Article Text

Abstract
The Antley-Bixler syndrome has been thought to be caused by an autosomal recessive gene. However, patients with this phenotype have been reported with a new dominant mutation at theFGFR2 locus as well as in the offspring of mothers taking the antifungal agent fluconazole during early pregnancy. In addition to the craniosynostosis and joint ankylosis which are the clinical hallmarks of the condition, many patients, especially females, have genital abnormalities. We now report abnormalities of steroid biogenesis in seven of 16 patients with an Antley-Bixler phenotype. Additionally, we identify FGFR2 mutations in seven of these 16 patients, including one patient with abnormal steroidogenesis. These findings, suggesting that some cases of Antley-Bixler syndrome are the outcome of two distinct genetic events, allow a hypothesis to be formulated under which we may explain all the differing and seemingly contradictory circumstances in which the Antley-Bixler phenotype has been recognised.
- Antley-Bixler syndrome
- FGFR
- congenital adrenal hyperplasia
- CYP21 deficiency
Statistics from Altmetric.com
In 1975 Antley and Bixler1 reported a patient with craniosynostosis, radiohumeral synostosis, and femoral bowing. A further 24 cases have since been added, showing that humeroulnar synostosis and straight femora may also be consistent with the condition,2 although the exact nosology can be controversial on occasion.3-5 An intriguing aspect of these reports is the frequency of genital abnormalities among affected subjects, 14 of whom were noted to have this feature. Of these 14 cases, 10 were females with clitoromegaly, fusion of the labia minora, and hypoplasia of the labia majora.2 6 Against this, eight females have been described in whom the genitalia were normal, or not thought worthy of comment by the authors.
The Antley-Bixler syndrome represents the severe end of the spectrum of syndromic craniosynostosis. Many such patients have choanal atresia and severe respiratory distress, often resulting in early death. In contrast with most clinically similar forms of syndromic craniosynostosis, which are transmitted in an autosomal dominant manner, Antley-Bixler syndrome has been thought to be an autosomal recessive disorder.7 This is based upon three reports of affected sibs8-10 and the birth of affected subjects to consanguineous parents.9 11 12 It should be noted that joint ankylosis in the context of craniosynostosis is not exclusive to Antley-Bixler syndrome and is also seen in some cases of Pfeiffer