


Abstract
Idiosyncratic drug reactions are a significant cause of morbidity and mortality for patients; they also markedly increase the uncertainty of drug development. The major targets are skin, liver, and bone marrow. Clinical characteristics suggest that IDRs are immune mediated, and there is substantive evidence that most, but not all, IDRs are caused by chemically reactive species. However, rigorous mechanistic studies are very difficult to perform, especially in the absence of valid animal models. Models to explain how drugs or reactive metabolites interact with the MHC/T-cell receptor complex include the hapten and P-I models, and most recently it was found that abacavir can interact reversibly with MHC to alter the endogenous peptides that are presented to T cells. The discovery of HLA molecules as important risk factors for some IDRs has also significantly contributed to our understanding of these adverse reactions, but it is not yet clear what fraction of IDRs have a strong HLA dependence. In addition, with the exception of abacavir, most patients who have the HLA that confers a higher IDR risk with a specific drug will not have an IDR when treated with that drug. Interindividual differences in T-cell receptors and other factors also presumably play a role in determining which patients will have an IDR. The immune response represents a delicate balance, and immune tolerance may be the dominant response to a drug that can cause IDRs.
I. Introduction
The term idiosyncratic drug reaction (IDR) has been used in various ways and has no clear definition, but the term is used in this review to designate an adverse reaction that does not occur in most patients treated with a drug and does not involve the therapeutic effect of the drug. IDRs are not the most common type of adverse drug reaction (ADR), but they are unpredictable and often life threatening. The propensity of a drug to cause an idiosyncratic reaction is dependent on its chemical characteristics, but individual susceptibility is determined by patient-specific factors, in particular the expression of immunologic receptors that display drug-derived antigens on the cell surface. IDRs represent a major problem for drug development because, unless the incidence is very high, they are usually not detected during clinical trials, and there are many examples where serious IDRs have led to the withdrawal of a drug from the market. Their unpredictable nature also makes prospective mechanistic studies in humans virtually impossible, and there are few valid animal models. Therefore, although progress is being made in understanding the mechanism of such reactions, they are quite complex and our understanding is still superficial. In addition, there are probably many different mechanisms by which a drug can induce an IDR. The aim of this article is to summarize the different types of IDR and explore the ways in which drugs and drug-derived products interact with immunologic receptors to stimulate T cells.
II. Types and Clinical Picture
Although most IDRs appear to be immune mediated, in most cases, definitive evidence is lacking, and certainly the details of how a drug can induce an immune response are unknown (Uetrecht, 2007). The clinical characteristics of IDRs provide mechanistic clues, and any mechanistic hypothesis should be consistent with these characteristics.
IDRs can affect virtually any organ, but the skin, liver, and blood cells are the most common targets. Some drugs cause IDRs that are limited to one organ, whereas many others can affect several organs, in some cases simultaneously. Different drugs can cause a similar pattern of IDRs, and there are certain characteristics common to most IDRs, but each drug causes a somewhat different spectrum of IDRs.
One characteristic that is common to most IDRs is a delay between starting the drug and the onset of symptoms. There is also a more rapid onset if a patient who has had an IDR to a specific drug is rechallenged (Uetrecht, 2007). This characteristic suggests an immune mechanism; however, there are rare exceptions to the delay in onset such as the liver injury associated with telithromycin, which can occur within a day of starting therapy (Clay et al., 2006). There are more exceptions to the lack of rapid onset on rechallenge (Uetrecht, 2009b). The lack of a rapid onset with rechallenge has been taken to indicate that the IDR in question is not immune mediated; however, there are several IDRs that are clearly immune mediated but without a rapid onset on rechallenge (Uetrecht, 2007). Although the delay in onset is almost universal, the delay varies with the type of IDR: mild rashes usually occur with a delay of about 1 week; more serious rashes usually occur a bit longer; and with liver injury and IDRs involving bone marrow, the delay is typically 1–2 months. These are typical times to onset; however, the delay can be less, and with some drugs, the delay can often be significantly longer. In particular, drug-induced autoimmunity usually occurs late, often after more than 1 year of treatment (Uetrecht, 2009a,b). In a few cases, the onset of the IDR occurs 1 month or more after the drug has been discontinued (Sharp, 1963; Keisu and Andersson, 2010; Tesfa and Palmblad, 2011).
Another characteristic of IDRs is that the risk often does not appear to increase with dose (Uetrecht, 2007). This has led some people to characterize IDRs as dose independent; however, no biologic effect is dose independent. What is true is that most patients will not experience an IDR at any dose, and because by the usual definition, the mechanisms of IDRs do not involve the therapeutic effect of the drug, there is no reason that the dose-response curve for the therapeutic effect and that for the IDR should be in the same range. The maximal incidence for the IDR is often at a dose below the therapeutic range so that the risk does not increase within the therapeutic range; however, by chance the two dose-response curves sometimes overlap, and an increase in IDR risk is apparent within the therapeutic range (Cameron and Ramsay, 1984). There are presumably IDRs that occur only above the therapeutic range, but such IDRs would not be observed. The therapeutic dose of most drugs is on the order of 100 mg, and the average molecular mass of drugs is approximately 400; therefore, given Avogadro’s number, the therapeutic dose of most drugs is on the order of 1020 molecules, and a dose can always be found below which no one will have an IDR. This is the basis for desensitization protocols, and even they typically involve at least 1/10,000 of the therapeutic dose or ∼1016 molecules of the drug.
The term idiosyncratic means specific to an individual, and in general, it is impossible to predict who will develop an IDR to a specific drug. In some cases, there is a strong genetic component, and when this has been observed, it is always a human leukocyte antigen (HLA) gene, i.e., major histocompatibility complex (MHC), either MHC-I or MHC-II (Daly, 2012). In most cases in which there is a strong gene association, most patients with the required genotype will not have an IDR if given the drug; however, an exception is abacavir hypersensitivity reactions in which approximately 50% of HLA-B*57:01 patients who are treated with abacavir will develop an IDR (Mallal et al., 2008). The mechanism by which this occurs will be discussed later. Other genes such as for glutathione S-transferase or other metabolic enzymes can be associated with an increased incidence of idiosyncratic drug toxicity, but to date, the relative risks of such gene associations are small. Other risk factors for IDRs exist but are also weak. For example, the risk of some IDRs is higher in women, but this is not true for all IDRs. The risk increases with age for many IDRs, such as isoniazid-induced liver injury; however, the liver injury associated with valproic acid is higher in infants (Zimmerman, 1999). The presence of a specific type of infection increases the risk of some IDRs, such as the increased risk of an amoxicillin-induced rash in patients with mononucleosis (Pullen et al., 1967) or the risk of a sulfonamide-induced hypersensitivity reaction in patients with AIDS (Mitsuyasu et al., 1983), but most IDRs occur in patients without any obvious interaction with a viral infection.
Another important characteristic of the drugs that are associated with a relatively high incidence of IDRs is that they are often associated with several different types of IDRs as described in more detail below. Although there are commonalities, each drug is associated with its own spectrum of IDRs. The IDRs associated with a few drugs such as halothane are limited to the liver, presumably because they are metabolized to a reactive metabolite by P450s in the liver, little if any reactive metabolite escapes the liver, and little reactive metabolite is formed in other organs. In contrast, carbamazepine can cause a wide variety of IDRs, including liver injury, mild skin rash, toxic epidermal necrolysis, agranulocytosis, aplastic anemia, autoimmunity, etc. (Jain, 1991; Syn et al., 2005), whereas many drugs can cause two or three different types of IDRs as described as follows: procainamide can cause autoimmunity or agranulocytosis, nevirapine can cause skin rash or liver injury, and felbamate can cause aplastic anemia or liver injury, etc. Some drugs such as amodiaquine can cause liver injury and agranulocytosis simultaneously (Neftel et al., 1986). On the other hand, with few exceptions such as fixed drug eruptions, other agents such as viruses can also cause the same adverse events as IDRs, which often makes causality difficult to determine.
A. Skin Rash
Skin rashes are the most common type of IDR. One simple reason is probably that even a very mild skin rash is visible, whereas mild IDRs affecting other organs such as the liver probably occur without the patient being aware of them. It is also likely that the skin is a common target because it is immunologically very active.
1. Maculopapular Rash.
Maculopapular or morbilliform rashes are the most common type of drug-induced skin rash comprising more than 90% of drug-induced skin rashes (Hunziker et al., 1997). The time to onset is typically after 1–2 weeks of treatment (Valeyrie-Allanore et al., 2007). In the absence of other manifestations, these drug rashes are not serious, and the rash often resolves despite continued treatment with the drug. Even if the drug is stopped, it is often possible to safely rechallenge patients (P-Codrea Tigaran et al., 2005). Cytotoxic CD4+ T cells are the dominant cell type (Pichler, 2003), and one reason that these rashes are mild is that most cells do not express high levels of MHC-II, to which CD4+ T cells bind. However, rashes that initially look like a mild maculopapular rash can progress into a more serious rash such as toxic epidermal necrolysis, which appears to be mediated by CD8+ cytotoxic T cells.
2. Urticaria.
The next most common type of drug-induced skin rash is urticaria, commonly called hives (Hunziker et al., 1997). Urticaria is typically an IgE-mediated allergic reaction to a drug such as penicillin; however, it is not always a true allergic reaction because some people have urticaria induced by cold or exercise (Mathelier-Fusade, 2006). It is characterized by relatively large, raised, pruritic skin lesions, any one of which does not last for more than 24 hours, although the urticaria can last for days as new lesions appear. Urticaria is classically part of anaphylactic reactions, which can be fatal. As with other IDRs, there is a delay between starting a drug and the onset of urticaria on initial exposure, but on rechallenge symptoms usually appear very rapidly—minutes to hours. Urticaria can be chronic and idiopathic. Chronic urticaria appears to be an autoimmune reaction (Vonakis and Saini, 2008), and it often responds to cyclosporine (Hollander et al., 2011). There are some cases of chronic urticaria that appear to start with an IDR and then later become independent of drug exposure (personal experience). Pseudoallergic reactions that include urticaria can also be caused by drugs such as nonsteroidal anti-inflammatory drugs and angiotensin converting enzyme inhibitors. In the case of aspirin, the mechanism involves diverting arachidonic acid metabolism toward the production of leukotrienes (Suzuki and Ra, 2009), and these reactions are not associated with the typical delay in onset of a true allergic reaction.
3. Fixed Drug Eruption.
This is an interesting type of drug rash that is always caused by drugs (Shiohara, 2009). It is composed of one or more lesions that recur at the same site every time a specific drug is administered. When the drug is stopped the lesions usually resolve with residual hyperpigmentation, which makes it easy to determine the affected area. On rechallenge, the time to onset is approximately 2 hours, and the number of lesions often increases with repeated exposures. It is mediated by CD8+ T cells with an effector-memory phenotype, and these cells are limited to the site of the lesion (Shiohara, 2009). Therefore, patch tests are usually positive, but only if applied to the site of a lesion. When a fixed drug eruption is limited to a single lesion it is usually mild, but when it is extensive it can be more serious with systemic symptoms such as fever and arthralgias, and it can even mimic Stevens-Johnson (SJS) syndrome.
4. Drug Reaction with Eosinophilia and Systemic Symptoms and Drug-Induced Hypersensitivity Syndrome.
The drugs first associated with these syndromes were the anticonvulsants, and the first term applied to the adverse reaction was anticonvulsant hypersensitivity syndrome (Shear and Spielberg, 1988). Then it was realized that other drugs could cause the same syndrome, and a more general term was drug-induced hypersensitivity syndrome (DIHS) (Walsh and Creamer, 2011). Another term that is used more or less synonymously with DIHS is drug reaction with eosinophilia and systemic symptoms (DRESS), and this term is now more commonly used. However, because of the variability of the syndrome there is not total agreement over the nomenclature (Roujeau, 2005; Kardaun et al., 2007; Shiohara and Kano, 2007). The clinical characteristics include an acute onset of rash, fever, and at least one of the following organ involvements: lymphadenopathy, hepatitis, nephritis, pneumonitis, carditis, thyroiditis, and hematologic abnormalities (eosinophilia, atypical lymphocytes, thrombocytopenia, or leukopenia) (Peyriere et al., 2006; Um et al., 2010; Walsh and Creamer, 2011). However, a rash is not always present, and the characteristics of the rash can vary significantly from one patient to another. The mortality can reach 10%, most commonly from liver failure. The usual delay in onset is 2–6 weeks, and this is an important diagnostic criterion (Cacoub et al., 2011; Creamer et al., 2012). The most common drugs associated with DRESS/DIHS are carbamazepine and other aromatic anticonvulsants, sulfonamides, allopurinol, and several of the anti-HIV drugs, especially abacavir and nevirapine. The onset of DRESS is associated with reactivation of herpes viruses (Descamps et al., 1997; Suzuki et al., 1998), and T cells from infected patients recognize herpes virus antigens (Picard et al., 2010). The usual treatment of severe DRESS is corticosteroids, and when they are discontinued, patients often relapse, possibly because the steroids prolong virus reactivation. There is clearly an association between reactivation of herpes viruses and DRESS, but the exact nature of this relationship is not clear. Specific HLA genotypes are major risk factors for DRESS/DIHS caused by specific drugs, and this will be discussed in more detail later.
5. Acute Generalized Exanthematous Pustulosis.
Acute generalized exanthematous pustulosis is characterized by an acute onset of a noninfectious pustular skin reaction, usually starting on the face, neck, groin and axillae, fever, and neutrophilia (Roujeau et al., 1991; Choi et al., 2010). The major drugs associated with this ADR are antibiotics. Patch tests with the offending agent are usually positive. The time to onset is shorter than with other serious skin rashes, often as short as 1 day, but this may be because of previous exposure to the drug (Roujeau et al., 1991).
6. Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis.
