oat2, Oat3 and Urat1. Two major regulatory mechanisms [i.e., regulation through growth hormone (GH) and regulation through sex steroids] are currently being investigated in an effort to elucidate the underlying mechanisms for the observed sex differences. GH exhibits distinct secretion patterns in men compared with women, such that it may influence sex differences in expression of specific OATs (Terry et al., 1977). In animal models, hypophysectomy or surgical removal of the pituitary gland followed by supplementation with GH is a common approach used to evaluate the role of GH secretion patterns in the regulation of renal and hepatic transporters (Cheng et al., 2007). Furthermore, gonadectomy is another common method used for evaluating the role of sex steroids in the regulation of transporter expression. Gonadectomy involves the surgical removal of the testes or ovaries, thus resulting in a reduction in circulating levels of sex hormones (Cheng et al., 2007). Evidence from such studies suggests that various members of the OAT family are regulated by sex hormones and GH, but display substantial isoform and species differences that are summarized below and in Table 3.
Sex differences in transporter expression in mouse and rat tissues
Reported sex differences in mRNA expression in selected OAT family members and the effect of hormonal regulation is summarized. Hormonal regulation was investigated through gonadectomy (GNX), hypophysectomy (HPX) and gonadectomy followed by hormone-replacement therapy (estrogen/dihydrotestosterone, DHT) in male and female animals.
1. Organic Anion Transporter 1.
Sex differences in renal expression of Oat1 mRNA have been observed in adult rats and mice. Oat1 expression levels gradually increased to an equal extent after birth in both male and female rats until approaching adult levels at approximately day 30 (Buist et al., 2002). At this stage, between days 40 and 45, the mRNA levels fell slightly in female rats, and greater expression was observed in male rats. Furthermore, protein studies using immunohistochemistry and Western blotting also indicated lower levels of Oat1 protein levels in female rats compared with male rats, characterized by weaker staining of Oat1 in the basolateral membranes of proximal tubules in kidneys of adult female rats compared with male rats (Ljubojevic et al., 2004). Similar sex differences have been reported in mice (Buist and Klaassen, 2004). Gonadectomy in male rats resulted in a disappearance of the observed sex differences, indicating that Oat1 expression is under positive control of testosterone (Buist et al., 2002). Such differences have not been observed in rabbits, indicating a species-dependent role of sex hormones (Groves et al., 2006). Hypophysectomy had no effect on Oat1 levels, an indication that GH does not play a significant role on the sex differences in renal Oat1 expression (Buist et al., 2003).
2. Organic Anion Transporter 2.
Although no clear-cut differences in Oat2 mRNA expression could be observed in mice or rabbits (Buist and Klaassen, 2004; Groves et al., 2006), clear sex differences have been reported in the abundance of Oat2 mRNA in rats. After puberty (postnatal day 30), renal mRNA expression has been found to sharply increase in female rats, whereas in adult male rats, greater mRNA abundance was reported in the liver compared with the kidneys (Buist et al., 2002; Kobayashi et al., 2002; Ljubojević et al., 2007). These sex differences have also been observed at the protein level, such that more Oat2 protein was found in the brush-border membrane of S3 cells in female rats compared with male rats (Ljubojević et al., 2007). Gonadectomy and hypophysectomy dramatically decreased renal expression of Oat2 in female rats without any effect on mRNA levels in male rats, suggesting that both androgens and female GH secretion patterns play a role in regulation of sex differences in renal Oat2 expression (Buist et al., 2003; Ljubojević et al., 2007). Furthermore, although no obvious sex difference has been observed in the mRNA levels of Oat2 in mice, immunohistochemical studies suggest sex differences in protein distribution similar to those observed in rats (Buist and Klaassen, 2004; Ljubojević et al., 2007).
