Article Text

Statistics from Altmetric.com
MDR1 (ABCB1), MRP2 (ABCC2), and BCRP (ABCG2) are members of the family of ATP binding cassette (ABC) transporters. These are plasma membrane transporters that are expressed in various organs. The role of MDR1 and MRP2 in the hepatobiliary system is well defined; both contribute to bile formation by transport of drugs, toxins, and waste products across the canalicular membrane. As they transport exogenous and endogenous substances, they reduce the body load of potentially harmful compounds. The role of ABCG2, which is also expressed in the canalicular membrane of hepatocytes, has not yet been fully characterised. All three proteins are also expressed in the apical membrane of enterocytes where they probably control oral availability of many substances. This important “gatekeeper” function of ABC transporters has been recognised recently and is currently under further investigation. Expression and activity of these transporters in the gut may differ between individuals, due to genetic polymorphisms or pathological conditions. This will lead to individual differences in bioavailability of different drugs, toxins, and (food derived) carcinogens.
Recent information on substrates, transport mechanisms, function, and regulation of expression of MDR1, MRP2, and BCRP in different species is summarised in this review.
INTRODUCTION
Detoxification of xenobiotics, including toxins, carcinogens, and drugs, is the central task of many metabolising enzymes in the body. Two groups of enzymes are known to handle metabolism of harmful compounds. The group of cytochrome P450 isoenzymes (CYPs) comprises numerous isoforms (approximately 60 are expected in humans1) leading to oxidation (mostly hydroxylation) of molecules (for an overview see Werck-Reichhart and Feyereisen2). Isoforms in the CYP groups 1, 2, and 3 mediate metabolism of many exogenous compounds. Many toxins and carcinogens require activation by CYP450 isoenzymes to obtain their reactive (that is, alkylating) properties in the body. Because CYP450 isoenzymes mostly catalyse the first step of biotransformation, this function is called phase I metabolism. Phase I metabolism often, but not always, precedes diverse conjugation steps, also called phase II metabolism. Phase II metabolism is mediated by several different enzymatic systems, the most important being the UDP glucuronosyltransferases.3 Isoforms of this enzyme class conjugate compounds to glucuronic acid, thereby making them more hydrophilic and suitable for excretion in urine or bile. Other important phase II systems are glutathione-S-transferases, sulphotransferases, and acetyltransferases.
More recent research has demonstrated that transport steps have to be added to our models of xenobiotic defence. Export pumps reduce the local cellular burden of toxic compounds giving the individual cell protection against toxic effects. These transport proteins are primarily expressed in the apical membrane of epithelial cells, such as enterocytes, which are exposed to exogenous xenobiotics. In these cells the same transporters function on the one hand to reduce the entrance of harmful substances and on the other hand to eliminate their detoxification products. The latter step has been called “phase III metabolism”,4 indicating the close connection to the oxidation and conjugation steps of detoxification. The first function (that is, direct elimination of xenobiotics on entrance into the cell) represents a first defence line against xenobiotics and likewise could be called “phase 0 metabolism”.
-
Metabolism comprises successive phases of modification and elimination (phase 0 to phase III); transporters accomplish phase 0 and phase III but can also act on phase I products.
All transporters involved in these mechanisms are members of the family of ATP binding cassette transporters. They mediate cellular efflux in an active ATP dependent manner against concentration gradients. Based on our present knowledge, at least two transporters play a prominent role in phase 0 and phase III defence against xenobiotics. They are the multidrug resistance transporter 1 (MDR1) and multidrug resistance associated protein 2 (MRP2). After the new nomenclature they were termed ABCB1 and ABCC2, respectively. However, a third transporter, more recently identified, called breast cancer related protein (BCRP) or ABCG2, is likely to be involved in this defence system also. All three transporters are present in both the intestine and liver and therefore can reduce oral bioavailability by two mechanisms: direct inhibition of uptake out of the gut and rapid elimination of xenobiotics and their metabolites via bile. Lack of intestinal ABC transporters leads to higher xenobiotic uptake into the portal vein and also results in higher systemic blood and organ levels. Lack of hepatic elimination results either in isolated high hepatic tissue levels (very rare) or, as a result of basolateral resecretion from the hepatocyte into the systemic circulation, higher systemic blood and also organ levels (fig 1).