Toxic epidermal necrolysis (TEN) is the most severe type of skin rash with a mortality rate of ~30% (Pereira et al., 2007; Downey et al., 2012). SJS appears to be a milder form of the same rash. The difference is the extent of skin involvement, with SJS involving less than 10%, TEN involving more than 30%, and SJS/TEN overlap involving 10–30%. The syndrome usually begins with a sudden onset of fever and malaise followed by a rash that is painful to the touch. Blisters form and the classic sign is Nikolsky’s sign, in which gentle lateral pressure results in sloughing of the epidermis. Histologically, this corresponds to widespread keratinocyte apoptosis with separation between the dermis and epidermis and a mild mononuclear infiltrate in the dermis. The mucus membranes of the mouth and genital area are involved early in the process, and intestine and eyes can be involved, sometimes resulting in blindness. The time to onset is usually a little shorter than for DRESS (14 ± 7 days), but it is greatly reduced if the patient is reexposed to the drug (Roujeau, 2005). These are clearly immune-mediated reactions, and again there are specific HLA associations with specific drugs; however, unlike DRESS, where the lymphocyte transformation test is often positive (Kano et al., 2007; Jurado-Palomo et al., 2010), it is typically negative in SJS/TEN (Tang et al., 2012). The cells that mediate the rash are reported to be cytotoxic T cells (Nassif et al., 2004; Wei et al., 2012), but other cells presumably play important roles (de Araujo et al., 2011; Tohyama and Hashimoto, 2012). The molecules that mediate the keratinocyte toxicity in SJS/TEN appear to include Fas (apoptosis antigen 1, CD95) ligand (Downey et al., 2012), granulysin (Chung et al., 2008), and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) (de Araujo et al., 2011). It is difficult to perform controlled trials to determine the best treatment of this rare rash. Intravenous immunoglobulin appeared to be beneficial in several studies, but the results are controversial, and other immunosuppressants such as cyclosporine have also been used (Paquet and Pierard, 2010a,b; Pierard and Paquet, 2010).
B. Liver Injury
Of the types of IDRs included in this review, idiosyncratic liver injury (IDILI) is the type that most commonly leads to drug withdrawal or black box warnings (Watkins, 2005). It is presumed that this is because the liver is the major site of drug metabolism, and this often leads to the formation of chemically reactive metabolites. The two most common types of IDILI are hepatocellular and cholestatic. Drugs can also cause other types of IDILI such as methotrexate -induced liver fibrosis, but they are less common.
1. Hepatocellular Liver Injury.
The most common serious IDILI involves the death of hepatocytes; this is referred to as hepatocellular IDILI. The time to onset is usually 1–3 months; however, sometimes the delay between starting the drug and the onset of IDILI can be more than 1 year (Bjornsson, 2010). In contrast, the time to onset of fluoroquinolone- and telithromycin-induced liver injury is short, often only a few days (Clay et al., 2006; Orman et al., 2011). As with other types of IDRs, drugs that cause serious IDILI are always associated with a higher incidence of mild IDILI that usually resolves despite continued treatment with the drug (referred to as adaptation), but the ratio of the incidence of mild DILI to serious DILI varies with the drug, and some drugs such as the statins rarely cause serious IDILI although they often (∼1% incidence) cause mild DILI. If, however, in addition to causing mild DILI, there are cases in which there is also an increase in serum bilirubin, the drug is likely to cause liver failure with an incidence of ~1/10 that of the cases of DILI that are associated with an increase in bilirubin. This is referred to as Hy’s rule or Hy’s law; this is very useful in catching drug candidates that are likely to cause liver failure (Temple, 2006). Temple’s corollary is that if a drug does not cause mild IDILI it is very unlikely to cause serious IDILI (Watkins et al., 2011). IDILI can be part of DRESS; in other cases it is more limited but still associated with fever and rash, but often there are no obvious symptoms of an “allergic” reaction. Drugs that cause IDILI with evidence of an immune response, such as halothane, are classed as immune idiosyncrasy, whereas those without obvious signs of an immune response have been classed as metabolic idiosyncrasy (Zimmerman, 1999). However, there are no examples in which a polymorphism in a metabolic pathway is sufficient to explain the idiosyncratic nature of IDILI, and these terms are used less often today. Even for drugs in which the IDILI has been classed as metabolic idiosyncrasy, there are usually specific cases with clear evidence of an immune mechanism such as a very rapid onset with rechallenge (Maddrey and Boitnott, 1973).
The liver histology can vary, but most typically there is a mild mononuclear infiltrate, and often eosinophils are present even in the absence of other signs of an allergic reaction (Zimmerman, 1999). In some cases the damage is greater in the centrilobular region of the liver, which is the area containing the highest concentration of cytochrome P450s, whereas in others it is more diffuse. IDILI is often associated with antidrug and/or autoantibodies, but in most cases the reagents for detection of antidrug antibodies are not available. The autoantibodies can be against the enzyme that formed the reactive metabolite such as in the case of tienilic acid and dihydralazine. Some drugs such as nitrofurantoin, α-methyldopa, and minocycline can cause IDILI that is indistinguishable from idiopathic autoimmune hepatitis except that it usually resolves when the drug is stopped; in such cases, the time to onset is usually greater than one year (Bjornsson et al., 2010; Czaja, 2011). Most drugs thatcause IDILI form reactive metabolites in the liver that are presumed to be responsible for the adverse reaction; however, some drugs such as ximelagatran and pyrazinamide do not appear to form reactive metabolites. In addition, biologic drugs such as infliximab also appear to cause autoimmune hepatitis even though their therapeutic effect involves immunosuppression (Doyle et al., 2011).
The risk of IDILI increases with age and female sex for many, but not all, drugs (Chalasani and Bjornsson, 2010). An exception is valproic acid-induced IDILI, which is more common in infants and often has features of mitochondrial dysfunction such as steatosis and hyperammonemia (Zimmerman, 1999). The incidence of valproate-induced liver injury is also much higher in patients with a mutation in POLG, which codes for mitochondrial DNA polymerase (Stewart et al., 2010). Drug-induced autoimmune hepatitis is definitely more common in females (Bjornsson et al., 2010; Czaja, 2011).
2. Cholestatic Liver Injury.
Cholestatic liver injury is characterized by a greater increase in alkaline phosphatase and bilirubin relative to alanine transaminase. Specifically, if the ratio in terms of the number of times the upper limit of normal of alanine transaminase/alkaline phosphatase is less than two, it is considered cholestatic IDILI, whereas if it is greater than five, it is considered hepatocellular IDILI; if the ratio is in between two and five it is considered mixed. The incidence of liver failure requiring transplantation is less than that of hepatocellular IDILI. It has been reported that the mortality rate in patients with cholestatic IDILI is similar to that of hepatocellular IDILI (Bjornsson and Olsson, 2005), but it appears that much of the mortality was not due to liver failure, possibly because the cholestatic pattern is more common in older patients (Lucena et al., 2009). In other studies hepatocellular IDILI led to death or liver transplantation more commonly than cholestatic IDILI (Chalasani et al., 2008). Although less likely to lead to liver failure, the course of cholestatic liver injury is often prolonged, with recovery taking more than a month (Hussaini and Farrington, 2007). Drugs associated with cholestatic IDILI include the phenothiazines, amoxicillin/clavulanic acid, and flucloxacillin.
C. Hematologic Adverse Reactions
Drugs can cause several types of IDRs involving blood cells either by affecting the production of blood cells or their destruction. They include agranulocytosis, thrombocytopenia, anemia, and aplastic anemia.
1. Agranulocytosis.
Agranulocytosis is characterized by a peripheral neutrophil count of less than 500 cells/µl blood. Agranulocytosis can be caused by cytotoxic drugs used to treat cancer, but it can also be idiosyncratic with noncytotoxic drugs. As with other IDRs, it usually takes 1–3 months of treatment before the onset of agranulocytosis. Although the drop in neutrophil count can be gradual, it is usually precipitous (Gerson and Meltzer, 1992). It is usually asymptomatic, and the first indication is an infection heralded by symptoms such as a sore throat and/or fever. Although agranulocytosis implies that there is an absence of all granulocytes, basophils can be spared (Besser et al., 2009), and lymphopenia can be present (Tesfa et al., 2009). This may be a reflection of the mechanism because basophils do not have the same peroxidase activity as neutrophils and eosinophils, whereas lymphopenia could be the result of an immune response. Agranulocytosis can result from peripheral destruction of neutrophils (Moeschlin and Wagner, 1952), from damage to neutrophil precursors in the bone marrow, or a combination of both (Tesfa et al., 2009). Typically, there is an absence of neutrophil precursors in the bone marrow back to the promyelocyte stage, and this has been termed “maturation arrest”; however, the appearance of the bone marrow is highly dependent on when in the course of the event the bone marrow was obtained, and it is difficult to be certain that the appearance truly represents an arrest in the maturation of neutrophils. At least in some cases, this appearance could be due to destruction of cells that have myeloperoxidase with sparing of any less mature cells lacking myeloperoxidase.
An interesting example is rituximab, which is associated with a late onset (1-9 months after the last treatment) granulocytopenia (Tesfa and Palmblad, 2011). Rituximab is an antibody that binds to CD20 leading to the depletion of B cells, and therefore, the mechanism presumably does not involve a reactive metabolite formed by myeloperoxidase. Several mechanistic hypotheses exist for rituximab-induced neutropenia, but the evidence for each is inconclusive.
2. Thrombocytopenia.
Virtually all idiosyncratic drug-induced thrombocytopenia appears to be immune mediated, but there are several different immune mechanisms (Aster, 2009; Aster et al., 2009). The classic drug associated with idiosyncratic thrombocytopenia is heparin (Warkentin, 2003). The incidence is somewhat lower with low molecular weight heparin. The more serious form is due to antibodies against the heparin-platelet factor 4 complex. It is interesting to note that although it is clearly an immune-mediated reaction, there is no immune memory. Specifically, if heparin is administered after the pathogenic antibodies are gone (~100 days after an episode of heparin-induced thrombocytopenia), there is usually no recurrence of thrombocytopenia, and if it does recur, it has a delay in onset (Warkentin and Kelton, 2001). The β-lactams act as haptens to produce antibodies that recognize modified platelet proteins, whereas quinine induces a conformation change in proteins that induces antibodies that only recognize the proteins in the presence of soluble drug (Aster et al., 2009). Still other drugs such as procainamide can induce autoantibodies that cause thrombocytopenia. The fibans, tirofiban and epitifibatide, appear to induce a conformational change in GPIIb-IIIa that is recognized by naturally occurring antibodies, leading to a rapid onset of thrombocytopenia on first exposure to the drug (Bougie et al., 2002).
3. Anemia.
Many of the same drugs that cause hemolytic anemia also cause thrombocytopenia or neutropenia, sometimes in the same patient (Garratty, 2012). It is also somewhat similar to drug-induced thrombocytopenia in that most of the cases are clearly immune mediated, some by drug-dependent antibodies and some involving autoimmune antibodies. One of the first drugs to be recognized as causing autoimmune hemolytic anemia was α-methyldopa. The antibodies induced by this drug do not require the presence of the drug to bind to red cells, but when the drug is stopped these autoantibodies also decrease to the point that the anemia resolves, usually with a significant titer of autoantibodies still present, As with other types of IDRs, the incidence of red cell autoantibodies associated with α-methyldopa is much higher than the incidence of significant anemia. Other types of autoimmune IDRs such as autoimmune hepatitis and a generalized autoimmune syndrome similar to lupus can also be caused by α-methyldopa.
The other type of antibody that causes hemolytic anemia requires the presence of drug. In some cases such as most of the β-lactams, the drug chemically reacts with the red cell membrane, thus acting as a hapten. In other cases, the interaction between the drug and red cell membrane is not covalent, but the drug changes the structure of the red cell membrane, leading to the binding of other proteins. Sera from some patients who have never taken the drug in question will bind to red cells in the presence of drug (Garratty, 2012).
Drugs such as procainamide and isoniazid can also cause pure red cell aplasia in which there is a decrease in the production of red cells, but this is less common than drug-induced hemolytic anemia (Giannone et al., 1987; Nakamura et al., 2010). In one case, the patient appeared to respond to cyclosporine, which suggests that it was immune mediated, and drugs that are associated with pure red cell aplasia are also associated with other autoimmune syndromes (Nakamura et al., 2010).
4. Aplastic Anemia.
Aplastic anemia is characterized by a lack of hematopoietic cells in the bone marrow (Young and Alter, 1994). Therefore, to make the diagnosis of aplastic anemia, a bone marrow biopsy is required to differentiate it from other syndromes causing pancytopenia. Unlike agranulocytosis, the cause of which is usually a drug, most aplastic anemia is idiopathic (Young et al., 2008). This can make it more difficult to determine if a specific case of aplastic anemia is drug induced. In addition, most of the cases of apparent chloramphenicol-induced aplastic anemia occurred a month, often several months, after the drug was stopped (Sharp, 1963; Wallerstein et al., 1969). There is both direct and indirect evidence that drug-induced aplastic anemia is immune mediated. The direct evidence is the finding of Th1 cells that produce IFN-γ in the bone marrow of affected patients that suppress hematopoiesis in vitro (Sloand et al., 2002). The indirect evidence is that most patients respond to immunosuppressants such as antithymocyte antibodies and cyclosporine whether the aplastic anemia is thought to be idiopathic or drug induced (Young, 2002; Young et al., 2006). Th17 cells also appear to be increased in aplastic anemia (de Latour et al., 2010). Many of the drugs that can cause agranulocytosis such as propylthiouracil and carbamazepine, can also cause aplastic anemia, but that does not always appear to be the case.
D. Drug-Induced Autoimmunity
There are many drugs that can cause various types of autoimmune syndromes, some of which such as autoimmune hepatitis and autoimmune hemolytic anemia have been described above. Other drugs can cause a generalized autoimmune syndrome that is similar to lupus. As mentioned earlier, the time to onset of drug-induced autoimmunity is usually longer than other types of IDRs, often more than a year (Uetrecht, 2009a). Drug-induced autoimmunity usually resolves when the drug is stopped, which is a key diagnostic feature; however, in rare cases what appears to be drug-induced autoimmunity does not resolve when the drug is stopped.
1. Drug-Induced Lupus-like Syndrome.
As mentioned, drugs can cause a generalized autoimmune syndrome similar to lupus (DIL). In the past, the two drugs that were associated with the highest incidence of a DIL were procainamide and hydralazine. However, for several reasons, these drugs are not commonly used today. The incidence of DIL in patients treated chronically with procainamide is ~20–30%, and almost all patients develop antinuclear antibodies even if they do not become symptomatic (Uetrecht et al., 1981a,b). Both of these drugs cause inhibition of DNA methylation, and there is strong evidence that this is involved in the mechanism (Richardson, 2003). DNA methylation in mature CD4+ T cells cause MHC-specific autoreactivity in vitro. Furthermore, T cells from patients with active lupus have hypomethylated DNA (Richardson, 2003; Richardson et al., 2012). Thus, although not discussed further, epigenetic effects may be quite important in the etiology of IDRs.