3. Organic Anion Transporter 3.
Significant sex differences have not been reported in the mRNA expression of this transporter in rat or rabbit renal tissues (Buist et al., 2002; Groves et al., 2006). Expression increases steadily in animals of both sexes until puberty, at which point it plateaus with no differences observed among male and female rats and rabbits. However, in contrast to mRNA expression in kidneys, a clear sex difference has been observed for Oat3 mRNA expression in the liver, where male rats display predominant hepatic expression of Oat3 mRNA (Buist et al., 2003). Gonadectomy reduced Oat3 mRNA levels in livers of male rats with no effect on livers of female rats, whereas hypophysectomy decreased Oat3 levels in male rats but led to an increase in female rats (Buist et al., 2003; Ljubojevic et al., 2004). These findings suggest that both androgens and female GH secretion patterns play a significant role in the sex differences in hepatic Oat3 expression. In mice, Oat3 mRNA expression levels showed sex differences, but not in all strains tested. Higher levels of kidney mRNA expression were observed in female 129J mice compared with male mice. Furthermore, similar male-predominant expression in the liver was also observed in mice, but to a lesser extent than that observed in rats (Buist and Klaassen, 2004).
4. Organic Anion Transporter 5.
Sex hormone-related differences in expression patterns of Oat5 in both rats and mice closely resemble those described for rOat2. In adult rats, rOat5 mRNA and protein were primarily detected in the outer stripe and medullary rays in the kidney, displaying female-predominant expression (Breljak et al., 2010a). Castration of adult male rats led to a strong up-regulation of the rOat5 expression, whereas ovariectomy did not display a significant effect in adult female rats. Likewise, testosterone treatment of castrated adult rats led to a strong down-regulation of expression, whereas treatment with estrogen and progesterone displayed only weak up-regulation (Breljak et al., 2010a). These observations suggest that androgen hormones strongly inhibit the expression of rOat5, whereas estrogen and progesterone display only weak stimulatory effects. Thus in adult male rats, androgen acts to keep the rOat5 expression levels low, whereas in adult female rats, factors additional to sex hormones, such as GH for example, may be responsible for the stimulation of rOat5 expression. In adult mice, immunohistochemical methods were used to characterize the expression of Oat5 and point to analogous sex-dependent expression patterns that may be regulated through the same mechanism (Breljak et al., 2010a).
5. Urate Transporter 1.
Clear sex differences have been reported in Urat1 mRNA and protein expression levels in mice. Male mice display 2.3-fold expression of this urate transporter in the kidney compared with female mice, indicating a sex-dependent regulation (Hosoyamada et al., 2004). Supporting this idea, an androgen-responsive element has been discovered in the promoter region of this gene, although the details of the underlying mechanism for this regulation have yet to be elucidated (Hosoyamada et al., 2004). In addition, it has been previously suggested that similar sex differences exist in humans, because as mentioned at the beginning of this section, men also display higher blood urate levels compared with women (Hediger et al., 2005).
Evidently, studies of sex differences in OAT expression have only involved animal models thus far. However, information from these findings may allow scientists to make some predictions regarding the clinical implications of sex-related transporter regulation in drug efficacy, toxicity, and ultimately personalized medicine.
B. Transcriptional and Post-Translational Regulation
Further interindividual variations in drug transport may be explained by up- or down-regulation of OAT expression as a consequence of transcriptional regulation or post-translational modifications, possibly as a result of prior exposure to drugs and xenobiotics. In 2003, through the use of comparative genomic approaches, binding sites for various transcription factors were identified in the noncoding regions of hOAT1 and hOAT3 (Eraly et al., 2003b). As a result, most studies investigating the regulational mechanisms affecting OATs have focused primarily on these two transporters. Furthermore, putative mechanisms for the functional regulation of these transporters and various other OAT family members have been proposed. These mechanisms can be divided into two broad categories based on the mode of regulation: long-term regulation, involving nuclear transcription factors and epigenetic mechanisms, and short-term regulation, involving various signaling pathways (Srimaroeng et al., 2008). Among these, the best-studied mechanisms implicated in the regulation of most OATs are post-translational mechanisms, especially N-glycosylation and phosphorylation, as they are both shared structural features of all members of the OAT family identified to date. In this chapter, these findings, in addition to other proposed modes of regulation will be discussed in the context of Oat1, Oat2, Oat3, Oat4, Oat7, and Urat1.