syndrome, many of which arise as new autosomal dominant mutations.13-15 However, unlike Antley-Bixler syndrome, genital malformations are not characteristic of Pfeiffer syndrome. Similarly, femoral bowing with craniosynostosis can be seen in some cases of thanatophoric dysplasia, again without accompanying genital anomalies. An important observation common to all three sibships with recurrence of Antley-Bixler syndrome is the concordance of genital abnormalities in all six cases.8-10
Mutation of members of the fibroblast growth factor receptor gene family (FGFR) has been established as the basis of several autosomal dominant forms of craniosynostosis.16-18 Other mutations of this gene family have been established as the cause of several variants of skeletal dysplasia, including thanatophoric dysplasia.19-21 As the effects ofFGFR mutation on receptor function have become clearer, it has been suggested that these mutant forms are viable only in the heterozygous state and that homozygosity forFGFR mutation would be lethal.18
New dominant mutations at the FGFR2 locus have been observed in patients with a phenotype very similar to that of Antley-Bixler syndrome,15 22-24 although the genitalia in these reports, which included three females, did not show evidence of clitoromegaly. Genital abnormalities are not characteristic of syndromes arising as a result of FGFRmutation and the high prevalence of such anomalies, especially among female infants, is an unexplained aspect of the Antley-Bixler syndrome. Another clue to the aetiology of Antley-Bixler syndrome might be the observation that the phenotype has been reported in the offspring of women taking high dose fluconazole, an antifungal agent, during pregnancy. To date, four instances of this have been recorded.25-27
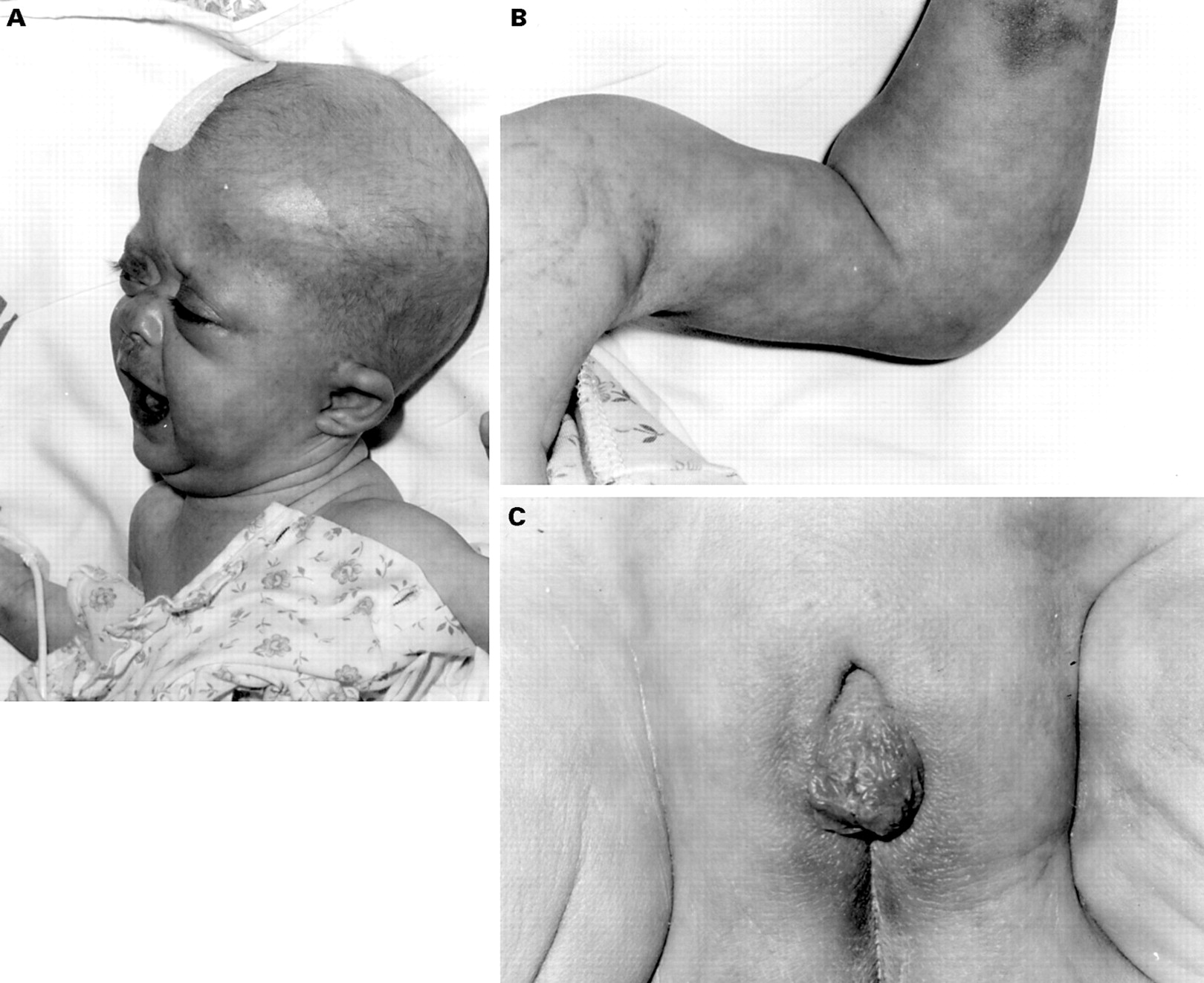
The observation of abnormalities of steroid biosynthesis suggestive of heterozygosity for a 21-hydroxylase (CYP21) deficiency in a patient with classical Antley-Bixler syndrome (fig 1) has led us to evaluate 15 further patients with craniosynostosis and joint fusion for abnormalities of steroid biosynthesis and forFGFR2 mutations. Based on our observations, we suggest that the genital malformations of Antley-Bixler syndrome may be the result of abnormalities of steroidogenesis and that the severe skeletal phenotype reflects an exaggerated response toFGFR mutation, at least in some cases. Finally, these observations offer a model under which the fluconazole induced cases of Antley-Bixler syndrome may be explained.