Determinants of oral bioavailability. Although transporters are only present in the intestine and liver, they influence almost all parameters of bioavailability (the large arrows indicate changes in the respective parameter in the absence of transporters). Effects can vary depending on the substrate and respective transporter. The importance of the kidney in systemic excretion of xenobiotics is mainly confined to glomerular filtration while active transporter mediated tubular secretion probably plays only a minor role.
-
This means that both elimination methods (intestinal and hepatic) reduce systemic blood levels and organ load of compounds and thereby decrease acute and chronic toxicity of toxins but also decrease the efficacy of (oral) drugs.
MDR1/ABCB1
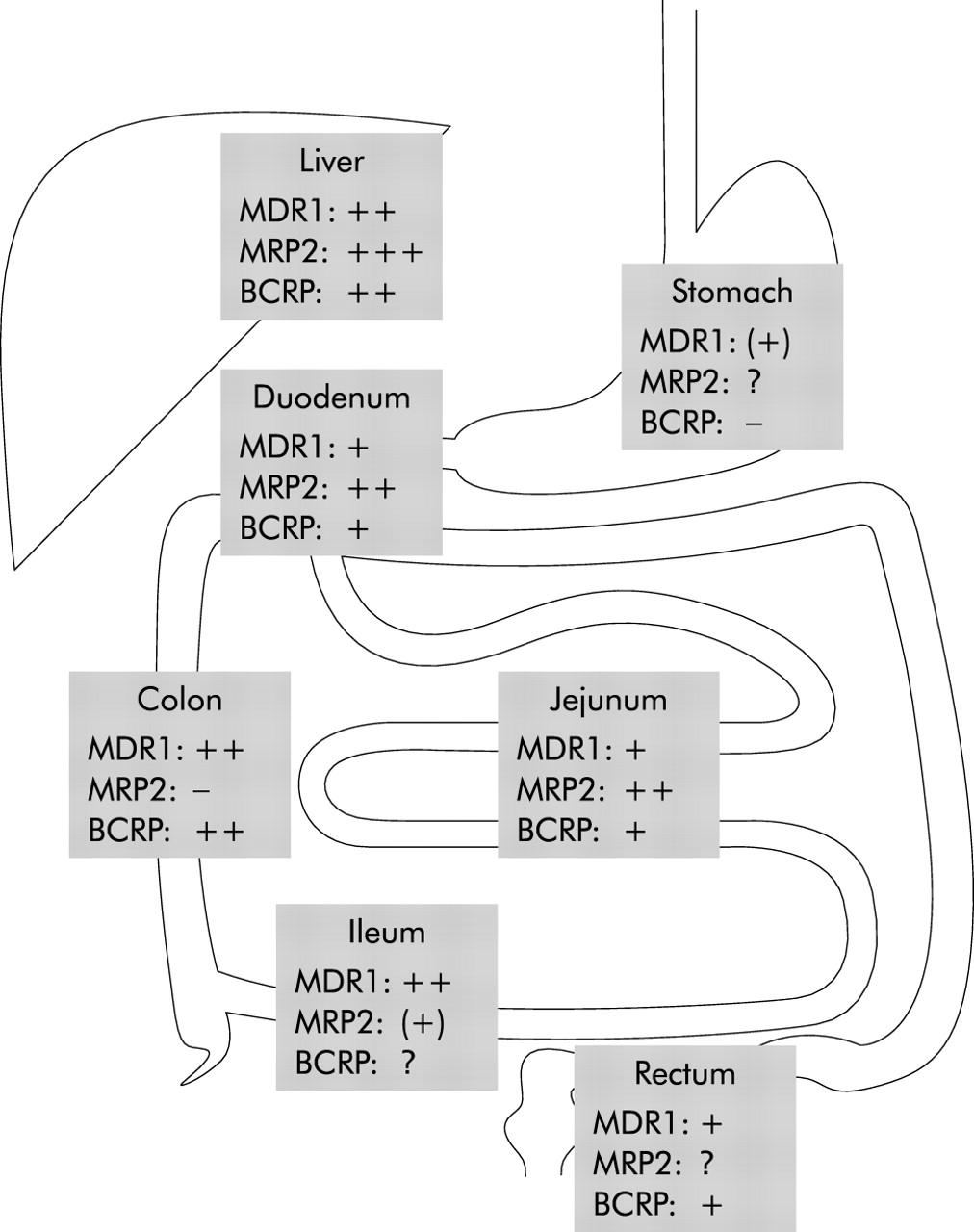
MDR1 was first described in cancer cells where it extrudes chemotherapeutic agents out of the cell thereby conferring multidrug resistance,5 a problem in the treatment of cancer. Its physiological function however only became fully clear on studies with several drugs in Mdr1a−/−, and later in combined Mdr1a/b−/−, mouse models.6,7 In contrast with humans, who have only one MDR1 gene, mice have two genes, Mdr1a and Mdr1b, with overlapping substrate specificity and tissue distribution. The above mentioned studies demonstrated that MDR1 functions as a gatekeeper against xenobiotics in the blood-brain barrier but also in the gut.8 Expression of MDR1 in the human intestine increases from proximal to distal, resulting in the highest expression levels in the colon (fig 2).9,10

Expression levels of transporters in different parts of the intestine and the liver (see text for details).
The gatekeeper function in the gut is desirable for toxins and carcinogens but was found to limit the oral availability of drugs. Individual differences in activity and/or expression of the protein were shown to lead to changes in drug bioavailability. In kidney transplant recipients, MDR1 protein concentration predicted intestinal absorption of cyclosporine11; in liver transplant recipients, it even correlated with poor