As mentioned above, OATs share a common 12-transmembrane domain structure, consisting of several potential N-glycosylation sites within the extracellular loop and various potential phosphorylation sites in the intracellular loop (Robertson and Rankin, 2006; Zhou and You, 2007). Both these structural features have been suspected to play a critical role in membrane trafficking of OAT family members, in addition to regulation of their transport function. In 1998, regulation of Oat1 by phosphorylation was first demonstrated in X. laevis oocytes, where the activation of PKC led to an inhibition of Oat1-mediated PAH transport (Uwai et al., 1998). This study was then followed by several others also demonstrating inhibition of PAH transport by PKC in various heterologous expression systems, including one in which additional inhibition of basolateral fluorescein uptake was also observed upon activation of PKC (Gekle et al., 1999; Lu et al., 1999; You et al., 2000; Wolff et al., 2003). Because both Oat1 and Oat3 transport fluorescein with high affinity, this finding suggested that phosphorylation has a similar effect on Oat3 as on Oat1, although much less is known about the effect of PKC activity on Oat3 function. Recent studies have demonstrated the significant role of different PKC isoforms on the regulation of OATs, such that different PKC isoforms may have different effects on transporter activity (Zhang et al., 2008; Barros et al., 2009; Li et al., 2009). One such example was demonstrated in a study carried out by Barros et al. (2009), where they showed that PKCς activation up-regulates OAT1 and OAT3 transport function. Given that these are quite recent observations, a complete understanding of the role of all PKC isoforms in the regulation of OAT function is still lacking. Furthermore, the molecular basis behind the regulational role of these proteins is also not yet fully understood. However, various studies have attempted to determine the underlying mechanism of PKC-mediated OAT regulation, including one published by Zhang et al. (2008) that demonstrated 1) that OAT1 intrinsically alternates between the plasma membrane and endosomes that carry out its internalization and 2) that PKC activation modifies this process by increasing internalization but not affecting its recycling back to the plasma membrane, hence resulting in a down-regulation of OAT1 activity. These findings explain the previously observed down-regulation of OAT1 and potentially OAT3, but still fail to explain the variable effects of different isoforms. Hence, many more studies are still necessary before a full understanding of the effect of PKC on OATs and its mechanism of action is achieved. Likewise, the molecular basis behind the role of N-glycosylation on OAT activity is still not very well understood, but it has been demonstrated that this enzymatic process plays a significant role in the regulation of protein folding and membrane sorting of mOat1, hOAT1, and hOAT4 proteins (Kuze et al., 1999; Tanaka et al., 2004; Zhou et al., 2005).
Another factor, involved in the post-translational regulation of OAT1 and -3 is epidermal growth factor, which, unlike PKC, has been shown to stimulate Oat1-mediated basolateral PAH uptake in rabbit tubules and Oat3-mediated ES uptake in their renal proximal tubules (Dantzler and Wright, 2003). These observations were further linked to a complex signaling cascade that involves activation of mitogen-activated kinases, phospholipase A2-induced release of arachidonic acid, conversion of arachidonic acid into prostaglandin E2 via cyclooxygenase 1, followed by prostaglandin E2 activation of adenylate cyclase, which subsequently results in cAMP production and activation of protein kinase A (Sauvant et al., 2002, 2003, 2006). Therefore, epidermal growth factor was later identified as an indirect activator of protein kinase A, which is now generally implicated in the stimulation of OAT1 and OAT3. Given these findings, it is of great interest to study the potential effect that drugs, such as NSAIDs, may have on this complex signaling mechanism and how they might eventually affect OAT1, OAT3, and other OAT family members' expression and function.