These and many other drugs that are associated with autoimmunity are oxidized by the myeloperoxidase system of leukocytes, and this may also play a role in their ability to cause autoimmunity (Uetrecht, 2005). The much higher incidence of lupus in women apparent with idiopathic lupus is not always apparent with DIL, and the prevalence can be affected by sex differences in drug use. The syndrome can be difficult to differentiate from idiopathic lupus, but DIL is usually milder and less likely to involve the central nervous system or kidneys. Both idiopathic and DIL are associated with antinuclear antibodies, but DIL is less likely to be associated with anti-double-stranded DNA antibodies and more likely to be associated with antibodies against histone proteins (Uetrecht and Woosley, 1981). Drug-induced lupus is often associated with antineutrophil antibodies, (Chang and Gershwin, 2011), which may be related to the oxidation of many of these drugs by myeloperoxidase.
Many biologic drugs can also cause a lupus-like syndrome (Chang and Gershwin, 2011). This occurs with drugs such as interferon-α (Borg and Isenberg, 2007), which is not surprising because type I interferons appear to be involved in the pathogenesis of lupus (Elkon and Wiedeman, 2012). However, it also occurs with drugs such as infliximab, which is an anti-tumor necrosis factor antibody that is used as an immunosuppressant (Costa et al., 2008; Williams and Cohen, 2011). This is a good example of the complexity of the immune system, and the effects of agents can be difficult to predict. In contrast to DIL caused by small molecules, DIL caused by biologics is often associated with anti-double-stranded DNA antibodies and anti-histone antibodies are less common. In addition, renal involvement has been reported (Costa et al., 2008). This suggests that the lupus-like syndrome caused by biologics is more like idiopathic lupus.
2. Drug-Induced Cutaneous Lupus.
A related syndrome is a cutaneous lupus-like syndrome, which as the name implies is largely limited to the skin (Callen, 2001). As with other drug-induced autoimmunity, the time to onset is long: months to years. The drug with the highest relative risk is terbinafine (Gronhagen et al., 2012a,b), and cases typically occur in the summer and affect sun-exposed areas, which suggests a photodermatitis component. RoSSA autoantibodies are characteristic of this syndrome (Callen, 2001).
3. Organ-specific Autoimmunity.
Several types of organ-specific autoimmunity were mentioned above under the target organ involved and will not be repeated here. The same drugs usually cause more than one type of autoimmunity, but the spectrum of autoimmune syndromes varies with the specific drug. One of the most common drug-induced autoimmune reactions is vasculitis, which can take the form of skin lesions in leukocytoclastic vasculitis usually affecting the lower extremities, or it can affect organs such as the kidneys or lungs (Wiik, 2008; Bukhari, 2012).
III. Mechanistic Aspects
The clinical characteristics of IDRs are most consistent with an immune-mediated reaction, and therefore the emphasis will be on immune mechanisms. IDRs are frequently linked to the chemical reactivity of a drug or a product of metabolic activation. In essence, a threshold level of a drug-derived product must interact with an endogenous target and activate an otherwise latent biologic process that brings about tissue injury in the host. Ever since the seminal work of Landsteiner and Jacobs (1935), who discovered a direct association between a chemical’s propensity to bind covalently to protein and immune sensitization, it has been presumed that the formation of chemically reactive metabolites is the first step in the development of an IDR. It has been demonstrated that the risk that a drug will cause IDRs is roughly related to the amount of reactive metabolite that it forms (Nakayama et al., 2009). Therefore, attempts have been made to design the ability of a drug or a drug candidate to form a reactive metabolite out of the structure. However, some drugs that are associated with an unacceptable risk of IDRs such as ximelagatran do not appear to form reactive metabolites (Uetrecht, 2008). In addition, some drugs such as β-lactams, proton pump inhibitors, and anti-platelet drugs that bind to the P2Y12 receptor require irreversible binding for their therapeutic activity. This type of mechanism is being exploited in several new classes of drugs, and as long as the daily dose is relatively low and the binding is reasonably specific, these drugs are not associated with an undue risk of IDRs (Kalgutkar and Dalvie, 2012). Most reactive metabolites do not reach sites distant from where they are formed, and therefore the site of reactive metabolite formation is likely to be an important determinant of what type of IDR occurs. The liver is the site of most reactive metabolite formation, and this is presumably why it is a common target of IDRs. The skin has much lower activity of most metabolic enzymes; an exception is sulfotransferase, which is responsible for bioactivation of nevirapine in the skin (Sharma et al., 2013). There are a few reactive metabolites such as acyl glucuronides that have low reactivity and freely circulate, and others such as aromatic nitroso metabolites and some glutathione conjugates (Baillie and Slatter, 1991), whose formation is reversible, can reach sites distant from where they are formed.
The purpose of this section is to review the immunologic basis of IDRs and describe the role of reactive metabolites play in the activation of immune cells. Furthermore, we discuss recent studies suggesting that the covalent modification of protein is not always needed to cause IDRs. The discovery of specific HLA alleles as important susceptibility factors for certain forms of IDRs suggests that the MHC molecule is an important target for drugs; thus, much of our discussion focuses on the interaction of drug-derived products with MHC.
Throughout our discussion we refer to the terms hapten, antigen, immunogen, and costimulatory agent. The meaning of each term in the context of IDRs is outlined below.
Hapten: a low molecular weight chemical that binds irreversibly to protein through the formation of a covalent bond.
Antigen: any drug or drug-derived product that interacts with high affinity with immunologic receptors.
Immunogen: any drug or drug-derived product that stimulates an immune response.
Costimulatory agent: any substance that interacts with dendritic cells, stimulating maturation and polarization of the immune response.
It is noteworthy that the terms hapten, antigen, and immunogen are drug dependent; however, the ultimate antigen/immunogen might not contain the drug (derived) product and that costimulatory agents can be drug or patient (disease) specific.
A. An Overview of the Immune Response
The immune system is thought (although not always proven) to amplify drug-derived signals in most forms of IDRs. Since immunology is a relatively new and emerging science, our knowledge of the cells and effector molecules involved in different forms of IDRs is still evolving. Thus, the following section briefly summarizes different components of the immune system that are thought to be centrally involved in IDRs.
1. Dendritic Cells.
Dendritic cells are the body’s immunologic sentinels. They act as a link between innate and adaptive immunity. Pattern recognition receptors expressed on the cell surface interact with specific pathogen components and endogenous molecules released from dead cells [e.g., uric acid (Shi et al., 2003), proinflammatory cytokines (Harris et al., 2012), and heat shock proteins (Tamura et al., 2012)], triggering differentiation and maturation. Activated dendritic cells migrate to local lymph nodes, transporting antigens for subsequent presentation to naive T cells. When activated, they express high levels of costimulatory receptors that interact with cell surface ligands expressed on T cells during antigen priming and secrete cytokines into the priming microenvironment and, as such, contribute to polarization of the immune response. Haptenic chemicals [e.g., dinitrohalobenzenes (Martin et al., 2011; Esser et al., 2012)] also trigger dendritic cell signaling via multiple pathways, including pattern recognition receptor triggering through the degradation of hyaluronic acid, the formation of reactive oxygen species, and/or the direct modification of cysteine-containing proteins. Moreover, independent studies by Pickard et al. (2009) and Watanabe et al. (2008) suggest that once a contact-sensitizing chemical passes through skin, its potential to cause strong immunologic reactions is determined by its ability to stimulate proinflammatory cytokine (IL-1β, IL-18) release through activation of the inflammasome (a protein complex composed of intracellular NOD-like receptors, the adaptor protein apoptosis speckled-like protein with a caspase recruitment domain and caspase-1). Modification of absorption and/or inflammasome signaling was found to convert a tolerizing chemical into a sensitizer. Recently, gene knockout mice were used to demonstrate that IL-1 receptor signaling is critical for the migration of antigen primed dendritic cells to draining lymph nodes, T-cell priming, and contact sensitization (Kish et al., 2012). The drugs amoxicillin (Rodriguez-Pena et al., 2006), sulfamethoxazole (Sanderson et al., 2007), and abacavir (Martin et al., 2007) have also been shown to at least partly activate dendritic cells; however, the cellular processes involved remain unresolved.
2. T Lymphocytes.
Naive CD4+ T lymphocytes differentiate into Th1, Th2, Th9, Th17, or Th22 effector cells after antigen exposure. The panel of cytokines naive cells are exposed to at the time of priming determines the nature of the effector T-cell response and the functional consequences of antigen exposure (Fig. 1) (Akdis and Akdis, 2009). The classification of CD8+ T cells is much simpler, based on the release of cytolytic molecules (Fas ligand, perforin, granzyme B, granulysin) after antigen stimulation. Cutaneous drug reactions have been classified according to the phenotype of drug-responsive T cells isolated from peripheral blood of sensitive patients and the cytokine secretion profile (Pichler, 2003). Keratinocyte damage in patients with maculopapular reactions involves CD4+ and CD8+ T cells, and Th1 and Th2 cytokine secretion is readily detectable (Kuechler et al., 2004; Yawalkar and Pichler, 2001; Rozieres et al., 2009). IFN-γ-secreting cytotoxic CD8+ T cells predominate in bullous skin reactions and DRESS (Naisbitt et al., 2003a,b; Nassif et al., 2004; Wu et al., 2007; Ko et al., 2011). IL-5, which is involved in eosinophil recruitment and activation, is readily detectable in drug-stimulated T-cell cultures from patients with DRESS. Pustular reactions involve CD8+-mediated cytotoxicity and secretion of the neutrophil chemoattractant IL-8 (Britschgi et al., 2001). Chung et al. (2008) showed that granulysin is a key cytotoxic molecule released from T cells in patients with Stevens-Johnson syndrome/toxic epidermal necrolysis and suggested that high expression of granulysin in this group of patients might explain the severity of the reactions that develop. However, a more recent report indicates that granulysin is secreted from drug-specific T cells isolated from patients with mild, moderate, and severe cutaneous reactions (Schlapbach et al., 2011). Although informative, this classification is largely based on a snapshot of the memory T-cell response, often many years after the clinical reaction subsides. Future studies are needed to compare the nature of the T-cell response at the time of drug exposure, during the IDR, and in the long term, as the patient recovers.
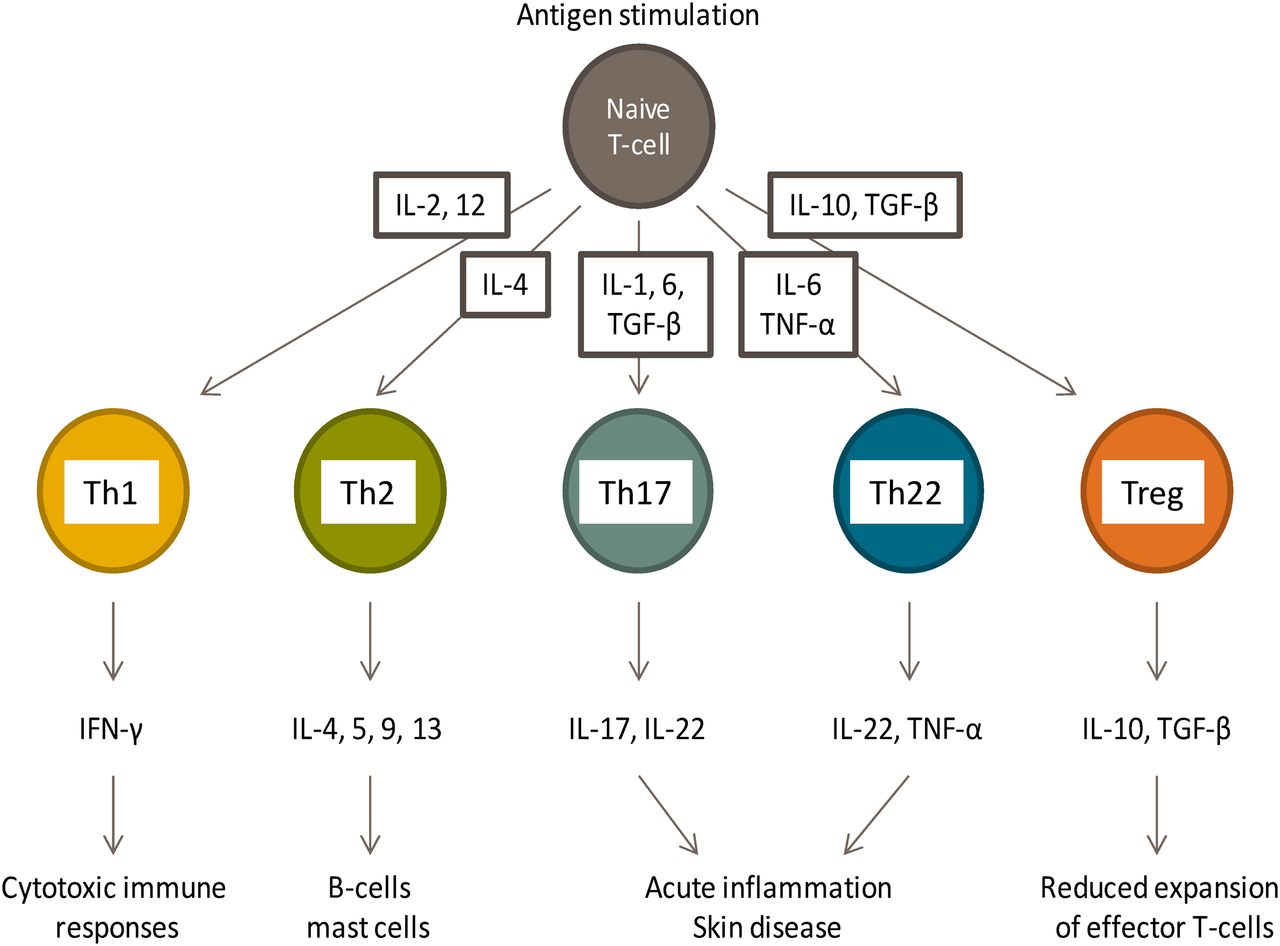
CD4+ T-cell subsets involved in immune regulation and pathology.