survival.12 The frequency of the first discovered polymorphism of MDR1, the C3435T polymorphism, was investigated recently in a large sample of Caucasian subjects; it was found that about 25% of subjects were homozygous for this polymorphism.13 Plasma levels of digoxin were elevated in healthy volunteers with the C3435T polymorphism of MDR1.14 In the latter study, reduced duodenal MDR1 expression was demonstrated in individuals homozygous for this polymorphism, indicating a direct influence of MDR1 function on oral bioavailability of digoxin. For nelfinavir, the same polymorphism was found to increase the immunological response (CD4 count) in human immunodeficiency virus positive patients,15 suggesting the clinical relevance of MDR1 activity. On the other hand, no effect of the C3435T polymorphism on the oral bioavailability of fexofenadine, a well established MDR1 substrate, could be found in vivo in healthy subjects.16 Another study identified several other polymorphisms in the MDR1 gene, partially linked to each other in the sense that C3435T did occur together with two other polymorphisms (C1236T and G2677T) in most patients.17 Interestingly, the C3435T polymorphism was recently linked to the development of ulcerative colitis,18 which is in line with results from Mdr1a−/− mice. These mice spontaneously develop a severe intestinal inflammation when kept in a pathogen free environment.19 There may also be a link of this polymorphism to renal epithelial tumours.20 At present, only data defining the impact of single polymorphisms on activity or expression exist. This complicates assessment of the clinical importance of MDR1 polymorphisms because additional parameters (for example, genetic linkage of polymorphisms or compensatory upregulation of other transporters) may contribute to overall bioavailability of xenobiotics. It seems clear that the influence of MDR1 activity on the effect of drugs is substrate specific and therefore has to be investigated for each drug.21
-
Polymorphisms of transporter genes influence the bioavailability of drugs and toxins and may also predispose to certain diseases.