Moreover, studies by Eraly et al. (2003b), which were mentioned at the beginning of this chapter, investigated binding sites for various transcription factors in the promoter regions of hOAT1 and hOAT3 genes (Table 4). Among these transcription factors were PAX1, PBX, WT1, and HNF1, indicating the involvement of one or more of these in the regulation of hOAT1 and hOAT3. After this discovery, studies focused on the functional analysis of transcription factors, initially mainly with respect to OAT1 and -3 and later with respect to other OAT family members. In this regard, OAT1 was found to be positively regulated by HNFs or hepatocyte nuclear factors, because HNF1α deletion in mice resulted in a significant reduction of renal mOat1 mRNA levels and transfection of HEK 293 parental cells with HNF1α or HNF1α and HNF1 resulted in the expression of hOAT1 (Maher et al., 2006; Saji et al., 2008). Furthermore, HNF4α was shown to activate hOAT1 when it was cotransfected with hOAT1 promoter into opossum kidney cells (Ogasawara et al., 2007). Likewise, OAT2 expression in the kidney and liver is also under the influence of HNF1 and -4. mOat2 expression in the kidney and liver was markedly reduced in HNF1α knockout mice (Maher et al., 2006). In addition, HNF4α was shown to activate the hOAT2 promoter when expressed in a human hepatoma cell line (Popowski et al., 2005). Of note is the observation that the interaction of farnesoid X receptor with a number of its ligands, including chenodeoxycholic acid, leads to a down-regulation of HNF4α and subsequently OAT2 expression (Popowski et al., 2005). This observation suggests that in disease conditions involving elevated intracellular levels of bile acids, hOAT2 expression and function may be noticeably reduced. HNF1α and -β homodimers, as well as HNF1α/β heterodimer, have also been shown to induce the expression of OAT3 when expressed in HEK 293 cells, although the heterodimer expression displayed less of a stimulatory effect (Kikuchi et al., 2006). Transcriptional regulation of URAT1 expression is very similar to those described for OAT3, in that HNF1α homodimer, HNF1β homodimer, and HNF1α/β heterodimer have all been found to increase the expression of the apically expressed URAT1 in liver hepatocellular carcinoma (HepG2) cells. However, HNF1β homodimer seems to exert a smaller effect than either the HNF1α homodimer or heterodimer (Kikuchi et al., 2007). Finally, hOAT7 and hOAT5 also seem to be under the regulation of HNF1α; Klein et al. (2010) identified two functional binding elements in hOAT5 proximal promoter and one functional binding element in hOAT7 promoter that are responsible for HNF1α-mediated regulation of promoter activities of these two proteins. Down-regulation of HNF1α in human Huh7 cells derived from liver led to a reduction in the endogenous expression of these two transporters, whereas hOAT5 and hOAT7 promoter regions were transactivated in the presence of HNF1α in HepG2 cells. In addition, mutations in both binding elements of hOAT5 led to a complete loss of HNF1α-mediated transactivation, whereas disruption of the single binding element of hOAT7 was sufficient to result in a loss of HNF1α-mediated transactivation. Furthermore, although a direct role of HNF4α in the regulation of these two transporters was not demonstrated, this group also observed a modest, yet significant, decline in hOAT5 and hOAT7 mRNA levels in small interfering RNA-mediated HNF4α knockdown liver-derived cells (Klein et al., 2010). These data are summarized in Table 4.
Transporter regulation by HNF1α, HNF1β, and HNF4α transcription factors
Transcription factors used for mRNA expression in mouse models (in vivo) and human cell models (in vitro). In all cases, mRNA levels increased.