Characteristic craniofacial features of Antley-Bixler syndrome in patient SP of this report (A). The characteristic fixed elbow joint of Antley-Bixler syndrome is shown (B). The genital malformations are illustrated to document the cliteromegaly and hooded prepuce observed in SP (C). (Photographs reproduced with permission.)
Materials and methods
Sixteen patients (4M, 12F) were available for study, all of whom had confirmed craniosynostosis and ankylosis of the elbow joints. Five of the 12 female cases showed variable degrees of malformation of the external genitalia (table 1). Two patients have been the subject of previous clinical reports.28-30
Summary of clinical, biochemical, and molecular findings in 16 patients with Antley-Bixler syndrome
Urine steroid metabolites were analysed by gas chromatography/mass spectrometry (GC/MS) after processing of the urine samples as follows. Solid phase C18 SEP-PAK silica cartridges (Waters Associates, Harrow, Middlesex) and ethanol were used for extraction of free steroids and steroid conjugates from the urine. The extract was dried and dissolved in 10 ml acetate buffer (0.5 mol/l, pH 4.6) to which was added 25 mg of enzyme powder, Sigma Sulfatase EC 3.1.6.1, type H-1 from Helix Pomatia (containing a sulphatase activity of 375-1000 units, and a β-glucuronidase activity of 7500 units). The sample was incubated at 37°C for 18 hours for hydrolysis to take place. After further extraction using an ethanol primed C18 SEP-PAK cartridge internal standards, androstanediol (A), stigmasterol (S), and cholesteryl butyrate (CB) were added, and the sample evaporated to dryness. Methyloxime derivatives of carbonyl groups were formed by the addition of 200 μl of 2% methoxyamine hydrochloride in pyridine (MOHCL) and incubation at 60°C for one hour. Thereafter, trimethylsilyl derivatives of hydroxyl groups were formed by the addition of 100 μl of trimethylsilylimidazole (TMSI) and incubation at 100°C for 16 hours overnight. After derivative formation the samples were placed under nitrogen gas to evaporate the pyridine and then 2 ml lipidex solvent (98:1:1 cyclohexane:hexamethyldisilazane:pyridine) was added and the sample sonicated. To remove excess silylating reagents, the sample was loaded onto a Lipidex 5000 column (hydroxyalkyl-Sephdex LH-20), followed by 4 ml lipidex solvent. The collected sample was then evaporated to dryness under nitrogen and dissolved in cyclohexane. The method has been described in detail elsewhere.31
FGFR2 mutations were sought using the previously published oligonucleotide primers and conditions.13 22 The CYP21gene was analysed for major deletions affectingCYP21 and the adjacent complement 4 gene by Southern blot analysis and for the P30L, intron 2 splice mutation, I172N, cluster of mutations in exon 6 (I236N + V237E + M239K), and V281L as previously described.32 In addition, sequence analysis of exons 1 to 8 was performed either after cloning into TA vector (Invitrogen) or by direct sequencing.
Results
Seven of the 16 patients we present showed evidence of altered steroidogenesis, one of whom has a documentedFGFR2 mutation (table 1). The details of the observed steroid abnormalities are documented in table 2 and include impaired adrenal and, in one case (DK), gonadal function. Of the remaining six patients with abnormal steroidogenesis, no mutation at the FGFR2 locus was found in three and no sample was available for examination in the other three cases. Genital anomalies were noted in all five female cases of Antley-Bixler syndrome with abnormal steroidogenesis (table 1), while normal genitalia were recorded in respect of both male cases with abnormal steroid profiles.