MDR1 transports a wide range of structurally diverse drugs, of which the most important are given in table 1. In the lower section of table 1, drugs known as inhibitors are specified. It is possible that some of these drugs will also be found to be substrates of MDR1 but until now only their potential in inhibiting MDR1 mediated transport of other substrates has been defined. The large number of drugs potentially influencing MDR1 activity makes interactions likely to occur in multidrug therapy, especially given the fact that some drugs (for example, doxorubicin57) also influence expression of MDR1 (see also below). One illustrative human study, assessing the effect of cotreatment with loperamide (a MDR1 substrate) and quinidine (a MDR1 inhibitor), demonstrated a possible impact. Loperamide, an opioid derivative given for diarrhoea, normally does not induce central effects. Under cotreatment with quinidine, healthy volunteers developed respiratory depression although plasma levels of loperamide remained unchanged.58 This study proved that the lack of central effects of loperamide is due to the gatekeeper role of MDR1 in the blood-brain barrier and represents a “proof of principle” for drug interactions by MDR1 although oral bioavailability was not influenced. Interactions between multiple drugs influencing the latter MDR1 function have not yet been established in humans but probably exist and may be of clinical importance.
Xenobiotics, used as drugs, which interact with MDR1 or MRP2 either as (predominantly) inhibitor and/or substrate (current state of knowledge)
-
Drug interactions occur when inhibitors and substrates (or two substrates) of the same transporter are given at the same time.
MDR1 blocking agents are used today in clinical trials of chemotherapy to reduce multidrug resistance. PSC-833, or valspodar,59 predominantly inhibits MDR1 but is also transported by it, albeit at an insignificant rate.60 Whether the inhibitory effect of PSC-833 on MDR1, discovered and characterised in vitro, can be fully translated to patients, is still not clear60 and requires further study. Agents such as PSC-833 not only increase the uptake of chemotherapeutic drugs in tumour cells but also their bioavailability, activity, and toxicity59 as well as that of comedications. Certainly, therapy with PSC-833 or other MDR1 inhibitors in the context of cancer chemotherapy cannot currently be recommended outside clinical trials.
Components of our daily nutrition can influence MDR1 activity also. Grapefruit juice has been shown to influence MDR1 activity, although results of in vivo and in vitro studies are conflicting with regard to the extent to which this happens.61–63 Methoxyflavones in orange juice have been shown in vitro to inhibit MDR1 activity64 but whether this is relevant in vivo needs further investigation. To date it seems too early to implement certain nutritional advice for patients taking MDR1 substrates in drug therapy.
For MDR1, links to intestinal carcinogenesis have been reported. β-Catenin/TCF4 binding elements in the MDR1 promoter were described previously and very recently it has been shown that intestinal tumorigenesis in ApcMin/+ mice lacking functional Mdr1 genes is suppressed, implicating a direct influence of Mdr1 transcription/expression on carcinogenesis.65 This may mean that the presence as well as the absence of MDR1 (by higher bioavailability of carcinogens) modulates development of malignancies but this result requires confirmation and mechanistic explanation in independent studies.
MRP2/ABCC2
MRP2 was first functionally characterised as a canalicular multispecific organic anion transporter in the apical membrane domain of hepatocytes.66 As such, it mediates transport of numerous organic anions, especially conjugated compounds, into bile and therefore out of the body.49 As a consequence, it influences the whole body load of endo and xenobiotics. MRP2 is not only expressed in the liver and kidney but also in epithelial cells of the intestine,67 the placenta,68 and at the blood-brain barrier.69 In the rat intestine, MRP2 expression is highest in the duodenum and subsequently decreases in direction to the terminal ileum and colon where it is only minimal.70 Our own unpublished human data show a similar distribution (fig 2).
Apart from conjugates, MRP2 also transports amphipathic uncharged compounds,27,71,72 indicating a much broader substrate spectrum of this transporter. Various studies show that MRP2 mediated transport of uncharged or cationic substrates can be stimulated by the presence of reduced glutathione (GSH).27,44–45,71 Labile complexing of substrates with reduced GSH or simple cotransport similarly seems to be a possible explanation for this phenomenon. The latter mechanism has also been described for transport of amphipathic compounds by another member of the MRP family, MRP1.73 Consequently, depletion of intracellular GSH inhibits MRP2 mediated export of uncharged compounds while that of anions is preserved.
Very recently it was reported that the transport mechanism of MRP2 is even more complex in that one substrate or modulator molecule of the transporter can stimulate the affinity of another. Thus it was found that transport of oestradiol-17β-glucuronide can be stimulated 30-fold by the presence of sulfanitran74 or sixfold in the presence of indomethacin.75 Kinetic analysis revealed that at least two binding sites are present in the transport protein that give rise to cooperative binding of substrates/modulators.