As briefly mentioned in the second chapter, an additional regulatory mechanism has been proposed to control the expression and function of URAT1. The C-terminal region of URAT1 contains a binding site for PDZ domain-containing proteins (Hung and Sheng, 2002). Anzai et al. (2004) studied this C terminus-associated protein interaction, which would act to alter the transport function of URAT1, and found the multivalent PDZ-domain containing protein PDZK1. Using a yeast-two hybrid assay, this group demonstrated an interaction of PDZK1 with URAT1 through its PDZ motif and a colocalization of the two proteins to the apical membrane of renal proximal tubules. Furthermore, coexpression of PDZK1 in hURAT1-expressing cells led to an increase in hURAT1 activity and surface expression, whereas deletion of the C-terminal region of URAT1 eliminated this effect (Anzai et al., 2004). These observations suggest that PDZK1 may be a scaffolding protein that plays a significant role as the physiological regulator of URAT1. Likewise, hOAT4 is possibly regulated in a similar manner as it interacts through its C-terminal region with PDZK1 and another multivalent PDZ protein [i.e., Na+/H+ exchanger regulator factor (NHERF-1) (Miyazaki et al., 2005]. Coexpression of either of these proteins in hOAT4-expressing cell lines also led to an increase in hOAT4 function and surface expression (Miyazaki et al., 2005). However, this observation seems to be tissue-specific, because hOAT4 function and surface expression were increased in kidney cell lines, but coexpression of either multivalent protein did not show an increase in hOAT4 transport activity or expression in placental cell lines (Zhou et al., 2008). These findings indicate that protein-protein interactions play a significant role in regulating OAT function; although similar to other aspects of OAT regulation, the precise mechanism is not completely understood and remains to be fully elucidated.
More recently, epigenetics have been more thoroughly investigated as another potential mechanism of OAT regulation. Kikuchi et al. (2006) identified multiple CpG dinucleotides, believed to be targets for DNA methylation, in the putative hOAT3 promoter region. They followed up these findings by inducing DNA methylation in vitro and found a decrease in hOAT3 promoter activity, whereas, when methyltransferase inhibitors were used to inhibit DNA methylation, hOAT3 transcription was restored. Hence, efficient hOAT3 expression correlates with a hypomethylated promoter (Kikuchi et al., 2006). Similar observations have been made with respect to URAT1 expression. In both liver and kidney medulla, where mUrat1 is not expressed, the promoter region is hypermethylated, whereas, in the kidney cortex, where mUrat1 is mainly expressed, the promoter region is hypomethylated (Kikuchi et al., 2007). Hence, sustaining low levels of promoter methylation seems to be of great importance in the expression of various OAT members, particularly in determining the tissue-specific expression of URAT1. In addition, this is of great clinical interest, especially in the use of cancer treatment with DNA-hypomethylating agents (such as decitabine and azacitidine) and DNA-methyltransferase inhibitors, which may produce nonspecific hypomethylation of genes.
IV. Genetic Variants and Clinical Impacts
Organic anion transporters interact with a myriad of toxins and foreign chemicals at an organism's interface; hence, it seems plausible that selective pressure would favor the existence of several different OAT variants in different human populations. After the cloning and characterization of the OAT family members, various SNPs began to be identified in many different populations, including whites, sub-Saharan Africans, Mexican Americans, and Japanese. Table 5 provides a summary of the nonsynonymous SNPs that have been found to occur in the coding regions of hOAT1, hOAT2, hOAT3, hOAT4, and URAT1 (Nishizato et al., 2003; Ichida et al., 2004; Fujita et al., 2005; Wakida et al., 2005; Xu et al., 2005; Bleasby et al., 2006; Erdman et al., 2006; Graessler et al., 2006; Urban et al., 2006). Owing to the critical role of OATs in drug disposition and elimination, the occurrence of SNPs in the coding region of OAT genes may result in interindividual variability in the bioavailability and excretion of many clinically important drugs. Clinical consequences for several of the reported SNPs have been described and will be summarized in this chapter.
Population variability of coding region polymorphisms in human organic anion transporters
The minor allele frequencies of reported nonsynonymous polymorphisms in selected ethnic groups are summarized. Population sizes are provided in parentheses after the allele frequencies.