The time-dependent recruitment of Th17 and/or Th22 secreting T cells into inflamed tissue has been observed in several types of skin disease including atopic dermatitis, allergic dermatitis, and psoriasis (Eyerich et al., 2009, 2010, 2011; Pennino et al., 2010). IL-17 promotes T-cell-mediated killing of keratinocytes by increasing membrane expression of adhesion molecules, whereas IL-22 exerts a protective effect by inducing keratinocyte proliferation and migration. Elegant studies by Pennino et al. (2010) have shown that IL-17 released by skin resident antigen-specific T cells from patients with allergic dermatitis direct non-antigen-specific Th1 secreting T cells to kill keratinocytes. Thus, the antigen-specific immune response is enhanced and sustained through IL-17 secretion and the bystander effect of non-antigen-specific T cells. Currently, Th17- and Th22-secreting drug-responsive T cells from patients with a history of an IDR have not been studied in detail.
The adaptive immune system is also implicated in IDRs that target the liver. However, the role of T cells in drug-induced liver injury is less well defined. In 1997, Maria and Victorino (1997) described lymphocyte responses to drugs in over 50% of patients with drug-induced liver injury. More recently, histologic examination of inflamed liver from a patient exposed to sulfasalazine revealed an infiltration of granzyme B-secreting T lymphocytes (Mennicke et al., 2009). To explore the phenotype and function of T cells from patients with drug-induced liver injury we recently focused on the β-lactam antibiotic flucloxacillin. Flucloxacillin exposure is associated with a high incidence of cholestatic liver injury. Peripheral blood mononuclear cell responses against the drug were detectable in 5/6 patients using an IFN-γ secretion (ELIspot) assay. T-cell cloning revealed that the majority of flucloxacillin-responsive T cells were CD8+. Drug stimulation resulted in the secretion of IFN-γ, and cytolytic molecules, including FAS ligand, perforin, and granzyme B (Monshi et al., 2013).
3. Natural Killer Cells.
Natural killer (NK) cells are a central component of the innate response. They secrete large quantities of granulysin after activation and are thought to act in unison with cytotoxic T cells to initiate tissue damage in patients (Tewary et al., 2010). NK cells respond rapidly after exposure to virally infected cells in the absence of MHC restriction and T-cell receptor signaling. Recently Schlapbach et al. (2011) showed that NK cells expressing granulysin make up a proportion of the cellular infiltrate in different forms of cutaneous ADR. Thus, it is possible that NK cells contribute toward the tissue injury in patients with an IDR.
4. B Lymphocytes.
Restimulation of antigen-specific memory B cells leads to a rapid increase in serum antibodies. Antigen-specific B cells also effectively present peptide fragments to specific T cells (Lanzavecchia, 2007). Enhanced antigen presentation is dependent on membrane-associated antibodies that sequester and concentrate the antigen prior to processing. Soluble antigen-bound antibodies have also been shown to modulate the presentation of peptide antigens to T cells. They influence the T-cell response by enhancing antigen capture and delivery by modulating processing pathways, thus suppressing the generation of dominant antigenic determinants and by preventing processing (Watts and Lanzavecchia, 1993). We and others have used drug-protein conjugates to detect anti-drug antibodies in certain tolerant and allergic patients (de Haan et al., 1986; Christie et al., 1988; Daftarian et al., 1995; Torres et al., 1997). However, the dynamics of the drug antigen-specific humoral response and the kinetics of antibody production have not been defined. Furthermore, we do not know whether antibody titers differ in patients that do and do not develop an IDR and how anti-drug antibodies modulate the drug-specific T-cell response.
It is now understood that IgG4 antibodies play a central role in immune regulation after grass pollen immunotherapy (James et al., 2011). It is believed that these IgG4 antibodies are produced by B cells under the guidance of allergen-specific regulatory T cells that appear during immunotherapy (Satoguina et al., 2008). Inhibitory IgG4 antibodies are thought to directly inhibit facilitated antigen presentation to T lymphocytes (van Neerven et al., 1999). It is relevant therefore to consider the roles played by specific IgG antibody subclasses in IDRs.
B. Antigen Processing, Presentation, and T Lymphocyte Activation
T cells are activated by peptides. For this to occur, proteins are degraded by protease enzymes to release peptides that associate with MHC molecules prior to display on the surface of antigen presenting cells. T cells subsequently bind to the MHC peptide complex through their T-cell receptor. This MHC peptide T-cell receptor binding interaction, at least in part, determines the nature of the induced T-cell response. The molecular pathways involved in the activation of CD4+ and CD8+ T cells are the subject of a recent review and beyond the scope of this document (Neefjes et al., 2011). Thus, we focus our brief discussion around drug distribution and how this might relate to the MHC antigen T-cell receptor binding interaction. Immunologic doctrine states that peptides derived from extracellular and intracellular proteins stimulate CD4+ and CD8+, respectively. This is because MHC class I (HLA-A, -B, and –C) and class II (HLA-DR, -DP, and -DQ) molecules display peptides originating from intracellular and extracellular compartments, respectively. Intracellular proteins are broken down by cytosolic and nuclear proteasomes. The derived peptides are transported to the endoplasmic reticulum where they bind to MHC class I molecules. MHC class I peptide binding grooves preferentially accommodate peptides of 8–9 amino acids. Once a peptide binds, the MHC molecule leaves the endoplasmic reticulum and transports the peptide to the cell surface for presentation to CD8+ T cells. MHC class I molecules are expressed on all nucleated cells (approximately 10,000–500,000 molecules per cell); thus, all tissues have the capacity to display drug-derived antigens on their surface. MHC class II molecules are similar in structure, but differ in terms of tissue distribution and the peptides that they display. MHC class II molecules are expressed on professional antigen presenting cells (dendritic cells, B cells), although expression can be upregulated on other cells during inflammation, and they present peptides derived from extracellular proteins. To enter the class II processing pathway proteins are internalized by phagocytosis. The membrane-derived endosomes fuse with lysosomes, which contain protease enzymes that digest the engulfed protein. MHC class II molecules assembled in the endoplasmic reticulum migrate to the endosomes where chaperone MHC-binding peptides are substituted with peptides derived from exogenous protein. The MHC class II molecule is then transported to the cell surface, and the peptides are displayed to CD4+ T cells. The MHC class II binding cleft displays longer peptides compared with MHC class I, because the binding grove is open at both ends.
Although in practice the division of protein processing into exogenous and endogenous pathways is a much simplified version of the processes that occur in living cells [presentation of peptides derived from extracellular proteins on MHC class I occurs through a process known as cross-presentation (Joffre et al., 2012), while autophagy delivers peptides derived from intracellular compartments to MHC class II (Munz, 2012)], it does provide a useful framework to explore whether compound distribution and the formation of exogenous/endogenous drug-derived antigens is related to the nature of the induced immune response. Kalish and Askenase (1999) were the first to link the distribution of drug-protein binding to immune polarization. They suggested that environmental chemicals (e.g., dinitrohalobenzenes) that cause CD8+-mediated contact sensitization and drugs susceptible to cytochrome P450-mediated metabolism (e.g., sulfonamides) likely form cell-associated antigens, whereas drugs that activate CD4+ T cells (e.g., β-lactam antibiotics) will preferentially form serum antigens.
Our discussion below reviews the literature describing patient studies on chemical sensitization and sulfonamide and β-lactam antibiotic reactions to provide an up-to-date commentary on the role of drug-protein binding in the activation of immune cells and whether the distribution of drug-protein adducts relates in any way to the nature of the induced response.
C. The Antigenicity and Immunogenicity of Directly Reactive Low Molecular Weight Drugs and Chemicals
Hapten dogma, originating from the studies of Landsteiner and Jacobs (1935), states that low molecular weight chemicals bind irreversibly to self protein to break immune tolerance. Subsequently, researchers in the field of contact allergy have shown that the reactivity of a chemical and its ability to bind covalently to protein is indisputably linked to the activation of immune cells and sensitizing potential (Meschkat et al., 2001a,b; Alvarez-Sanchez et al., 2003). In fact, these observations have resulted in the development and use of chemical reactivity measurements for the predictive identification of skin sensitizing chemicals (Divkovic et al., 2005; Gerberick et al., 2007, 2008). The development of sophisticated protein mass spectrometry methods to measure the binding of sensitizing chemicals to protein has greatly assisted analysis of chemical protein interactions (Jenkins et al., 2008). Protein binding has been found to vary in terms of protein and amino acid specificity, reaction mechanisms, and rates of reaction (Aleksic et al., 2008, 2009). Furthermore, the protein binding profile cannot be predicted through studies with isolated amino acids or simple peptides because binding is restricted to sites in the protein accessible to the chemical and is also affected by neighboring amino acids. Despite this complexity, interdisciplinary studies involving analysis of protein binding in parallel with assessment of human T-cell responses have shown that protein adducts activate T cells from sensitized patients and prime highly purified naive T cells. Below, we discuss the exemplars 2,4-dinitrochlorobenzene (DNCB), p-phenylenediamine (PPD), and β-lactam antibiotics.
1. Dinitrochlorobenzene.
DNCB belongs to a large class of nitrohalobenzenes, which all form the same dinitrophenyl-modified protein adduct. Irreversible binding of DNCB occurs at cysteine and lysine residues on protein and peptides (Kitteringham et al., 1985; Maggs et al., 1986). Protein modifications are selective and dependent on the inherent reactivity of individual amino acids within a protein (Aleksic et al., 2007). Topical DNCB exposure activates a cellular immune response in 100% of subjects that is readily detectable after skin challenge (Friedmann et al., 1983; Pickard et al., 2009). Both CD4+ and CD8+ T cells are stimulated to secrete IFN-γ in the presence of DNCB. The proliferative response of DNCB-responsive T-cell clones is blocked by fixation of antigen presenting cells with glutaraldehyde, which inhibits metabolic activity including the processing of protein antigens (Pickard et al., 2007). Collectively, these data indicate that DNCB binds to multiple cellular and extracellular proteins that generate CD4+ and CD8+ T-cell antigens through protein processing. Several groups have demonstrated that DNCB-treated antigen presenting cells readily prime T cells isolated from naive human subjects (Dai et al., 1993; Dai and Streilein, 1998; Vocanson et al., 2008). Moreover, dinitrophenyl-modified human serum albumin can be used to prime T-cell responses when processed by dendritic cells (Dietz et al., 2010). Thus, naive T cells are also activated and respond to dinitrophenyl-modified extracellular and cellular protein. Groundbreaking studies by Weltzien and co-workers (Martin et al., 1992, 1993; Martin and Weltzien, 1994; Preckel et al., 1997) were the first to show that MHC-associated dinitrophenyl-conjugated peptides are major antigenic determinants for T cells. Designer peptides with known MHC binding motifs were synthesized and selectively haptenated to study their capacity to stimulate T cells. The presence of a bound dinitrophenyl moiety was found to be critical for the activation of T cells. Furthermore, two forms of T-cell receptor triggering were identified. One set of T-cell receptors were activated in the presence of the hapten, irrespective of the makeup of the peptide carrier. The other required two apparently independent signals from the hapten and carrier peptide. These studies are the closest researchers have come to defining hapten theory; however, antigenic MHC-associated dinitrophenyl-modified peptides eluted from DNCB-treated dendritic cells have not been characterized. Thus, researchers in the field are still waiting on definitive evidence to support the hapten hypothesis. Fig. 2 shows to structure of the compounds that we focus on in this article, the principal pathway of antigen presentation, and the phenotype of the drug-responsive T cells.

Drug-specific T cell responses. HLA-restriction and mechanisms of antigen presentation. AMajor pathway of antigen presentation listed. Hapten-specific T cells are often stimulated with drug-derived antigens binding directly to MHC. For carbamazepine and abacavir, drug-protein antigens have not been characterized. Thus, the hapten hypothesis has not been tested.
2. p-Phenylenediamine.
PPD is used as a primary intermediate in many hair dye formulations, and more recently it has been added to henna dyes. PPD is not directly protein reactive. However, in solution and on contact with skin, PPD undergoes sequential oxidation and self-conjugation reactions that produce a monomeric quinone diimine intermediate, products of dimerization and trimerization, and the trimer rearrangement product Bandrowski’s base. We have shown that the quinone diimine binds to selective cysteine residues in peptides and proteins, including glutathione S-transferase π and human serum albumin (Jenkinson et al., 2009, 2010). More recently, the binding of PPD oxidation products to protein was studied using an electrochemical oxidation step prior to protein modification. The findings largely support our study with human serum albumin as a protein target (i.e., only the monomeric quinone diimine modifies free cysteine groups). However, oligomeric oxidation products of PPD, including Bandrowski’s base, were found to modify cysteine groups on other proteins including β-lactoglobulin and hemoglobin (Jahn et al., 2012). The nature of the PPD-derived antigen that interacts with MHC and stimulates T cells is intriguing given its unique chemistry. PPD and Bandrowski’s base are classified as sensitizers in the mouse local lymph node assay (Warbrick et al., 1999; Aeby et al., 2009); however, the oxidation products formed after topical application cannot be assessed in this assay. To address this, we sensitized mice against either PPD or Bandrowski’s base through systemic injection and assessed antigen specificity by measurement of ex vivo T-cell proliferation after antigen recall (Farrell et al., 2009). This study confirmed that Bandrowski’s base-derived antigens were potent immunogens in the mouse. PPD-specific T-cell responses were not detected. Bandrowski’s base-responsive T cells are also detected in hair dye-exposed human subjects; however, their presence seems to reflect an acquired immune response that does not translate into an allergic reaction (Sieben et al., 2002; Coulter et al., 2008, 2010). A second population of Th2 secreting T cells, which are stimulated with PPD-derived primary oxidation products but not Bandrowski’s base, are detected in patients with hair dye allergy, and their presence seems to represent an important discriminator between allergic and tolerant patient groups. A synthetic albumin conjugate modified at the single available cysteine residue with PPD quinone diimine was used to demonstrate that lymphocytes and T-cell clones from allergic patients were stimulated with a protein conjugate (Jenkinson et al., 2010). The T-cell response could be blocked by fixation of antigen presenting cells with glutaraldehyde, indicating that peptides derived from the conjugate are likely antigenic determinants.
3. β-Lactam Antibiotics.
The term hapten has been adopted by researchers exploring mechanisms of IDRs. Entering the search terms “drug” and “hapten” into the PubMed database (http://www.ncbi.nlm.nih.gov/pubmed; searching years 1964–1st Oct 2012) displayed 3800 results. From 1964 to 1975, use of the terms “drug” and “hapten” together increased rapidly to approximately 90 outputs per annum. From 1975 onward, use of the two search terms has remained fairly constant (60–121 publications annually), and hapten theory remains the most widely accepted mechanism by which drugs generate antigens to prime naive T cells. Although intrinsic reactivity is designed out of most drugs during the drug development process, the β-lactam class of antibiotic represents an important exception. They bind irreversibly to bacterial penicillin binding proteins (proteins involved in the synthesis of peptidoglycan) to exert their pharmacological effect and at least have the potential to modify other proteins in the same way to generate T-cell antigens. β-Lactam antibiotics are a common cause of both immediate (IgE mediated) and delayed type (T-cell mediated) IDRs. For protein conjugate formation, the β-lactam ring is targeted by nucleophilic lysine residues. Nucleophilic attack leads to ring opening and binding of the penicilloyl group (Batchelor et al., 1965). The penicilloyl antigen can also be formed through binding of the reactive degradation product penicillenic acid (Levine, 1960). Furthermore, drug-protein antigens derive from spontaneous conversion of β-lactam antibiotics into penicilloic acid and penicilloate (Levine and Redmond, 1969). Using novel mass spectrometric methods, we recently characterized β-lactam-albumin conjugates in patient plasma and defined the profile of drug-protein conjugation at specific lysine residues with respect to dose and incubation time (Meng et al., 2011; Whitaker et al., 2011). Furthermore, using piperacillin-induced immune reactions in patients with cystic fibrosis as a model to study antigenicity, we characterized for the first time the minimum levels of modification associated with the stimulation of a clinically relevant drug-specific T-cell response. Immunochemical methods using a specific anti-drug antibody to visualize drug-protein adducts revealed that albumin is the only detectable protein modified with the drug in culture. Selective modification of Lys541 was observed at low piperacillin concentrations, whereas at higher concentrations up to 13 lysine residues were modified, four of which (Lys190, 195, 432, and 541) were detected in patients’ plasma (Whitaker et al., 2011; El-Ghaiesh et al., 2012). These data are in line with early studies that show albumin conjugates account for over 90% of covalently bound penicilloyl groups in serum (Lafaye and Lapresle, 1988). A synthetic piperacillin-albumin conjugate mirroring that seen in patients was generated and shown to stimulate lymphocytes and 100% of piperacillin-responsive T-cell clones to proliferate and secrete cytokines. Collectively, these data are consistent with the previous reports showing that penicillin-albumin constructs can stimulate T cells (Brander et al., 1995), but crucially here relate to antigens that are formed under physiologic conditions. The T-cell response to β-lactam albumin conjugates is inhibited when antigen processing is blocked, indicating the antigenic peptides are liberated from the modified protein. Using a strategy similar to that described above with DNCB, Weltzien and co-workers generated designer MHC binding peptides modified with penicillin to show that hapten-modified peptides are recognized as antigenic determinants by T cells (Padovan et al., 1997). Interestingly, β-lactam antibiotics also directly modify MHC and/or embedded MHC binding peptides to stimulate drug responsive clones via a pathway that avoids the need for protein processing. It is possible that this pathway of drug-specific T-cell activation is an in vitro artifact mimicking the action of processed hapten-modified peptides. Importantly, several groups have used β-lactam albumin conjugates to detect anti-drug antibodies in tolerant and allergic patients (de Haan et al., 1986; Christie et al., 1988; Torres et al., 1997). Thus, hapten theory is thought to accurately describe the drug-specific activation of B cells that recognize protein antigens directly through their B cell receptor.
It is interesting to consider whether the targeting of specific organs by β-lactam antibiotics might relate to the distribution of protein binding in vivo. In this respect, we recently compared the clinical and chemical characteristics of two very different forms of immunologic drug reaction: piperacillin-induced skin rash and flucloxacillin-induced liver injury (Fig. 3). Drug-responsive T cells are readily detectable in patients with both forms of immunologic reaction. Piperacillin- and flucloxacillin-specific T cells are, for the most part, CD4+ and CD8+, respectively (El-Ghaiesh et al., 2012; Monshi et al., 2013), and can be activated via a hapten mechanism involving protein binding and processing of the derived conjugate. Both drugs bind readily to selective lysine residues on human serum albumin in exposed human subjects (Jenkins et al., 2009; Whitaker et al., 2011), and similar exogenous protein adducts are thought to be important sources of antigen for CD4+ T cells. This, however, does not explain the predominant CD8+ T-cell response observed in patients with flucloxacillin-induced liver injury. One possible explanation may reside within the hepatocyte, a liver cell that synthesizes proteins including albumin. Thus, it is possible that unlike other β-lactam antibiotics, flucloxacillin accumulates and binds to intracellular albumin in hepatocytes, generating a protein conjugate that will liberate peptides expressed on the surface of hepatocytes by MHC class I molecules and ultimately provide a pathway for flucloxacillin-responsive T cells to target liver. In support of this theory, Carey and van Pelt (2005) found that flucloxacillin binds in a highly selective fashion to liver protein in rats. Fractionation of liver cytosol and analysis of binding using an anti-drug antibody revealed the modification of a single protein with a molecular weight comparable with albumin.

Clinical and immunologic features of two forms of β-lactam hypersensitivity reaction.
D. The Antigenicity and Immunogenicity of Drugs that Acquire Protein Reactivity through Metabolism
Formation of reactive species through metabolic activation (bioactivation) is implicated in many forms of IDR. If inadequately detoxified the reactive species has the potential to (1) bind to protein as a hapten, generating antigenic determinants for the adaptive immune system, and (2) stimulate stress-related signaling pathways and activate the innate immune response. The human safety risks posed by reactive metabolites and how formation of reactive metabolites affects the drug development process are the subject of a recent review (Park et al., 2011). This article, which summarizes the views of industrial and academic researchers in the field, states from the outset that although many drugs associated with a high incidence of IDRs have been shown to form reactive metabolites, there is no simple correlation between drug bioactivation in vitro or in patients and the development of reactions in the clinic. Furthermore, the authors emphasize that (1) covalent binding should be regarded as a marker of metabolic activation, (2) not all IDRs involve the formation of reactive metabolites (this is discussed in detail below), and (3) we do not have the tools available to define precisely the role of reactive metabolites in IDRs. With this in mind, the following section reviews the evidence available that supports a role for metabolism in drug-induced immune hepatitis and sulfamethoxazole (SMX)-induced skin reactions.
1. Drug-Induced Immune Hepatitis.
Hepatic protein adducts have been detected in patients with liver injury associated with exposure to drugs such as diclofenac, halothane, tienilic acid, and SMX (Pumford et al., 1993; Aithal et al., 2004; Cribb et al., 1996; Eliasson and Kenna, 1996; Robin et al., 1996), which demonstrates that metabolic activation leads to protein binding in the target organ. For each drug, circulating IgG antibodies that bind to drug (metabolite)-modified hepatic protein are detectable, which confirms that adduct formation results in a drug-specific immune response (Eliasson and Kenna, 1996; Lecoeur et al., 1996; Bedard et al., 2000; Aithal et al., 2004). For halothane and tienilic acid, antibody binding proteins have been identified as the cytochrome P450 enzymes involved in the drug’s metabolism, indicating that reactive metabolites interact with proteins in the vicinity of where they are formed. The role of bioactivation in the hepatic injury associated with halothane is also illustrated by a consideration of the relationship between the in vivo metabolism of general anesthetics and the observed incidence of liver injury in humans. Halothane is metabolized by CYP2E1 to yield trifluoroacetyl chloride, which binds covalently to proteins. The level of metabolites detected in human urine is around 10-fold lower for the drugs enflurane and isoflurane, and these drugs are associated with only rare cases of hepatic injury (Park et al., 1998).
2. Sulfamethoxazole.
It is difficult to comprehend how liver-derived metabolites participate in extrahepatic reactions because they are unlikely to escape the liver’s detoxification mechanisms, circulate around the body, and target selective tissue proteins. In the following discussion we use the drug allergen SMX to describe whether cell/tissue-selective metabolism generates reactive metabolites in sufficient quantities and also in the correct location to activate the innate and adaptive immune response and discuss the alternative P-I (pharmacological interaction of drugs with immunologic receptors) concept, which hypothesizes that drugs bind directly to MHC molecules through a readily reversible (pharmacological) interaction to activate T cells.
Most SMX is metabolized by hepatic N-acetyltransferase enzymes to an acetylated derivative that is readily eliminated from the body. However, a small quantity of SMX is converted to a hydroxylamine intermediate, a reaction catalyzed by CYP2C9 (Cribb et al., 1995). SMX hydroxylamine is not protein reactive (Naisbitt et al., 2001; Castrejon et al., 2010b); it is sufficiently stable to circulate in the body and is excreted unchanged in urine (Cribb and Spielberg, 1992; van der Ven et al., 1994b; Gill et al., 1996), which suggests that most tissues are exposed to the hydroxylamine metabolite after a therapeutic dose. NADH cytochrome b5 reductase and cytochrome b5 contribute toward the enzymatic reduction of the hydroxylamine (Kurian et al., 2004; Sacco and Trepanier, 2010), potentially restricting the formation of sulfonamide-derived protein adducts and direct sulfonamide toxicity. However, oxidation of SMX hydroxylamine occurs spontaneously, generating nitroso SMX, which has been shown to modify selective cysteine residues expressed on both cellular and extracellular protein (Naisbitt et al., 1999, 2001, 2002; Manchanda et al., 2002; Summan and Cribb, 2002; Callan et al., 2009; Eyanagi et al., 2012 ). Modification of cell surface proteins on immune cells occurs rapidly; protein conjugates are then internalized via caveolae-dependent endocytosis (Manchanda et al., 2002; Elsheikh et al., 2010).
It is possible that metabolic intermediates (i.e., the hydroxylamine) transport haptens around the body in an inactive form, with the reactive hapten only being released locally under conditions of oxidative stress. This scenario might at least partly explain why (1) genetic polymorphisms in hepatic metabolizing enzymes are not major predisposing factors (Pirmohamed et al., 2000; Alfirevic et al., 2003; Wolkenstein et al., 2005) and (2) sulfonamide reactions are detected in a higher number of subjects with AIDS and cystic fibrosis (van der Ven et al., 1994a; Lavergne et al., 2010) where oxidative stress plays an important role in the disease pathogenesis (van der Ven and Boers, 1997; Walmsley et al., 1997; Lezo et al., 2013). Nitroso SMX stimulates innate immunity through the activation of dendritic cells (Sanderson et al., 2007) and adaptive immunity through the generation of protein adducts. In rodent models, topical and systemic exposure to nitroso SMX primes naive CD4+ and CD8+ T cells. The T-cell response is dependent on protein processing, and liberated peptide antigens are presented to specific T-cell receptors in the context of MHC molecules (Choquet-Kastylevsky et al., 2001; Naisbitt et al., 2001, 2002; Farrell et al., 2003; Hopkins et al., 2005; Castrejon et al., 2010b). In contrast, administration of SMX does not activate immune cells. In vitro studies with peripheral blood mononuclear cells from drug naive subjects show the activation of T cells against nitroso SMX in almost 100% of individuals (Engler et al., 2004). We have recently shown that nitroso SMX also primes naive T cells, which change to a memory phenotype after drug stimulation (Faulkner et al., 2012). The reason why nitroso SMX is able to prime immune cells so readily has not been defined, but it might relate to its ability to modify cysteine residues on multiple proteins, hence generating many hapten-modified peptides with binding affinity for multiple MHC molecules. This in turn might explain why strong associations between expression of specific HLA alleles and SMX reactions have not been identified (Lonjou et al., 2008; Alfirevic et al., 2009). Independent research groups have shown that skin- and blood-derived T cells from all SMX hypersensitive patients are activated by nitroso SMX, which indicates that drug metabolism and the formation of hapten-modified proteins are relevant in the clinical setting (Schnyder et al., 2000; Burkhart et al., 2001; Nassif et al., 2004; Elsheikh et al., 2011 ). Furthermore, recent studies show that nitroso SMX stimulates the majority (~90%) of drug responsive T-cell clones isolated from patients with a history of SMX-induced skin injury (Castrejon et al., 2010a).
It is possible that T-cell stimulatory drug-protein adducts are formed, not as a consequence of hepatic metabolism, but by localized metabolic transformations in target tissue (e.g., skin). If metabolic activation takes place in the target tissue and adducts are restricted to the site of formation, then tissue-specific metabolism might contribute toward the tissue selectivity of certain IDRs. Skin cells are known to express patterns of CYP enzymes that differ from those seen in liver (Baron et al., 2008), and although metabolism in the skin represents only a fraction of that seen is hepatic tissue, the metabolites formed might be involved in the localized generation of antigens for T cells or direct toxicity that promotes the innate immune system. In terms of SMX-induced skin injury, Svensson and co-workers conducted a series of elegant experiments to demonstrate that sulfonamides are metabolized by flavin-containing monooxygenase 3 and peroxidases expressed in human epidermal keratinocytes into metabolites that bind covalently to cellular protein (Reilly et al., 2000; Vyas et al., 2006a,b). Exposure of keratinocytes to SMX promoted the release of proinflammatory cytokines and increased expression of heat shock protein 70 (Khan et al., 2007). We have shown that (1) SMX metabolism and protein adduct formation above a threshold stimulates cell death and (2) drug-metabolite modified necrotic cells provide a powerful activation signal to dendritic cells (Naisbitt et al., 2002; Elsheikh et al., 2010). Thus, metabolism of SMX might indirectly activate cutaneous dendritic cells and support the presentation of skin-derived drug-protein adducts.
Using a multidisciplinary approach with samples from animal and human experimental systems, we have also been able to characterize SMX metabolism-derived protein adducts in immune cells and define the relationship between adduct formation, costimulatory signaling, and stimulation of a T-cell response (Sanderson et al., 2007; Lavergne et al., 2009, 2010; Elsheikh et al., 2010, 2011). The presence of various pathologic factors [e.g., lipopolysaccharide (LPS), viral proteins, and cytokines] increased the formation of protein adducts in SMX-treated dendritic cells and reduced the time needed to detect adducts. Interestingly, in contrast to nitroso SMX, which forms cell surface adducts, SMX adducts formed through metabolism are detected intracellularly. SMX metabolism and the subsequent irreversible modification of intracellular protein is associated with partial maturation of dendritic cells, detected by increased expression of the activation marker CD40, and by the activation of nitroso SMX-responsive clones from hypersensitive patients. When SMX-treated mouse dendritic cells are adoptively transferred to drug naive recipients, antigen-specific T-cell priming is readily detectable; however, the T cells are nitroso SMX specific. The only possible explanation for these data are that peptides derived from intracellular drug-protein adducts, formed through intracellular metabolism, are important drug-derived antigens that prime naive T cells in vivo.
Collectively, these data show that haptens generated through drug metabolism bind to protein, and the derived adducts are involved in the immune response detected in certain patients with an IDR. The lack of relevant drug-derived antigens (reactive metabolites, protein, and peptide conjugates) and analytical methods to quantify low levels of metabolism in target tissue/immune cells, for the most part, prevent a more global analysis of the hapten theory.
E. The Antigenicity and Immunogenicity of Drugs That Do Not Form Covalent Bonds with Protein
The detection of T-cell responses against drugs bound noncovalently to MHC molecules has added a new layer of complexity. The following section summarizes evidence originating from the influential studies of Pichler and colleagues (Adam et al., 2011) that support the pharmacological activation of T cells by drugs. Throughout this discussion, readers must be aware that, as yet, it is difficult to define the contribution of hapten/pharmacological pathways of T-cell activation in patients, and indeed whether the noncovalently bound parent drug mimics the action of hapten-peptide conjugates by surmounting the binding energy needed for T-cell activation in an in vitro model. In this respect, Chen et al. (2009) have shown in an animal model that once an immune response is induced by a reactive metabolite, the response can spread to recognize the parent drug (discussed in detail in Section IV.A).
The P-I concept states that “a drug is able to stimulate T cells directly without forming a hapten, in a HLA-dependent manner” (Adam et al., 2011; Yun et al., 2012). In vitro studies using peripheral blood lymphocytes and T-cell clones from patients with a history of an IDR provide strong evidence to support this concept. First, drugs that do not themselves bind covalently to nucleophilic amino acids stimulate T cells via their T-cell receptor in an MHC restricted manner; second, fixation of antigen presenting cells, which blocks protein processing, does not prevent the drug-specific activation of certain clones; third, the kinetics of drug-specific T-cell receptor triggering and calcium signaling are too quick to allow protein processing; and finally, the removal of soluble drug through repeated washing of drug-treated antigen presenting cells prevents MHC-restricted drug presentation and T-cell activation. These findings have been replicated by several independent research groups using an increasing number of drugs (Brander et al., 1995; Schnyder et al., 1997; Hashizume et al., 2002; Naisbitt et al., 2003b, 2005; Nassif et al., 2004; Wu et al., 2006; Keller et al., 2010 ).
More controversially, the P-I concept proposes that drug-responsive T cells derive from the memory pool, and clinical signs of an IDR develop even in the absence of an innate immune response (the activation of monocytes/macrophages and dendritic cells and inflammatory cytokine release) (Pichler, 2005). It is of course possible that drug (metabolite) exposure is not the major contributory factor that leads to the triggering of innate signaling pathways in patients with an IDR; however, the increased frequency of reactions in patients with known “danger signals” (i.e., HIV infection, cystic fibrosis) argues that the innate immune system participates in some way in the disease pathogenesis. Furthermore, through the development of in vitro T-cell priming methods using peripheral blood mononuclear cells from healthy drug-naive subjects, it is possible to demonstrate that drugs (e.g., sulfamethoxazole metabolites, flucloxacillin, carbamazepine) stimulate naive CD4+ and/or CD8+ T cells (Martin et al., 2010; Faulkner et al., 2012). T cells that divide in the presence of the drug-derived antigen change from a naive (CD45RA+) to a memory phenotype (CD45RO+) and secrete cytokines and cytolytic molecules when activated. Importantly, T-cell priming is dependent on the presence of dendritic cells and the presentation of the drug-derived antigen in a microenvironment rich in costimulatory signals. Blockade of the interaction of inhibitory B7 family ligands expressed on dendritic cells with their T-cell counterparts increases the quality of the drug-specific T-cell response (unpublished data), which shows that innate signaling is involved in priming of the naive T cells. These studies show for the first time that drug-derived antigens drive naive T cells to an antigen experienced “memory” phenotype, which express T-cell receptors in oligomeric complexes on their surface that account for their increased sensitivity to antigen and ability to respond in the absence of costimulatory signals (Kumar et al., 2011).
F. Viral Infection and IDRs
In certain patients, the onset of an IDR coincides with an acute viral infection. As mentioned above, the viral infection may activate the innate immune system through the provision of danger signals, thus priming the immune system against the drug-derived antigen. However, in patients with DRESS, there seems to be a more intimate relationship between reactivation of human herpes viruses and the development of clinical features of the drug reaction. Clinical studies pioneered by Hashimoto, Shiohara, and Kano (Tohyama et al., 1998; Kano et al., 2006; Shiohara et al., 2007; Shiohara and Kano, 2007) indicate that several features of the reaction [e.g., the deterioration in clinical status several days after drug withdrawal and sequential appearance of clinical symptoms (fever, hepatitis, eosinophilia)] actually correlate with the reactivation of herpes viruses for 2–3 weeks after the reaction onset. These data raise the intriguing possibility that virus-specific CD8+ T cells are at least partly responsible for the development of the IDR. In 2005, Hashizume and Takigawa (2005) reported a case of tribenoside-induced hypersensitivity syndrome associated with cytomegalovirus reactivation and found that skin-infiltrating CD4+ T cells were drug-reactive, whereas CD8+ T cells were activated by cytomegalovirus. Moreover, Shiohara and Kano (Shiohara and Kano, 2007; Shiohara et al., 2012) suggest that the anti-viral immune response might be activated by cross-reacting drug-derived antigens. In this respect, Picard et al. (2010) conducted a detailed analysis of the anti-viral T-cell response in 40 patients with DRESS. Epstein-Barr virus or human herpes virus reactivation was detected in 76% of patients, and almost half of expanded CD8+ T cells were found to be activated with known viral peptide epitopes. The authors also demonstrated that drugs associated with DRESS increased production of Epstein-Barr virus in transformed B cells and used this data to argue that drug-dependent virus production triggers the clinical features associated with DRESS. Although this study clearly outlines a role for virus-specific T cells in DRESS, new studies are needed to dissect the role drug- and virus-specific T cells play in the different features of the disease.
G. HLA Class I-Associated IDRs
The discovery of surprisingly strong associations between expression of particular HLA alleles and susceptibility to different forms of IDRs has changed the way in which researchers in the field define immunologic reactions; some reactions are no longer completely unpredictable. The association between abacavir hypersensitivity and HLA-B*57:01 has become a paradigm for mechanistic studies to characterize pathways of drug-specific T-cell activation and the relationship between drug exposure and the development of an IDR. Genetic screening in the clinic for expression of the HLA risk allele has resulted in the effective elimination of this form of iatrogenic disease, and as such, abacavir hypersensitivity represents a major pharmacogenomics success story and one of the first examples of a personalized medicine (Phillips and Mallal, 2009).
Abacavir is a nucleoside analog reverse transcriptase inhibitor that is used for the treatment of HIV infections. Although highly effective when used in combination with other antiretroviral drugs, abacavir is associated with the development of a hypersensitivity syndrome, characterized by fever, gastrointestinal symptoms, and internal organ involvement in 5–8% of patients (skin reactions develop in approximately 70% of patients with hypersensitivity). Two independent studies in 2002 identified the association between HLA-B*57:01 and abacavir hypersensitivity (Hetherington et al., 2002; Mallal et al., 2002). The subsequent program of research demonstrated that abacavir stimulates (1) an innate immune response activating antigen-presenting cells via the endogenous HSP70-mediated Toll-like receptor pathway (Martin et al., 2007), (2) an adaptive immune response activating patient peripheral blood mononuclear cells to secrete IFN-γ and TNF-α (Almeida et al., 2008), and (3) a positive patch test, which is effective for the diagnosis of immunologically mediated abacavir reactions (Phillips et al., 2005). A randomized double-blind controlled trial that enrolled approximately 2000 patients and conducted real-time (excluding abacavir from subjects positive for B*57:01) and retrospective HLA-B*57:01 screening alongside clinical monitoring and patch testing to diagnose abacavir hypersensitivity [PREDICT-1 (Mallal et al., 2008)] demonstrated a 100% negative predictive value of HLA-B*57:01 for abacavir hypersensitivity and thus provided the evidence needed for HLA-B*57:01 screening to avoid abacavir hypersensitivity. Interestingly, almost 50% of patients positive for HLA-B*57:01 are able to tolerate abacavir. Thus, factors, discussed in greater detail below, in addition to drug exposure and HLA-B*57:01, are required for the development of an IDR.
There are now around 40 HLA risk allele-linked IDRs or drug-induced syndromes (Phillips et al., 2011; Pavlos et al., 2012). If one accepts that the drug-derived antigen binds selectively to the MHC molecule and that this is an important step in the activation of T cells that participate in the IDR, then one would expect HLA risk allele-linked reactions to be drug specific; indeed, this generally seems to be the case. However, HLA-linked reactions are also disease phenotype specific and dependent on the study population. The best example of a disease phenotype and study population-specific HLA risk allele-linked reaction to consider is HLA-B*15:02 and Stevens-Johnson syndrome triggered by the anticonvulsant carbamazepine. Carbamazepine exposure is associated with a variety of immune-mediated reactions including, but not limited to, mild/moderate maculopapular skin eruptions; hypersensitivity syndromes presenting with rash, fever, and internal organ involvement; and severe skin reactions (e.g., Stevens-Johnson syndrome/toxic epidermal necrolysis). Chen et al. (2011) identified a strong association between HLA-B*15:02 and carbamazepine-induced Stevens-Johnson syndrome. The initial study found that HLA-B*15:02 was present in 100% (44/44) of patients with Stevens-Johnson syndrome but only 3% of tolerant and 8.6% of the general population. More recently, 4877 subjects were genotyped to determine whether they expressed HLA-B*15:02 prior to carbamazepine exposure. The data generated found that genetic testing significantly decreased the incidence of carbamazepine-induced Stevens-Johnson syndrome (Chen et al., 2011). Carbamazepine was subsequently found to interact with HLA-B*15:02 and other HLA-B75 family members to activate drug-responsive CD8+ T cells (Wei et al., 2012) that release granulysin, a key mediator in keratinocyte death in patients with Stevens-Johnson syndrome (Chung et al., 2008). These data effectively link the HLA risk allele to the disease pathogenesis; however, the HLA association is disease specific (i.e., HLA-B*15:02 is not associated with other forms of carbamazepine adverse reactions), and it is restricted to patients of Asian ancestry (Lonjou et al., 2006, 2008).
To evaluate the global applicability of these findings with abacavir and carbamazepine, one needs to consider earlier studies characterizing the nature of the induced drug-specific T-cell response in hypersensitive patients. Drugs of different chemical class, including carbamazepine, activate CD4+ and CD8+ T cells from patients with Stevens-Johnson syndrome, hypersensitivity syndromes, and maculopapular eruptions (Mauri-Hellweg et al., 1995; Schnyder et al., 1997, 2000; Hashizume et al., 2002; Naisbitt et al., 2003a,b; Lerch et al., 2007; Wu et al., 2007). In fact, in several studies, activation of CD4+ and CD8+ T cells from the same patient was found to be HLA class II and I restricted, respectively, indicating that the drug-derived antigen interacts with numerous HLA molecules. Thus, it seems that for many of the HLA risk allele-linked IDRs, the immune response will likely be much more heterogeneous with many HLA molecules displaying the antigen. Indeed, this seems to be the case in patients with flucloxacillin-induced HLA-B*57:01 associated liver injury. We have shown that cytotoxic CD8+ T cells are preferentially activated with flucloxacillin, and the response is HLA-B*57:01 restricted. However, HLA-class II restricted CD4+ T cells were also readily detectable (Monshi et al., 2013).
It is possible that future research will reveal a similar picture with carbamazepine reactions in Caucasian patients. HLA-A*31:01 has been shown to be associated with a full range of reactions; the presence of the allele increases the risk of tissue injury from 5.0% to 26.0%, whereas its absence reduces the risk to 3.8% (McCormack et al., 2011). The association suggests that carbamazepine binds selectively to the class I molecule to activate CD8+ T cells. Although this may be the case, one should note that (1) the majority of patients with carbamazepine-induced idiosyncratic reactions do not express HLA-A*31:01 (McCormack et al., 2011), (2) HLA class I- and class II-restricted responses are detectable in hypersensitive patients (Mauri-Hellweg et al., 1995; Naisbitt et al., 2003a; Wu et al., 2006, 2007), and (3) T-cell proliferative responses are readily detectable in hypersensitive patients with and without the HLA risk allele (Niihara et al., 2012). Thus, the relationship between the expression of the HLA risk allele, activation of HLA-restricted T cells and the disease is far from clear. Interestingly, HLA-A*31:01 is not a predisposing factor for lamotrigine- and phenytoin-induced skin injury (McCormack et al., 2012), and peripheral blood mononuclear cells and T-cell clones are not stimulated with the related drugs (Naisbitt et al., 2003a,b).
H. Lessons to be Learned from the Study of HLA Class I-Associated IDRs
Characterization of highly HLA class I-restricted T-cell responses against abacavir and carbamazepine prompted researchers to study in detail the nature of drug MHC binding interactions. Using abacavir-responsive CD8+ T-cell clones isolated from drug-naive subjects expressing HLA-B*57:01, Adam et al. (2012) demonstrated that T cells could be activated with the drug bound directly to surface MHC molecules. Analysis of calcium influx as a marker of T-cell activation revealed that the drug-specific T-cell response was rapid, and the ability of individual clones to react was determined by T-cell receptor avidity. Increasing the abacavir concentration accelerated the activation kinetics. Interestingly, antigen presenting cells cultured overnight with abacavir, prior to repeated washing to remove soluble drug, additionally activated the clones. The time dependency of the drug antigen presenting cell pulse is intriguing as we recently showed that (1) abacavir accumulates rapidly in antigen presenting cells and (2) intracellular concentrations remain constant for up to 24 hours (unpublished data). Furthermore, 24 hours is the time needed to detect high levels of abacavir oxidative metabolites in antigen presenting cells.
A novel pathway of abacavir-specific activation of T cells has been proposed by several groups (Illing et al., 2012; Norcross et al., 2012; Ostrov et al., 2012). They have shown that abacavir binds directly to endogenous MHC molecules independent of drug metabolism and hapten formation. Abacavir binding alters the peptides that can be accommodated within the binding groove, and as such, novel peptide sequences are loaded onto HLA-B*57:01. The MHC abacavir peptide complex is then transported to the cell surface and displayed to T cells. The authors suggest that the T-cell response might be triggered by the novel self peptides, but their origin, structure, and indeed the role of the drug in the T-cell response is yet to be completely resolved. Norcross et al. (2012) demonstrated that abacavir alters the binding of fluorescently labeled self peptides to HLA-B*57:01 and adopted a mass spectrometry approach to show that abacavir alters the repertoire of peptides that bind to HLA-B*57:01. Interesting, flucloxacillin (associated with HLA-B*57:01-restricted DILI) did not have this effect. Illing et al. (2012) conducted a range of functional studies to show the importance of specific amino acid residues located within the peptide binding groove of the HLA molecules for abacavir binding and used mass spectrometry to show the characteristics of peptides eluted from HLA-B*57:01 was altered in the presence of the drug. Transporters associated with antigen processing-deficient antigen presenting cells were used to show that protein processing was important for activating abacavir-specific T cells. A key aspect of these studies was the elucidation of a crystal structure of a self peptide bound to HLA-B*57:01 in the presence of abacavir. Similar results were obtained with carbamazepine and cells expressing HLA-B*15:02. Ostrov et al. (2012) used computer modeling techniques based on peptide libraries to screen HLA-B*57:01 binding peptides and predict how abacavir could influence peptide binding. The modeling studies suggested a potential binding site for abacavir within the F pocket of HLA-B*57:01 close to Ser116. A crystal structure of HLA-B*57:01, peptide, and abacavir was solved, and in agreement with Illing et al. (2012), abacavir was found to make extensive contacts with HLA-B*57:01 but fewer contacts with the peptide. Blood lymphocytes from abacavir hypersensitive patients were used to investigate T-cell responses with peptide libraries and/or abacavir.
As we move forward, it is important to investigate whether (1) these finding are relevant in other forms of IDR and (2) the majority of drug antigen-specific T cells isolated from patients can be activated via hapten, P-I, and altered peptide pathways under the correct experimental conditions. In respect to the latter, T-cell receptors will interact with irreversibly and reversibly bound drug MHC peptide complexes in a very similar fashion. Hapten binding to protein might also alter the natural processing of endogenous protein, meaning that the prediction of peptide sequences displayed by MHC molecules would be difficult.
I. HLA Class II-Associated IDRs
Our discussion on HLA-associated IDRs has thus far focused around HLA class I alleles, because a functional role has been demonstrated for the receptors. Interestingly, a number of HLA class II associations have also been described, most commonly with drug-induced liver injury [e.g., ximelagatran (Kindmark et al., 2008), amoxicillin-clavulanate (Lucena et al., 2011), lapatinib (Spraggs et al., 2011), and lumiracoxib (Singer et al., 2010)], which suggests that drug-specific CD4+ T-cell responses underlie the disease pathogenesis. For ximelagatran, lymphocyte proliferative responses have been detected in a small number of patients with liver injury (Kindmark et al., 2008), but the nature of the T-cell response and the MHC restriction has not been studied. Thus, a causal role for the reported HLA class II associations has still to be defined. It is possible that other, as yet undetermined HLA alleles that reside on an extended haplotype are involved in the disease. To explore possible haplotype associations, we have recently used an in silico approach to analyze data stored in our database and in public repositories. We found that (1) different HLA alleles associated with drug-induced liver injury are in strong linkage disequilibrium in populations of Caucasian and non-Caucasian ancestry, and (2) HLA risk alleles associated with unrelated drugs, such as flucloxacillin, ximelagatran, lapatinib, and antituberculosis drugs, reside on two main haplotypes (Alfirevic et al., 2012). Investigations are now clearly needed with lymphocytes from HLA-typed patients to investigate the immune mechanism(s) in different forms of HLA class II-associated IDRs.
J. Additional Factors Involved in Susceptibility to HLA-Associated IDRs
With the exception of abacavir hypersensitivity reactions, the majority of patients carrying HLA risk alleles do not develop an IDR when exposed to the drug. Thus, it is likely that additional, for the most part unresolved, patient- and drug-dependent factors contribute toward individual susceptibility in subjects carrying HLA risk alleles. The following section summarizes three recently described areas of research.
In view of the fact that drugs show exquisite selectivity in their binding to MHC molecules, it seems rational to anticipate that they will also interact with and stimulate a limited repertoire of T-cell receptors. The T-cell receptor is a heterodimer composed of alpha and beta chains. Analysis of Vbeta chains is regularly used to analyze the expansion of specific T-cell clonotypes. Early studies characterizing drug-responsive T-cell clones describe the preferential usage of a limited repertoire of Vbeta chains (Hashizume et al., 2002; Sieben et al., 2002; Naisbitt et al., 2003b). Until recently, however, no clear association between Vbeta usage and drug class or disease was identified. Innovative studies by Ko et al. (2011) analyzed the carbamazepine-specific T-cell receptor repertoire in HLA-B*15:02 positive patients with carbamazepine-induced Stevens-Johnson syndrome by spectratyping the complementarity determining region 3 (CDR3; the most variable region in the T-cell receptor). They found a restricted and common T-cell receptor usage of CD8+ T cells in patients with Stevens-Johnson syndrome. The clonotype Vbeta-11-ISGSY was present in 16 out of 19 patients with Stevens-Johnson syndrome and absent in all drug-tolerant patients. Might a restricted T-cell receptor usage be important in other forms of immune-mediated IDRs? The obvious next step: exploration of T-cell receptor usage in the HLA-B*57:01 abacavir hypersensitivity model, has recently been published (Illing et al., 2012). A polyclonal T-cell receptor usage was noted in seven subjects, indicating that abacavir-responsive T-cell clones express a variety of different T-cell receptors.
Mallal and co-workers (Ostrov et al., 2012) point out that the altered peptide repertoire model of T-cell activation discovered with abacavir might be more accurately described as drug exposure-dependent heterologous immunity against preexisting class I-restricted effector memory T-cell responses to prevalent viral infections. They also postulate that the peptides recognized by drug-responsive T cells might derive from proteins that are genetically polymorphic and only present in a portion of individuals. In this respect, abacavir readily activates T cells from peripheral blood of drug-naive human subjects with a memory (CD8+CD45RO+) phenotype. However, whether this model is widely applicable to other HLA-associated IDRs is still open to question.
Regulatory T cells (Tregs) are believed to regulate allergic disease; thus, it is possible that they also participate in the regulation of immunologic IDRs. One report has shown an increased frequency of Tregs in patients at the acute phase of mild reactions (Takahashi et al., 2009), raising the possibility that the development of clinical signs of an IDR is governed by altered immune regulation. However, at present, it is not known whether Tregs prevent effector T-cell responses in tolerant patients. A recent report indicates that PD-L1 expressed on dendritic cells plays a critical role in allergic reactions to nickel by controlling the nature of the antigen-specific T-cell response (Hitzler et al., 2012). Thus, regulatory receptor ligand binding interactions between dendritic cells and naive T cells likely also play an important role in HLA-restricted drug-specific T-cell priming.
K. Mitochondrial Injury Hypothesis
Many drug (metabolites) that cause adverse reactions in patients are thought to target mitochondria. Mitochondria are the main cellular producers of energy through fatty acid oxidation and ATP formation. Drugs cause mitochondrial toxicity through a variety of mechanisms, including direct inhibition of mitochondrial respiration or mitochondrial β-oxidation of fatty acids. Alternatively drugs can target mitochondrial DNA, mitochondrial transcripts, or mitochondrial protein synthesis to indirectly inhibit mitochondrial respiration. A detailed discussion of drug-dependent mitochondria toxicity is beyond the scope of our review [see Pessayre et al. (2012) for a detailed account of mitochondria biology and mechanisms of drug-specific mitochondria disruption]. We will simply highlight one area in which mitochondria damage might relate to the drug-specific immune response. Mitochondrial toxicity is known to release damage-associated molecular patterns into the extracellular environment. These structures activate the innate immune system through ligation of Toll-like receptor 9 (Zhang et al., 2010). Recently, systemic release of mitochondrial DNA has been shown in patients with drug toxicity (McGill et al., 2012). Thus, drug metabolite-mediated mitochondria damage potentially provides a link between drug exposure, innate signaling, and drug-specific T-cell priming.
IV. Animal Models
There are significant limitations to the studies that can be performed in humans, not least of which is that it is important to obtain samples before the onset of an IDR so that the events leading up to the IDR can be studied, but in general it is impossible to predict which patients will have an IDR. Animal models are essential for most areas of biomedical research because they permit control of the parameters that may be involved, and the study of IDRs is no exception. However, to be useful, the mechanism of the IDR in the animal model must be the same, or at least very similar to the IDR that occurs in humans. Most animal models to date represent acute toxicity and are unlikely to represent the mechanism of the IDR that occurs in humans. We have reviewed animal models of IDRs recently (Ng et al., 2012); a more limited discussion will be presented here along with some new data that are soon to be published.
A. Nevirapine-Induced Skin Rash in Female Brown Norway Rats
Nevirapine is a non-nucleoside reverse transcriptase inhibitor used for the treatment of HIV infections. Its use is limited by a relatively high incidence of skin rash, some of which are life threatening, and IDILI. It also causes a rash in female Brown Norway rats (Shenton et al., 2003). The rash is clearly immune mediated in rats, and the characteristics of the rash in the rat model are very similar to those in humans; therefore, it is likely that the mechanisms are also similar (Shenton et al., 2005). Specifically, it takes about 3 weeks of treatment before the onset of the rash in both humans and rats. In addition, the rash in rats is dependent on CD4+ T cells in the rat, and patients with a low CD4 T-cell count have a lower incidence of rash. Furthermore, incubation of lymphocytes from affected patients or rats produce IFN-γ (Keane et al., 2007; Chen et al., 2009). Although the rash can be induced in other strains of rats, the incidence is much lower, and it can only be induced in male rats by using a cotreatment with aminobenzotriazole. Nevirapine does not cause significant liver injury in rats.
We were able to demonstrate that most of the covalent binding of nevirapine in the liver is due to a quinone methide metabolite formed by oxidation of the methyl group (Sharma et al., 2012). A benzylic sulfate is responsible for covalent binding of nevirapine in the skin. It is formed by oxidation of the same methyl group to a benzylic alcohol in the liver followed by sulfation of the alcohol in the skin. Inhibition of sulfation in the liver by depletion of the cofactor, 3′-phosphoadenosine-5′-phosphosulfate with salicylamide decreased blood levels of the benzylic sulfate, but it did not prevent the rash or covalent binding of nevirapine in the skin (Sharma et al., 2013). In contrast, inhibition of sulfotransferase by topical administration of 1-phenyl-1-hexanol prevented covalent binding and the rash but only where it was applied (Sharma et al., 2013). We were not able to induce a rash in mice, and in contrast to experiments with rat and human skin, incubation of the benzylic alcohol with mouse skin and 3′-phosphoadenosine-5′-phosphosulfate did not lead to covalent binding (manuscript in preparation). These data provide conclusive evidence that it is the benzylic sulfate formed in the skin that is responsible for the skin rash. It would have been virtually impossible to determine what metabolic pathway is responsible for the rash with studies in humans. These pathways are summarized in Fig. 4.

The intermediate formed by oxidation of nevirapine partitions between loss of another hydrogen atom to form a quinone methide and oxygen rebound to form a benzylic alcohol. The quinone methide covalently binds in the liver, inactivates P450, and is presumably responsible for idiosyncratic liver injury. The benzylic alcohol travels to the skin where sulfotransferases in the epidermis form a sulfate conjugate that covalently binds and leads to a skin rash.
This model also made it possible to test the basis for the P-I hypothesis. The unstated assumption upon which the P-I hypothesis is based is that what T cells from patients with a history of an IDR respond to is what initially induced the immune response. However, in the nevirapine model, T cells from affected animals responded better to nevirapine than they did to the benzylic alcohol metabolite, although we can be sure that it was the sulfate of the benzylic alcohol that induced the skin rash (Chen et al., 2009). Thus it appears that once an immune response is initiated by a reactive metabolite, the immune response can spread to recognize the parent drug. It would not have been possible to do these experiments in humans.
This is one model that does appear to represent the same mechanism as the IDR in humans, and it will continue to be used to study how the reactive sulfate metabolite of nevirapine induces an immune response that leads to a skin rash.
B. Penicillamine-Induced Autoimmunity in Brown Norway Rats
Penicillamine is used to treat Wilson’s disease; however, it causes a variety of autoimmune syndromes in humans (Chalmers et al., 1982). It also has efficacy for the treatment of other diseases such as rheumatoid arthritis, but the high incidence of adverse reactions limits its use. It also causes a lupus-like syndrome in Brown Norway rats with an incidence of ~50%, but other strains are resistant (Donker et al., 1984; Tournade et al., 1990). It is interesting that although Brown Norway rats are highly inbred and therefore genetically essentially identical, only ~50% of the animals develop autoimmunity. Although it takes about 3 weeks for the autoimmune syndrome to become clinically evident, 24 hours after the first dose of penicillamine, a spike in IL-6 predicts which animals will develop autoimmunity (Zhu et al., 2011). Th17 cells appear to be a key cell involved in the pathogenesis of the autoimmunity. It is also of note that penicillamine causes autoimmune syndromes although it is used for the treatment of autoimmune diseases such as rheumatoid arthritis.