-
Mutual substrates or modulators of MRP2 can strongly modify each others’ affinity for the transporter and thereby influence the efficacy of clearance.
The functional importance of MRP2 in the intestinal epithelium has not yet been clearly defined. In the rat, bioavailability of an abundant food derived carcinogen (PhIP) is reduced by MRP2.72 MRP2 expression in the human duodenum is inducible by rifampicin,67 indicating possible interactions in multidrug therapy. This induction has been shown to be the result of signalling by nuclear receptors which are induced by a variety of xenobiotics (see below) and represents one underlying mechanism for acquired differences in protein expression. Polymorphisms, as described for MDR1, have now also been found in the MRP2 gene76 but neither frequency nor influence on transporter activity or expression have yet been defined. Another possibility of acquired individual differences in protein expression is the prevalence of certain intestinal diseases (Crohn’s disease, coeliac sprue). MRP2 is expressed in the tips of the intestinal villi.67,77 These are atrophic in coeliac sprue, which represents a mechanism for reduction of intestinal MRP2 with possibly increased oral bioavailability as a consequence (own unpublished results). Substrates and inhibitors of MRP2 are included in table 1. Note that MDR1 and MRP2 share some substrates as well as inhibitors, which may lead to broader interactions in the oral bioavailability of drugs. In addition, there are drugs (for example, tamoxifen78) which influence expression of the protein thereby influencing bioavailability, activity, and toxicity of the substrates. In contrast with MDR1, systematic clinical investigations regarding the influence of MRP2 inhibition on oral bioavailability of substrates are lacking.
Components of our daily diet are also substrates for MRP2, such as the flavonoid epicatechin in tea,79 chrysin and its metabolites,80 and the meat derived heterocyclic amine PhIP.71 While the first two compounds are thought to have antitumorous effects, PhIP is a carcinogen with genotoxic properties. Further studies are necessary to define the role of MRP2 in the defence against food derived xenobiotics.
Transcripts in the human jejunum have been analysed, showing higher levels for MRP2 transcripts than for MDR1.81 Thus MRP2 may be at least as important in the gut as MDR1. It has to be kept in mind however that transcription analysis is not representative for protein expression, especially because MRP2 expression can be regulated translationally.82 Additionally, protein levels will not be representative of actual transport activity.
The human Dubin-Johnson syndrome is associated with nonsense mutations in the MRP2 gene resulting in truncation and degradation of the protein. In patients with this syndrome, MRP2 is completely absent in canalicular membranes of hepatocytes and apical membranes of enterocytes. To date, this syndrome is considered to be benign and does not have a clear influence on the health or lifespan of affected individuals. It is tempting to speculate whether in the light of the gatekeeper function of MRP2, patients with Dubin-Johnson syndrome might be at risk for toxic effects of drugs or, even worse, for the carcinogenic effects of food derived xenobiotics. The rarity of this acquired syndrome precludes epidemiological studies. All other causes for inherited and acquired reductions in activity or expression of MRP2, as described above, will probably be investigated in the future.
-
By limiting uptake of food components, transporters may modulate toxic and carcinogenic effects of our nutrition. Consequently, individual differences in transporter expression or activity can contribute to overall cancer and disease susceptibility.
BCRP/ABCG2
Breast cancer resistance protein (BCRP) was originally discovered, as implied by its name, in breast cancer cells.83 It was also termed mitoxantrone resistance protein (MXR) because of one of its substrates.84 On the basis of homologies in sequence and domain arrangements, it has been added to the ABCG group.
BCRP has a relatively broad tissue distribution; the transporter was found in the small intestine, colon, and in the canalicular membrane of hepatocytes (fig 2).85 This supports the hypothesis that BCRP plays a similar role in the body as MDR1 and MRP2. Transcription of BCRP in the human jejunum is higher than that of MDR1 and comparable with that of MRP2.81 In mice, BCRP reduces the oral bioavailability of topotecan,86 a topoisomerase inhibitor used in cancer chemotherapy.
BCRP is also expressed in the breast and placental syncytiotrophoblast.85 In the study on topotecan disposition in mice cited above, it was shown that BCRP influences the fetal penetrance of topotecan.86 Other topoisomerase inhibitors such as irinotecan and its metabolite SN3887 also belong to the substrates of this protein. Differential phenotypes of BCRP mediated multidrug resistance, shown as differences in substrate specificity, can be the result of single amino acid mutations in the protein.88 While the wild-type protein with an arginine on position 482 confers resistance to mitoxantrone and irinotecan, R482T or R482G mutations (arginine replaced by threonine or glycine, respectively) result in additional outward transport of rhodamine and doxorubicin by BCRP.
-
Single amino acid alterations do not result in only loss of substrates but also in acquired transport properties (“gain of function”).
Currently, a novel topoisomerase inhibitor (ST 1481), designed to overcome BCRP mediated multidrug resistance in tumour cells, is in clinical evaluation and has been shown in vitro not to be a substrate for BCRP.89
Recently, knockout mice for the Bcrp/Abcg2 gene have been produced.90,91 As might have been expected, these animals are hypersensitive towards mitoxantrone.91 The importance of Bcrp function in the gut was highlighted by the studies of Jonker and colleagues90 who showed that Bcrp−/− mice develop phototoxic lesions on light exposed areas of the skin. This phenotype only developed when the animals were fed with lab chow and not on a synthetic diet. It could be demonstrated that the phototoxicity was caused by pheophorbide, a chlorophyll breakdown product that occurs in various plant derived foods and food supplements. Bcrp transports pheophorbide and is highly efficient in limiting its uptake from ingested food.
REGULATION OF MDR1 AND MRP2
The human promoter sequences of MDR1(GenBank accession No M 29423,92), MRP2(GenBank accession No AJ 005200,93 see fig 3), and BCRP(GenBank accession No AF 35634794) have been cloned and allow studies concerning regulation of gene expression. Regulation of expression of MDR1 and MRP2 in several experimental animal models such as bile duct ligation (extrahepatic cholestasis), endotoxin treatment (intrahepatic cholestasis), and partial hepatectomy (proliferation) has been defined extensively in the liver (reviewed by Müller95). Most of the data come from rodent models; in vivo data for the human situation are rare. For MDR1, Sp1 and factors interacting with the Y box element (NF-Y, YB-1) seem to play a role in transcriptional activation. Interestingly, YB-1, a Y box binding protein, has the opposite suppressive effect on rat MRP2 transcription.96MRP2 is activated (fig 3) through binding of a heterodimer formed by the retinoid X receptor (RXR, NR2B1) and the retinoic acids receptor (RAR, heterodimer abbreviated as RXR:RAR). Downregulation of MRP2 expression in inflammatory responses has been shown to be due to interleukin 1 mediated suppression of RXR:RAR binding,97 a result obtained in the liver, but we have shown also involvement of both proteins in intestinal MRP2 expression (manuscript submitted). Tissue specific mechanisms of regulation are obviously important; RXR:RAR mediated suppression of MRP2 has been shown to be absent in the kidney.98 The fact that MRP2 is also induced by binding of the farnesoid X receptor FXR (NR1H4; as heterodimer FXR:RXR), a nuclear receptor in the liver and gut involved in bile acid homeostasis, suggests a role for MRP2 in enterohepatic cycling of bile acids.99 This may relate to the ability of MRP2 to transport glucuronidated and sulphated bile salts.

Scheme of the rat Mrp2 promoter, showing overlapping binding sites for different nuclear receptors, which activate transcription. ER8 and DR5 denote the respective binding sites (see text for abbreviations of the nuclear receptors). HNF, hepatocyte nuclear factor. Note that several different heterodimers can bind to the ER8 element. Binding of different receptors to their respective binding site at the same time can result in complex regulation events.
It is easy to hypothesise that there should be one mechanism in the body which, as a response to xenobiotic exposition, activates the whole set of enzymes necessary for detoxification and elimination of this xenobiotic. As the transporters for elimination of the unmetabolised parent compound and for elimination of the detoxification products are the same, this reaction can build up a coordinated defence system with at least three lines of defence. Rifampicin (a drug) and 2-acetylaminofluorene (2-AAF, a carcinogen) both have been characterised as inducers of MDR1 and MRP2.67,100,101 The newly discovered orphan receptor, pregnane X receptor (PXR, in humans sometimes termed steroid X receptor SXR, gene nomenclature NR1I2), has been identified as the cause for upregulation of both transporters in human cell lines and in rodents.99,102 Apart from rifampicin and 2-AAF, hyperforin (from St John’s wort), taxol, clomitrazole, phenobarbital, ritonavir, and dexamethasone as examples of exogenous ligands, but also lithocholic acid, ursodeoxycholic acid, and C21 steroids, called pregnanes, activate PXR as endogenous ligands.103–106 All of these molecules exhibit diverse structures and differ greatly in size and shape but most are potentially harmful either directly or when accumulating as a result of impaired metabolism or excretion. PXR also activates the most important drug metabolising cytochrome isoform, 3A4, and isoforms of the 2B and 2C class107 as well as UDP-glucuronosyltransferase isoforms,108 all involved in metabolism of steroids and xenobiotics in the intestine and liver. Additionally, the constitutive androgen receptor CAR (NR1I3), another CYP inducing receptor, can replace PXR in the binding together with RXR to the respective response element and also induce MRP2 expression,99 which creates more ligands for regulation of this transporter gene.
-
Endogenous and exogenous compounds can activate all phases of metabolism via nuclear receptors, such as PXR and CAR.
Both PXR and CAR regulate overlapping sets of genes in a tissue specific fashion. In mice, common activators of PXR/CAR upregulate Mrp2 in the liver via CAR and in the intestine via PXR, while Mdr1a/b are upregulated via PXR in the liver and intestine, with an additional effect via CAR in the intestine confined to Mdr1a.109 Taken together, this establishes a defence system with a very broad specificity, which also includes endogenous substances. However, this system can cause a new type of drug interaction. In healthy human volunteers, cotreatment with rifampicin and talinol resulted in significant reduction of oral bioavailability of the latter compound, caused by PXR mediated induction of MDR1 by the former drug.42 Here, excretion of the MDR1 substrate talinol was altered although the drug itself cannot influence expression of MDR1.
-
Networks of nuclear receptors broaden the defence against xenobiotics but can also promote drug interactions.
CONCLUSIONS