Within the coding region of hOAT1 (SLC22A6), eight nonsynonymous SNPs were identified. Six of these SNPs were initially reported by Fujita et al. (2005) and resulted in amino acid changes R50H, P104L, I226T, A256V, R293W, and R454Q. Xu et al. (2005) and Bleasby et al. (2005) each identified an additional SNP (i.e., L7P and K525I, respectively), that resulted in amino acid changes. R50H-hOAT1 mutation, which has been reported in subjects of Mexican-American and African descent, is associated with significantly higher transporter affinity for nucleoside phosphonate analogs (e.g. adenovir, cidofovir, and tenofovir), which suggests a potential for adverse drug reactions in response to antiviral drugs in these patients (Fujita et al., 2005; Xu et al., 2005). Furthermore, Cheong et al. (2011), using linkage disequilibrium analyses, identified a haplotype block containing this hOAT1 SNP in an Asian study cohort including Korean, Han Chinese, and Japanese populations, but not in African American or European American cohorts. K525I-hOAT1 mutation was also investigated for altered affinity toward these nucleoside phosphonate analogs, but this mutant seemed to have transport properties similar to those observed in wild type (Bleasby et al., 2005). Moreover, none of the reported SNPs conferred any changes in the transport of PAH, ochratoxin A, or MTX (Fujita et al., 2005). In addition to nonsynonymous SNPs, Bhatnagar et al. (2006) have identified an additional SNP in the conserved 5′ regulatory region of SLC22A6 in an individual of Pacific Islander descent. This SNP is located downstream from the consensus Wilm's tumor gene recognition site, which is believed to be an important gene in the development of a functional kidney.
In the coding region of hOAT2 (SLC22A7), three nonsynonymous SNPs have been identified that result in amino acid changes: T110I, V192I, and G507D. However, the clinical consequences of these SNPs have not yet been investigated (Xu et al., 2005). Moreover, Shin et al. (2010), investigating the association between genetic polymorphisms in the SLC22A7 gene and hepatic hOAT2 protein expression, found that polymorphisms in the coding region of this gene do not contribute significantly to the interindividual variations observed in protein expression or transport activity.
Studies by Erdman et al. (2006) and Urban et al. (2006) identified 10 nonsynonymous SNPs in the coding region of the SLC22A8 gene, resulting in amino acid changes F129L, R149S, Q239Stop, I260R, R277W, V281A, I305F, A310V, A399S, and V448I. Nishizato et al. (2003) and Xu et al. (2005) each identified one additional nonsynonymous SNP, which resulted in the amino acid changes A389V and I175V. F129L, V281A, A310V, A399S, and V448I did not seem to result in a change of transport function of the organic anion substrates that were tested (Nishizato et al., 2003; Erdman et al., 2006). In addition, A389V was identified only in a Japanese population but did not seem to have an effect on the transport of the substrates tested (e.g., pravastatin) (Nishizato et al., 2003). On the other hand, R277W-hOAT3 and I305F-hOAT3 mutants resulted in a slower transport of ES and a slight increase in preference for the transport of cimetidine compared with wild type. R277W is found among African Americans, whereas I305F is found in Mexican Americans and Asian Americans (Erdman et al., 2006; Urban et al., 2006). Furthermore, R149S-hOAT3, Q239Stop-hOAT3, and I260R-hOAT3 mutations resulted in a loss of transport function with respect to the transport of the prototypical OAT3 substrate estrone sulfate. R149S-hOAT3 was found among Europeans and Asian Americans, whereas Q239Stop-hOAT3 and I260R-hOAT3 SNPs were found only among Asian Americans (Erdman et al., 2006; Urban et al., 2006). Hence, three SNPs that were identified in Asian Americans resulted in a loss-of-function mutation that could have a potential clinical impact with respect to the transport and excretion of OAT3-specific substrates, discussed in section II. Bhatnagar et al. (2006) further identified seven SNPs in the conserved 5′ regulatory regions of the hOAT3 gene, two of which were found in all the ethnic groups that were tested and were located downstream of the consensus steroid hormone recognition element.