C. Halothane-Induced Liver Injury
There have been many studies that have tried to reproduce halothane-induced liver injury in animals. Early studies used hypoxia to increase the toxicity of halothane in rats (McLain et al., 1979), but this is unlikely to represent the mechanism of halothane-induced IDILI in humans. Investigators were able to induce an immune response to halothane in Guinea pigs with mild liver injury, but the response decreased with repeated challenges (Furst and Gandolfi, 1997; Furst et al., 1997a,b). Intraperitoneal injection of halothane in oil induced liver injury in mice and generated an innate immune response (You et al., 2006). Mouse strain-, age-, and sex-dependent variations in the severity of this injury were observed, including female sex and older age (Cheng et al., 2009, 2010; You et al., 2010), which are analogous to the risk factors in humans. Moreover, this injury was increased by agents such as poly-IC that act through Toll-like receptors (Cheng et al., 2009). It was recently shown that injury in this model is associated with an infiltrate of eosinophils, which is also typical of clinical halothane-induced hepatitis, and depletion of eosinophils decreased the injury (Proctor et al., 2012). It is likely that this represents the initial innate immune response to halothane in humans, but severe idiosyncratic liver injury in humans also appears to involve an adaptive immune response that progresses over a period of days to weeks and can lead to liver failure.
D. Acetaminophen-Induced Liver Injury
Probably the most studied ADR is acetaminophen-induced liver injury. It is both clinically important and easy to reproduce in mice. It is caused by a reactive imidoquinone metabolite that appears to cause liver injury by damage to mitochondria. Instead of being mediated by the immune system, it appears that the innate immune system exerts a protective effect (Jaeschke et al., 2012). However, acetaminophen-induced liver injury is not idiosyncratic, and therefore this animal model is not a good model for IDRs.
E. Inflammagen Model
It has been postulated that IDRs are caused by a chance combination of an inflammatory stimulus and administration of a drug (Roth et al., 2003). This is based on an animal model in which the treatment of rats with a combination of ranitidine and LPS caused liver injury that did not occur with either agent alone at the dose administered (Luyendyk et al., 2003). This model was repeated with several other drugs. There are many features of this model that are not consistent with IDILI. It is an acute model, and IDILI almost always requires more than a week of treatment before the onset of the liver injury. If the delay were caused by the random occurrence of an inflammatory stimulus, the time to onset should also be random; however, although the time to onset can vary somewhat between individuals and with different drugs, it is not random. More important, the response to Toll-like receptor agonists such as LPS is very rapidly downregulated (Fan and Cook, 2004); if it were not, patients with inflammatory bowel disease would have a very high incidence of IDILI. In fact, serious IDILI leading to liver failure progresses over a period of a week or more, often after the drug has been discontinued, and this is incompatible with the inflammagen model. In addition, the pathology observed in the animal model is not typical for IDILI. Specifically the pathology in the animal model is dominated by neutrophils (Luyendyk et al., 2003), which is typical for LPS-induced liver injury. In contrast, the typical pathology in IDILI is dominated by lymphocytes, sometimes with eosinophils (Zimmerman, 1999). Given the disparity between this animal model and IDILI, it is very unlikely that the mechanisms are similar. This model also appears to be of limited practical value because ranitidine is a safe over-the-counter drug.
F. Mitochondrial Superoxide Dismutase-deficient Model of IDILI
A homozygous deficiency in mitochondrial superoxide dismutase (SOD2) is lethal in mice but the heterozygote, SOD2+/−, is phenotypically normal. It was observed that treatment of SOD2+/− animals with troglitazone induced mild liver injury that did not occur in the wild-type animals (Ong et al., 2007). This model is very attractive because, in contrast to most animal models, the onset of liver injury was delayed, much as it is in humans. However, although the laboratory that developed this model has found other drugs to cause enhanced liver injury in these animals, other laboratories have been unable to reproduce the troglitazone results (Fujimoto et al., 2009). Therefore, it is unclear whether this model represents the mechanism involved in troglitazone-induced IDILI or whether it can readily be reproduced.
G. IDRs in Pets, e.g., Sulfonamide-Induced Hypersensitivity in Dogs and Propylthiouracil-Induced Autoimmunity in Cats
Although most attempts to develop practical animal models of IDRs have been unsuccessful, it is clear that animals are susceptible to IDRs, and veterinarians often see examples of IDRs in pets. A good example is that the use of sulfonamide antibiotics in dogs is associated with a significant incidence of a hypersensitivity syndrome similar to that which occurs in humans (Trepanier, 2004). However, the incidence is only about 1%, and it is more frequent in large breed dogs. Another example is propylthiouracil-induced autoimmunity in cats; propylthiouracil can also cause a lupus-like syndrome in humans (Aucoin et al., 1985; Waldhauser and Uetrecht, 1996). Unfortunately, the low incidence of these IDRs and the species involved make them impractical for extensive mechanistic studies.
H. Toxic Epidermal Necrolysis Mouse Model Generated Using Blood Cells and Skin from Patients
Saito et al. (2013) recently established a humanized model of drug-induced toxic epidermal necrolysis through the intravenous injection of human peripheral blood mononuclear cells from a patient who had recovered from an IDR into immunocompromised mice and the grafting of human skin (Saito et al). After exposure to the causative drug, skin-grafted mice showed clear darkening of the grafted areas that was accompanied by the presence of dead keratinocytes. It should be noted that human cells also infiltrated the conjunctiva of grafted mice and the animals developed ocular damage. Additional studies are clearly needed to characterize the mechanism of tissue injury in mice and to explore how the model can be applied more generally to investigate the human disease mechanism.
I. Why Is it Difficult to Develop Valid Animal Models of IDRs?
We have made many attempts to develop animal models of IDRs, and with the exception of the nevirapine-induced skin rash in Brown Norway rats, these attempts have ended in failure (Ng et al., 2012). The attempts not only included treatment of several species with a variety of drugs that cause IDRs in humans, they also included cotreatment with agents such as those that activate the immune system through binding to Toll-like receptors, use of animals such as Cbl-b knockout mice that have impaired immune tolerance, cotreatments with agents to deplete glutathione, immunization with drug-protein conjugates, etc. In addition we have tried to repeat published work describing animal models, and in general, we were unable to reproduce their results.
Why is it so difficult to develop animal models of IDRs? The answer to this question would have very important implications for the mechanisms of IDRs. There are several possible reasons. Given the strict MHC requirement for some IDRs such as abacavir hypersensitivity discussed above, the animals used in the studies may simply not have the requisite MHC, and even if they do, they might not have the required T-cell receptors. However, not all IDRs appear to have such strict MHC requirements. Another possible reason is that the animal simply does not form sufficient reactive metabolite. An example of this lack of bioactivation is that, in contrast to rats and humans, mice lack the sulfotransferase in their skin that is required to form the reactive sulfate metabolite of nevirapine that is responsible for the skin rash induced by nevirapine (Sharma et al., 2013). However, this is unlikely to be the major reason for the difficulty in developing animal models of IDRs. The fact that many patients have a very mild adverse reaction to a drug that can cause a serious IDR and this adverse reaction resolves despite continued treatment with the drug may represent the development of immune tolerance as discussed above. If this is the default response of the immune system to drugs that can cause IDRs, overcoming this tolerance may be a key to the development of useful animal models and understanding a major reason why IDRs are idiosyncratic (Uetrecht, 2009a).
A related question is why it was relatively easy to develop the rat model of nevirapine-induced skin rash. The immune response to modified proteins in the skin is very different from the immune response to modified proteins in the liver and most other organs, because the skin represents the major barrier to the outside world and is immunologically very active. In contrast, the major immune response in the liver is immune tolerance. However, the skin has very limited ability to metabolize drugs to reactive metabolites. The one metabolic enzyme that the skin has in abundance is sulfotransferase (Anderson et al., 1998), and the final step of activation of nevirapine leading to a skin rash is the formation of a sulfate conjugate.
V. Summary and Conclusions
The adaptive immune system participates in many forms of IDRs, with drug antigen-responsive T cells controlling the effector and regulatory processes that determine the nature of the clinical response. It is likely that the innate immune system also plays an important role in the immune response. There appear to be many different mechanisms by which drugs can induce an immune response, but with few valid animal models it is difficult to study mechanistic details and rigorously test hypotheses. There is strong evidence that reactive metabolites are responsible for many, but not all, IDRs. The discovery of HLA molecules as important risk factors for abacavir hypersensitivity and carbamazepine-induced Stevens-Johnson syndrome means that it is now possible to predict susceptible patient groups and restrict drug use. Additional HLA risk alleles have been identified and associated with specific forms of IDRs, often in restricted ethnic groups. It is not clear yet what fraction of IDRs has a strong HLA dependence, and T-cell receptor repertoire is likely to be an additional risk factor for many IDRs. Mechanistic studies are now required to (1) relate carriage of the HLA molecule to the pathogenic T-cell response (especially for the HLA class II associations) and (2) explain why drugs can be tolerated by certain patients carrying HLA risk alleles, and conversely, why certain patients develop reactions without the risk allele(s). Our understanding of the chemical basis of T-cell antigenicity and immunogenicity has progressed rapidly. We now have three well-characterized models that describe the way in which drugs interact with immunologic receptors and stimulate patient cells ex vivo. Viewed from the simplest chemical perspective, the hapten, P-I, and altered peptide models each describe the interaction of a drug, a peptide, and two receptors. The challenge as we move forward is to explain how each model contributes to the pathogenic immune response in patients that leads to an IDR.
Author Contributions
Wrote or contributed to the writing of the manuscript: Uetrecht, Naisbitt.
Footnotes
This work was supported by the Canadian Institutes of Health Research [Grants MOP-84520, MOP-93647] (to J.U.); and the Medical Research Council UK [Centre for Drug Safety Science Grant G0700654] (to D.J.N). J.U. holds the Canada Research Chair in Adverse Drug Reactions.
Abbreviations
- ADR
- adverse drug reaction
- DIHS
- drug-induced hypersensitivity syndrome
- DIL
- drug-induced lupus
- DNCB
- dinitrochlorobenzene
- DRESS
- drug reaction with eosinophilia and systemic symptoms
- HLA
- human leukocyte antigen
- IDILI
- idiosyncratic drug-induced liver injury
- IDR
- idiosyncratic drug reaction
- IFN
- interferon
- IL
- interleukin
- LPS
- lipopolysaccharide
- MHC
- major histocompatibility complex
- NK
- natural killer
- P-I
- pharmacological interaction
- PPD
- p-phenylenediamine
- SMX
- sulfamethoxazole
- SJS
- Stevens-Johnson Syndrome
- SOD
- superoxide dismutase
- TEN
- toxic epidermal necrolysis
- Th
- helper T cell
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





{kind=link}
{kind=link}
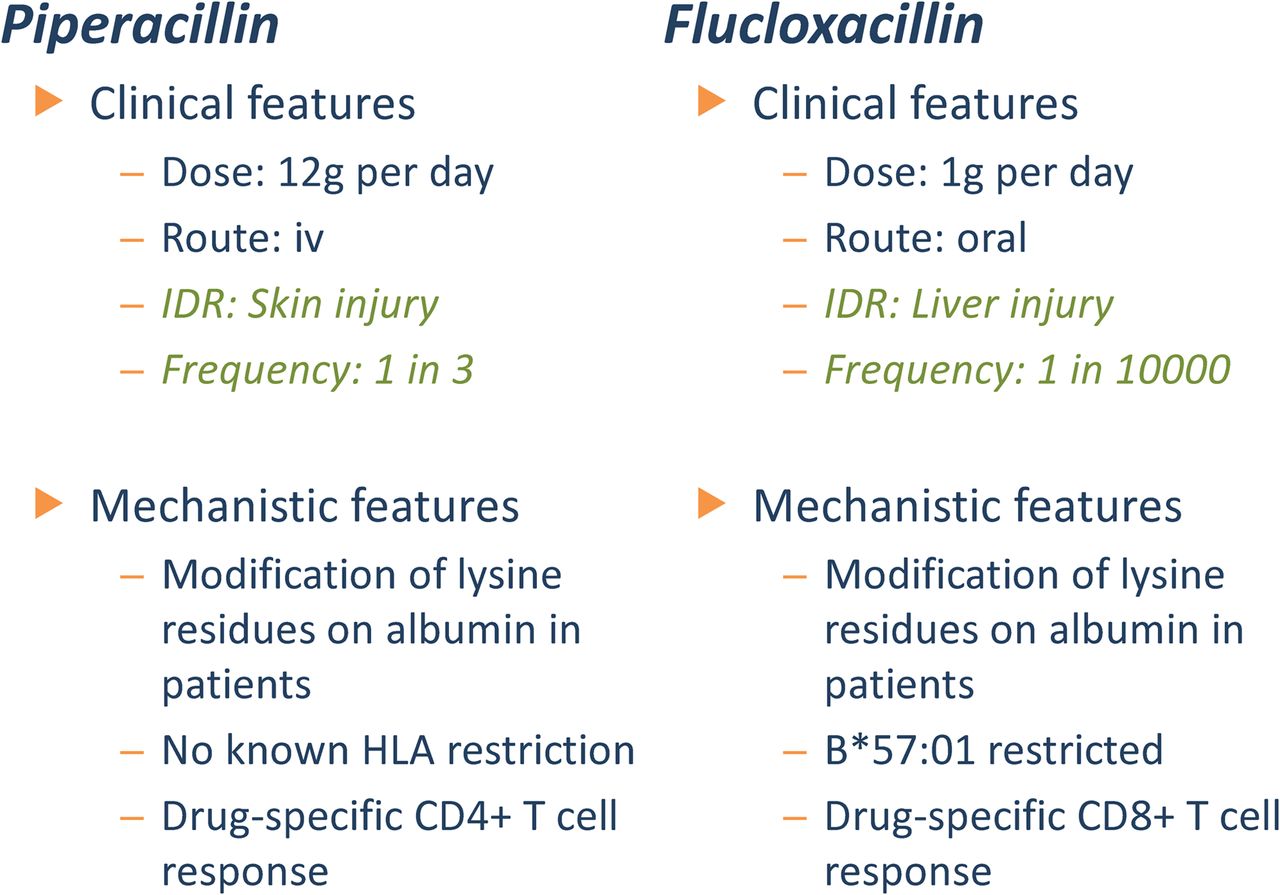{kind=link}
{kind=link}