The recent discoveries inspire the concept of a pharmacogenetic barrier in the intestine, built to protect the body from toxic or carcinogenic compounds. At the same time, it is one of the major determinants in pharmacotherapy and should draw the attention of research into the development of new drugs.
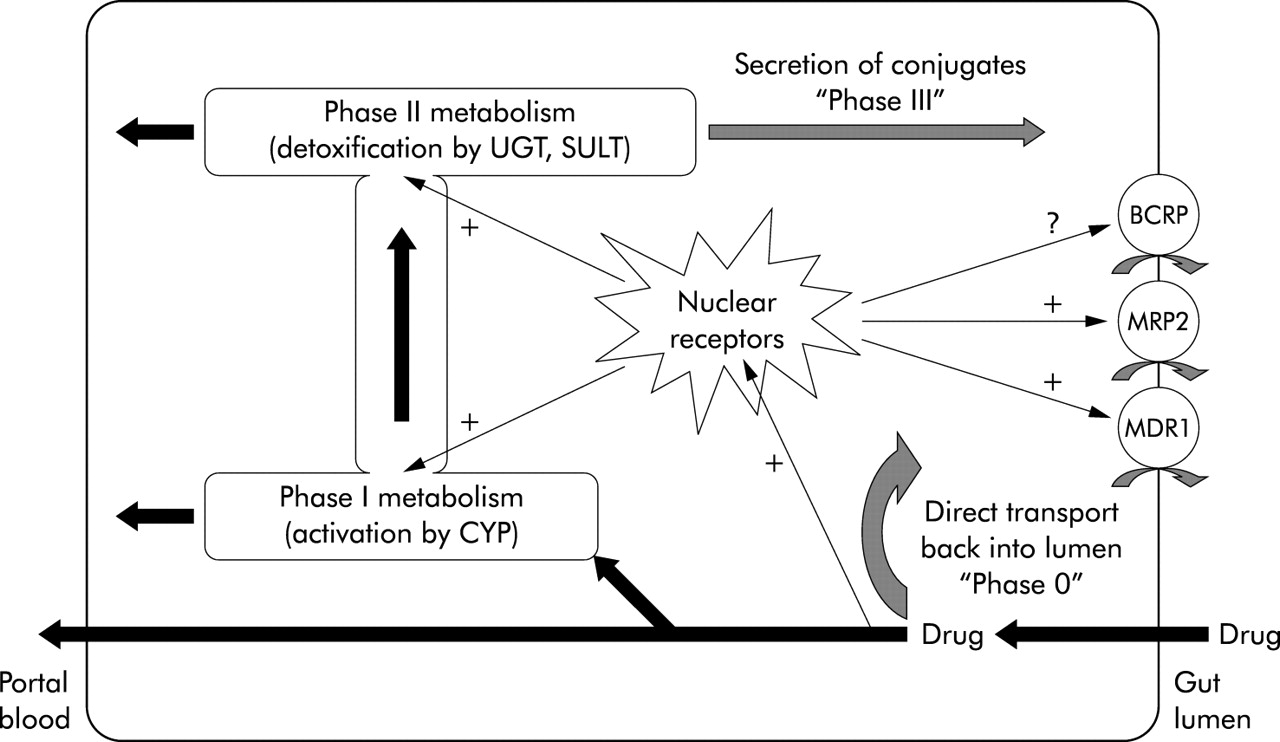
In fig 4, a scheme of this multi-enzymatic barrier with several lines of defence is depicted. In this defence model, the gatekeeping transporters in the intestinal epithelium reduce the cellular burden of the xenobiotic (first line of defence); the remaining intracellular molecules activate all lines of defence by common nuclear receptors and undergo CYP 3A mediated metabolism (as a second line of defence). Afterwards, they are excreted by transporters (third line of defence) again. In such a concept, the liver only remains a “backup” metabolic site for xenobiotics, reaching this organ via the portal blood.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic representation of the defence system in enterocytes. The different lines of defence are further explained in the text. Note that nuclear receptors as common activators can upregulate enzymes of all metabolic steps. CYP, cytochrome P450; UGT, UDP-glucuronosyltransferases; SULT, sulphotransferases;+, upregulation, ?, unknown effect.
Projects regarding the identification of polymorphisms, their clinical relevance, and the influence of intestinal disease on transporter expression are currently in progress and are expected to define the extent of interindividual differences in bioavailability and cell toxicity of drugs and toxins.
GLOSSARY
-
Apical membrane. In polarised cells (for example, epithelial cells, hepatocytes), two membrane domains exist. The apical membrane domain faces the lumen of the intestine, the renal tubule, or the biliary duct, respectively, the basolateral membrane domain faces the blood vessels.
-
Expression. In the strict sense of the word, expression is the collective term for all steps necessary to produce protein from DNA and therefore in most genes denotes transcription (see below)and translation (conversion from mRNA to protein via ribosomes).
-
Knockout mice. Animal model, where targeted mutations result in inactivation of genes so that no protein expression takes place or the resulting protein is degraded or has no activity.
-
Phenotype. Morphological and/or physiological alterations which are directly or indirectly caused by mutations or polymorphisms (genotype).
-
Polymorphism. Alterations of the nucleotide sequence, normally termed mutations, are called polymorphisms if their frequency in the general population is above 1%.
-
Promoter. DNA region upstream of the transcription initiation site (starting point of transcription) where regulatory elements bind and thereby activate or inhibit transcription of the respective gene.
-
Transcription. Conversion of DNA to RNA and later to messenger RNA (mRNA, after splicing, which involves removal of intronic (non-coding) DNA).
Acknowledgments
The work of Christoph G Dietrich and Andreas Geier is supported by the Deutsche Forschungsgemeinschaft (grant DI 729/3-1 and SFB542, TPC1).