Within the coding region of hOAT4 (SLC22A11), eight nonsynonymous SNPs have been identified by Xu et al. (2005), which translated into amino acid changes of V13M, R48Stop, T62R, V155M, A244V, E278K, V339M, and T392I. These SNPs were reported in populations of Japanese, white, African, and Indo-Pakistani origins, individuals from African origin being affected by more than one reported SNP. However, the functional impact of these mutations has not yet been investigated, and the clinical impact of these SNPs remains largely unknown. However, one of the few in vivo studies investigating the functional significance of OAT polymorphisms found a significant association (P = 0.002) between renal torasemide clearance and an SLC22A11 SNP (rs11231809) located 20 kilobases upstream of the SLC22A11 transcriptional start site. This finding suggests that only the genetic variations in the hOAT4 gene, and not those in hOAT1 or hOAT3, may affect the fraction of torasemide that is excreted (Vormfelde et al., 2006). More recently, genome-wide association (GWA) studies have further identified novel loci associated with uric acid concentrations, one of which lies within the SLC22A11 gene and corresponds to a novel SNP (rs17300741) (Kolz et al., 2009). Another study replicated this finding and in investigating the mechanism behind the observed association between the novel loci and serum uric acid levels also found a strong correlation between SLC22A11 locus and waist circumference, a factor closely related to serum uric acid levels (van der Harst et al., 2010).
The first nonsynonymous mutation in the coding region of hURAT1 (SLC22A12) was identified by Enomoto et al. (2002a) and was found to result in W258Stop. This SNP results in a loss-of-function mutation and was linked strongly with familial idiopathic hypouricemia, which is a disease commonly detected in Japanese and Korean populations, associated with below normal levels of plasma urate. Approximately 74% of patients diagnosed with this disease were found to carry this mutation (Enomoto et al., 2002a; Ichida et al., 2004). Since then, additional SNPs in the coding region of this gene have been further identified in Japanese patient populations, which result in amino acid changes of R90H, R92C, V138M, G164S, T217M, Q297Stop, E298D, Q382L, M430T, and L418R (Enomoto et al., 2002a; Ichida et al., 2004; Wakida et al., 2005; Xu et al., 2005; Graessler et al., 2006; Komatsuda et al., 2006). The strong correlation between these mutations and disease status sheds more light on the physiological role of URAT1 and can be used as a predictive clinical tool for identifying predisposed individuals. Furthermore, similar to SLC22A11, one of the novel loci associated with serum uric acid levels, which was identified through GWA studies also lies within the SLC22A12 gene and is represented by SNP rs505802 (Kolz et al., 2009). Another GWA study of serum urate levels in African American populations, carried out by Tin et al., identified a novel rare nonsynonymous SNP rs12800450, which resulted in the amino acid change G65W (Table 5). Functional studies further showed reduced urate transport in these G65W-hURAT1 mutants in vitro (Tin et al., 2011). Furthermore, GWA studies identified an additional locus that is associated with uric acid plasma levels in humans, known as SLC2A9 or GLUT9 (Li et al., 2007). In addition to its association with uric acid levels, mutations in the SLC2A9 gene have been significantly associated with gout and monogenic forms of hypouricemia (Döring et al., 2008; Matsuo et al., 2008; Dinour et al., 2010).
V. Conclusions
In the past decade, much has been elucidated with respect to the role of OATs in renal and extrarenal organic anion secretion and the effect of SNPs on their function. Substrate studies have revealed the promiscuity of OATs, as they are capable of transporting a wide range of compounds, including a large number of clinically relevant xenobiotics and endogenous chemicals. It is now clear that OATs are not exclusively renal transporters and that, in fact, several members of this family are expressed only in extrarenal tissues. Furthermore, many regulatory processes, both short and long term, have been identified that may potentially have very detrimental effects on the expression and function of OATs. Yet, the mechanisms underlying the regulation of these transporters are still not largely understood, and further investigations in this field are required for a deeper understanding of the regulation of OATs, which undoubtedly would be integral in allowing us to link these findings to pathophysiological conditions. Moreover, various SNPs have been identified in coding and noncoding regions of OATs, some of which have been linked to potential functional changes and pathologic conditions. However, most studies are still in their preliminary stages, focusing only on a limited number of clinically relevant compounds. Given the diversity and the pivotal role of OATs in the secretion of a wide variety of clinically relevant compounds, it is now of great value to investigate OAT variability with respect to a broad selection of pharmaceuticals. The importance of studying the mechanisms of regulation and pharmacogenetics of OATs lies in the vulnerable position of OATs as they interact with a wide variety of toxins, making them primary targets for toxicity. As an example, several nephrotoxins have been identified as substrates for OATs, including ochratoxin A, tetracycline, mercuric conjugates, and the antiviral drugs adefovir and cidofovir. Hence, mutations in OAT sequences may, for example, result in an accumulation of toxins at the proximal tubules and result in further extrarenal toxicity because of substrate competition for OAT transport. Extrarenal toxicity may also result from delayed clearance of substrates as a result of mutations that lead to decreased or loss of OAT function. Further investigation of OAT SNPs and their functional consequences would allow a better prediction of predisposition to adverse reactions to these and other clinically relevant drugs.