Investigations in seven Antley-Bixler patients with abnormal steroid profiles. Cortisol and 17-hydroxyprogesterone values are in nmol/l
FGFR2 mutations were established in seven cases (table 1), one of these, MA, also showing evidence for steroid abnormality (fig 2). None of the common mutations in theCYP21 gene were identified in the 11 cases from whom DNA was analysed, although the parent of one patient (case BM) unavailable for study was heterozygous for a minor deletion ofCYP21. Sequence analysis failed to identify any additional mutations.

{kind=link}
{kind=link}
FGFR2 mutation in MA is shown. The mutation G882C causes Trp 290Cys substitution. An MboI digest site is lost because of the mutation, which is confirmed in the digest. Wild type cut with MboI gives bands of 118, 118, and 75 bp. G882C gives bands of 236 and 75 bp. Lane 1, 1 kb ladder; lane 2, PCR product uncut; lane 3, patient product digested with MboI; lane 4, control product digested with MboI; lane 5, control product digested with MboI; lane 6, undigested PCR product.
Discussion
The paradoxes surrounding the aetiology of Antley-Bixler syndrome are multiple. Firstly, there are the reports ofFGFR2 mutations, thought to represent new dominant events, in cases with this apparently autosomal recessive syndrome.15 22-24 Secondly, the observations of abnormal female genitalia in reported cases of Antley-Bixler syndrome2 and in our own cases (table 1) are unlikely to represent phenotypic sequelae of FGFRmutation in that such features are uncommon in those conditions which are caused by FGFR mutation. Finally, the undeniable phenotypic overlap between Antley-Bixler syndrome and multiple malformations recorded in four infants born to mothers on high dose fluconazole during the early stages of pregnancy is a confounding observation and one which does not easily lend itself to explanation, given the other information available currently with respect to the cause of Antley-Bixler syndrome.25-27 We suggest that the data we now present go some way towards resolving these apparently disparate findings.
We present evidence for abnormality in steroid biochemistry in seven of 16 cases with Antley-Bixler syndrome (table 2). Several of these patients are dead and, owing to the severe nature of their illness, systematic evaluation of adrenocortical function was not possible in all cases. As a consequence, many of the patients we report have not had a formal challenge of the adrenal axis to evaluate steroid biosynthetic effects. However, it is noteworthy that those in whom steroid biochemistry was abnormal were all deemed to require oral steroid supplementation. Moreover, all five female cases with altered steroidogenesis had abnormalities of the genitalia (tables 1 and 2). Further evidence that these indicators of aberrant steroid biochemistry are relevant stems from our observations of corroborative findings in the families of three of these seven cases whom it was possible to investigate, such as a relative with CYP21 deficiency or a parent with similar steroid profile (table 2). Notwithstanding these deficiencies in data collection, which do make comparison between cases difficult, we submit that there is evidence for a degree of adrenocortical/gonadal compromise in these seven cases, although it is not clear from the data we present that the compromise shares identical pathology in all cases. The 17-hydroxyprogesterone concentrations observed were consistent with carrier status for CYP21 deficiency.32 However, with one exception, genetic analysis failed to identify any of the common mutations which account for approximately 90% of cases of CYP21 deficiency. In the exception, the child was dead and no sample was available, but the biochemistry was suggestive of a CYP21 abnormality and the father was shown to be heterozygous for a minor deletion (case BM).
Our findings offer a new avenue to address the overlap of Antley-Bixler syndrome with the multiple malformations seen in offspring of mothers on high doses of fluconazole in pregnancy. Fluconazole is a potent antifungal agent particularly in immunocompromised patients.33 The therapeutic antifungal effect of the azole drugs is mediated through the cytochrome P450 enzyme C-14 alpha demethylase, principally inhibiting the demethylation of lanosterol, the predominant sterol of the fungal cell wall. The resulting depletion in ergosterol leads to increased permeability and inhibition of cell growth and replication.34 In contrast with fluconazole, earlier generations of azole drugs were somewhat non-specific in their affinity for cytochrome P450 enzymes, inhibiting both fungal and human enzymes.34 35 As a result, ketoconazole, first introduced in 1981, came to have a recognised therapeutic value as an inhibitor of human steroid biosynthesis.36 More importantly in the present context, ketoconazole was known to cause clinically significant endocrine abnormalities in humans owing to this non-specific inhibition of the cytochrome P450 enzyme system, including enzymes necessary for the synthesis of adrenal and gonadal steroid hormones.34 37 One of the therapeutic advances represented by the introduction of fluconazole in 1990 was relative specificity for the fungal cytochrome P450 enzyme system.33 34
Studies of fluconazole use have not shown a systematic teratogenic link in humans. Inman et al 38retrospectively reviewed 239 women who had taken fluconazole at the time of conception and did not identify any malformation attributable to the drug. Another study, evaluating the use of single dose fluconazole for vaginal candidiasis during the first trimester of pregnancy, reported no excess of fetal anomalies or of miscarriages in the 226 fetuses exposed to fluconazole.39 In reviewing the phenotype of those four cases with an Antley-Bixler phenotype born to mothers on long term, high dose fluconazole throughout pregnancy, Aleck and Bartley27 draw attention to subtle differences between the four reported cases and “typical” Antley-Bixler syndrome. None of the fluconazole exposed cases had frontal bossing, choanal atresia, or camptodactyly. Additionally two of these cases had minor terminal limb deficiency.25 26 Aleck and Bartley27went on to speculate that “Antley-Bixler syndrome may represent a defect of sterol metabolism and that fluconazole inhibition of cytochrome P450 may mimic that defect”. The observations of abnormal steroid biochemistry in seven cases with Antley-Bixler syndrome in this report lend further strength to their hypothesis and have caused us to consider this matter further.
Clinical pharmacology studies of fluconazole are reported to confirm its selectivity for fungal as opposed to mammalian cytochrome P450 linked enzymes.33 At doses of 50 mg/day, no “clinically important effect” on cortisol response to ACTH or levels of gonadal hormones was noted in healthy females.33 Similarly, healthy males showed small inconsistent effects in testosterone levels.33 35 However, the usual dose of fluconazole is 100 to 200 mg/day40 and the dosage in the mothers of the four cases in question was 400 mg/day in two and 800 mg/day in the others. Published data are largely unhelpful in addressing the possible effects of such a high dosage on the human cytochrome P450 enzymes. Clinical trials data, in which 15 healthy human male volunteers took fluconazole at doses of 200 mg, 300 mg, and 400 mg daily for 14 consecutive days showed no significant difference in plasma cortisol levels in those on high dose (400 mg). However, those patients on 400 mg/day did show a statistically significant (p<0.001) reduction in cortisol response to ACTH (short tetracosactrin stimulation test) on day 10 as compared to their responses when measured on day 1.41 Thus, the very limited data available do suggest that ACTH responsiveness is impaired in patients on high dose fluconazole over prolonged periods of time and this effect is likely to be magnified further in mothers on maintenance high dose fluconazole over a period of several weeks, a clinical situation for which no clinical trials are available. We would therefore suggest that a situation of relative adrenal insufficiency may have occurred in these mothers and their infants during the pregnancy while the fluconazole continued, an analogous situation to the relative deficiency of cortisol recorded in several of our patients (table 2).
Accordingly, we raise the question whether disordered steroidogenesis, intrinsic or extrinsic, induced by drug interactions, might contribute to Antley-Bixler syndrome in at least some cases. The genital malformations recorded in Antley-Bixler syndrome are more significant in affected females2 and usually comprise fused labia and clitoromegaly. The genital malformations seen in the most commonly occurring cytochrome P450 enzyme defects, such as CYP21 deficiency, also result in virilisation of the female genitalia with a similar outcome.42 While abnormal steroidogenesis would not result in craniosynostosis or joint ankylosis, it could certainly offer a plausible explanation for the abnormal genital development so often seen in female patients with Antley-Bixler syndrome, while the presence of a mutation at the FGFR2 locus could account for the skeletal features observed, including the variability with respect to femoral bowing, in reported cases.
Three of our patients, in whom we have no reason to suspect adrenal insufficiency, have an Antley-Bixler phenotype with normal genitalia and a documented FGFR2 ser 351cys mutation. This particular mutation has previously been recorded with a very severe phenotype of craniosynostosis and joint ankylosis.22-24 The substitution of a cysteine for a conserved amino acid is one of the most frequently observed patterns of mutation in FGFR mutants and is suggested to result in ligand independent inactivation.18 43 It seems likely that the most probable interpretation is that this particular mutation is associated with an especially severe phenotype independent of steroid abnormality.
We also observed a trp290cys mutation in one of our patients, in whom low cortisol was recorded. This mutation has only been reported on two previous occasions, both associated with a severe phenotype, including elbow ankylosis.15 44 A similar argument could be mounted, that this is an especially deleterious mutation associated with a very severe phenotype overlapping clinically with Antley-Bixler syndrome. Alternatively it might suggest that the slight genital abnormality seen in the one female case with this mutation15 and the low cortisol recorded in our male case with this mutation both indicate subtle alterations in adrenal steroid activity and that the effects of the FGFR2mutation are modified in such an environment.
We also report a cys342ser substitution in one of our cases. This substitution was one of those recorded in the initial report ofFGFR2 mutation causing Crouzon syndrome and has subsequently been noted by other authors in familial Crouzon45 and in Pfeiffer syndrome.46However, the very extreme phenotype observed in our patient exceeds that noted in those cases.
Our inability to establish an FGFR2 mutation in six cases from whom samples were available is by no means unusual. Meyers et al 46 reportedFGFR2 mutations in 11 of 17 unrelated cases with Crouzon, Pfeiffer, and Jackson-Weiss syndromes and we have previously recorded several instances where noFGFR2 mutation could be established in classical cases of Crouzon syndrome.47
The clinical diversity resulting from FGFR2mutation is well established and even extends to distinct phenotypes resulting from identical mutations.13 45 46No clear explanation for these varying clinical outcomes has been established. However McKeehan et al 48 have proposed a structural model of the FGFR complex which suggests that phenotypes are mutation site dependent because each mutation results in a different degree of abnormal activity which is independent of activating FGF. Their model proposes that FGFR normally exist as preformed dimers by interaction of their extracellular domains.49 However, since transphosphorylation between kinases in a dimer is the key activating event caused by FGF, the proximity of the kinases must be restricted in the absence of FGF. This restriction is imposed by a specific interaction of the FGFR ectodomain with rigid heparan sulphate chains of pericellular matrix proteoglycan, which is mediated by divalent cations and which in turn restricts transphosphorylation between the intracellular kinase domains.50 This is normally overcome by FGF, which binds into the binary complex of FGFR and heparan sulphate chain transmitting conformational change that brings the intracellular kinases into proximity for transactivation. Mutations inFGFR subvert this conformational restriction and result in gain of function of FGFR. A heterozygote will have 25% of the dimeric binary complexes comprised of homodimers of normal FGFR, 50% comprised of heterodimers of mutant and wild type FGFR, and 25% comprised of homodimers of mutant FGFR. Since the model predicts that both partners of the FGFR dimers must be occupied by FGF to trigger optimum intracellular transphosphorylation, 75% of the unliganded complexes will be inactive and requiring FGF for activation, while up to 25% (the mutant homodimers) may exhibit constitutive, FGF independent activity. The percentage of unregulated activity within the possible 25% fraction depends on the site of mutation, in essence the severity of the impact of the structural mutation in subverting restrictions which then allows transphosphorylation in the absence of FGF. This model also explains why specific mutations, which cause a particular combination of abnormalities in specific tissues, have no effect in many other tissues where the FGF system also appears to play a role. In some tissues or cells, a small gain of function of up to 25% causes abnormality, but that level is of no clinical consequence in other tissues where response remains dependent on activation by FGF. More recent work suggests that the interaction between heparan sulphate and FGFR, which mediates both restriction of activity and subsequent activation by FGF, exhibits stringent structural requirements in respect of composition of the heparan sulphate chains, particularly with regard to sulphation pattern.51-52 This model predicts that a variety of factors which perturb the restrictive conformational relationship within the FGFR complex may cooperate with gain of function mutations to modify the quantitative spectrum of FGFR activity and, therefore, the severity of the phenotype. Preliminary experiments in vitro indicate that the alteration of the composition of heparan sulphate or its relationship with the FGFR ectodomain through perturbation with chelating agents, heparinases, and proteases can trigger FGFR activity independent of FGF (McKeehan and Kan, unpublished data). It has also been shown that in cells in vitro, steroids alter the composition of heparan sulphate proteoglycans, dependent on cell type (McKeehan and Kan, unpublished data).53-55 Indeed, tentative links between abnormal steroid biogenesis andFGFR mutation already exist in the case of acne vulgaris. Heterozygotes for CYP21mutations are more common in acne vulgaris than in the general population, although factors other than CYP21 are thought to contribute to the pathology of this skin disorder.56 Similarly, acne vulgaris is known to be common in patients with Apert syndrome and localised acne has been associated with somatic mosaicism for theFGFR2 C934G mutation in skin scrapings.57
The observations we report on the Antley-Bixler phenotype enable a series of possible hypotheses to be formulated which might explain the different circumstances in which that phenotype is observed. Firstly, some FGFR2 mutations appear to cause this phenotype independent of other factors. There is an undeniable clinical overlap between Pfeiffer syndrome in the severe form and Antley-Bixler syndrome without genital anomaly. Our findings of S351C mutation in three of our patients with bony ankylosis and craniosynostosis allied to those published cases with this mutation support this clinical overlap, which arises as a result of new dominant mutation at the FGFR2 locus. Secondly, a more innocuous FGFR2 mutation, acting in an abnormal steroid environment (specifically low cortisol/glucocorticoid) may have an exaggerated effect resulting in the Antley-Bixler phenotype and, in this situation, genital anomaly may be a feature, particularly in female cases. Case MA in our study represents a likely example of this, and we would cite the genital malformation reported in another female case with this mutation (T290C)15 as supportive evidence. Thirdly, it may be that abnormality of steroid biogenesis can unmask an FGFR sequence change which is usually not deleterious enough to cause abnormal phenotypic features but becomes so in the abnormal state of steroidogenesis. This model offers a possible explanation for the fluconazole related cases, two of whom were born to the same mother. In this situation, it might be expected that the genitalia might be abnormal, particularly in female cases. Of the four cases with the fluconazole related phenotype born to date, two have been female and the genitalia were not remarked upon.25 26 This latter model of Antley-Bixler syndrome would also offer a possible explanation for the recurrence in sibships in three instances and is supported by the observation of genital malformation in all six affected cases in those three families.8-10 In this situation a non-pathologicalFGFR2 sequence change would be coinherited with a heterozygous change in a CYP related gene to give the phenotype. The chance of this digenic inheritance occurring would be 1 in 4 for susceptible couples.
Examples of digenic inheritance causing human disease are uncommon. Kajiwara et al 58 presented cases of retinitis pigmentosa where the phenotype was the result of mutation at two independent loci. Similarly, a subtype of Waardenburg syndrome type II, associated with ocular albinism, was shown to be the clinical expression of mutation at the MITFlocus and a functionally significant polymorphism at theTYR locus.59 The evidence we now present suggests that some cases of Antley-Bixler syndrome may represent a similar phenomenon. Irrespective of whether the hypotheses we propose for Antley-Bixler syndrome are refuted or not by future work, we submit that there is now a gathering body of evidence that Antley-Bixler syndrome represents a phenotype which may be the product of dysfunction in FGFR2 signalling exacerbated by abnormal steroid biogenesis and, as such, represents a further example of a human disorder resulting from digenic events.
Acknowledgments
Grant funding from Action Research is gratefully acknowledged (SM, RMW, and WR). We are pleased to acknowledge the assistance of Drs J Montalto, S Brown, and Neil Green.