Until recently, the focus of most studies have been on gaining a deeper understanding of the implication of OATs in drug disposition and excretion in humans, yet the true physiological roles of most OATs remain to be understood. Studies have shown evolutionary conservation of SLC22 transporters and the existence of endogenous substrates for most OATs. Nevertheless, it is still believed by some scientists that the physiological substrates that would be most vital for the discovery of the physiological role of these transporters have yet to be discovered. As discussed in this review, OATs are believed to be part of a larger metabolic network, involved in “remote sensing.” This hypothesis requires further investigation, but if established, it could offer avenues for the development of much more beneficial therapeutics with more targeted modes of action.
Much has been done in the field of OATs to increase our understanding of expression, function, regulation, and variability of these transporters; however, with the cloning of new members of this family in the past decade, much research remains to be done to be able to completely understand the OATs and their specific functions within the body and their role in disease states and interindividual response to therapeutic treatment.
Acknowledgments
This work was supported by the Wilhelm-Sander-Stiftung [Grant 2010.059.1]; the Bundesministerium für Bildung und Forschung [Grants 03 IS 2061C, 0315755]; the Deutsche Forschungsgemeinschaft [Grant SCHW 858/1-1]; the FP7 EU Initial Training Network program “Fighting-Drug-Failure” [Grant PITN-GA-2009-238132]; and the Robert Bosch Foundation. We thank Claudia Resch for providing help in finding some relevant references.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Emami Riedmaier, Nies, Schaeffeler, and Schwab.
Footnotes
This article is available online at http://pharmrev.aspetjournals.org.
↵1 Abbreviations:
- OAT
- organic anion transporter
- PKC
- protein kinase C
- PAH
- para-aminohippurate
- NSAID
- nonsteroidal anti-inflammatory drug
- URAT1
- urate transporter 1
- TM
- transmembrane
- DHEAS
- dehydroepiandrosterone sulfate
- ES
- estrone sulfate
- KO
- knockout
- HR2
- histamine receptor 2
- ACE
- angiotensin-converting enzyme
- ARB
- angiotensin II receptor blocker
- OCT
- organic cation transporter
- MTX
- methotrexate
- NLT
- novel liver-specific transporter
- HEK
- human embryonic kidney
- AZT
- zidovudine
- BBB
- blood-brain barrier
- PDZK1
- PDZ domain containing 1
- NHERF
- Na+/H+ exchanger regulatory factor
- NHE3
- Na+/H+ exchanger 3
- ORCTL3
- organic cation transporter 3
- Rst
- renal specific transporter
- GH
- growth hormone
- HNF1
- hepatocyte nuclear factor 1
- SNP
- single nucleotide polymorphism
- GWA
- genome-wide association
- h
- human
- r
- rat
- m
- mouse.
- © 2012 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue





{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. Localization, Substrate Specificity and Molecular Pharmacology of Organic Anion Transporter Family Members
- III. Regulation Mechanisms of Organic Anion Transporters
- IV. Genetic Variants and Clinical Impacts
- V. Conclusions
- Acknowledgments
- Authorship Contributions
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters