Article Text

Statistics from Altmetric.com
SUMMARY
Chemoprevention has been considered as a possible approach for cancer prevention. A significant effort has been made in the development of novel drugs for both cancer prevention and treatment over the past decade. Recent epidemiological studies and clinical trials indicate that long term use of aspirin and similar agents, also called non-steroidal anti-inflammatory drugs (NSAIDs), can decrease the incidence of certain malignancies, including colorectal, oesophageal, breast, lung, and bladder cancers. The best known targets of NSAIDs are cyclooxygenase (COX) enzymes, which convert arachidonic acid to prostaglandins (PGs) and thromboxane. COX-2 derived prostaglandin E2(PGE2) can promote tumour growth by binding its receptors and activating signalling pathways which control cell proliferation, migration, apoptosis, and/or angiogenesis. However, the prolonged use of high dosages of COX-2 selective inhibitors (COXIBs) is associated with unacceptable cardiovascular side effects. Thus it is crucial to develop more effective chemopreventive agents with minimal toxicity. Recent efforts to identify the molecular mechanisms by which PGE2 promotes tumour growth and metastasis may provide opportunities for the development of safer strategies for cancer prevention and treatment.
INTRODUCTION
The most effective treatments for cancer, including various combinations of surgical resection, radiation, and/or chemotherapy, depend on the detection of cancer at a very early stage. Unfortunately, it has not been possible to identify all individuals who are at the highest risk for developing cancer. Most patients present to their physician with advanced cancer when standard treatment regimens for solid malignancies result in a much lower five year survival. It is generally agreed that an effective way to control cancer is to find better ways of preventing it. Chemopreventive approaches are definitely worth considering for healthy persons who have a strong family history of cancer or those who are particularly susceptible for other reasons. One promising group of compounds with cancer preventive activity includes NSAIDS.
A large body of evidence from population based studies, case control studies, and clinical trials indicate that regular use of NSAIDs over a 10–15 year period reduces the relative risk of developing colorectal cancer by 40–50%.1 Furthermore, use of NSAIDs leads to regression of pre-existing adenomas in patients with familial adenomatous polyposis (FAP).2 As many other human cancers are reported to have elevated levels of COX-2 and overproduce PGs, there is great interest in evaluating the role of NSAIDs for prevention and treatment strategies for other cancers such as breast, stomach, pancreas, urinary tract, lung, and prostate. However, the prolonged use of NSAIDs is associated with side effects such as nausea, dyspepsia, gastritis, abdominal pain, peptic ulcer, gastrointestinal bleeding, and/or perforation of gastroduodenal ulcers.3 It was hypothesised that NSAIDs exert their anti-inflammatory and antitumour effects through inhibition of the inducible COX-2,4,5 while unwanted side effects of these drugs such as damage to the gastric mucosa and gastrointestinal bleeding are thought to arise from the inhibition of the constitutive COX-1.6 This hypothesis led to the development of COXIBs, such as celecoxib, rofecoxib, and valdecoxib. Indeed, highly selective COX-2 inhibitors retain the anti-inflammatory and antitumour effects of the NSAIDs while not interfering with COX-1 responsible for protection of the gastroduodenal mucosa from the effects of acid from the stomach.5,7–9 Therefore, these drugs were approved by the FDA, and as novel anti-inflammatory agents their use is associated with about a 50% reduction in gastrointestinal toxicity. Moreover, these agents have some potential for use as chemopreventives.10–12 One of them, celecoxib, was approved in December 1999 by the FDA for use in the prevention of colorectal polyp formation of patients with FAP. Unexpectedly, the prolonged use of higher doses of COX-2 selective inhibitors is associated with increased thrombotic events in humans.13–15 We know that this original hypothesis was overly simplistic because of our lack of knowledge of the importance of signalling pathways which are affected downstream of COX-2. Thus it has become essential for us to understand how NSAIDs interfere with COX-2 mediated cellular functions in both normal physiology and pathological conditions.
NSAID TARGETS
Inflammation and cancer
NSAIDs are chemically distinct compounds that share a common therapeutic action. The best known of these are aspirin, ibuprofen, piroxicam, indomethacin, and sulindac. NSAIDs are generally prescribed to ameliorate symptoms associated with acute pain and chronic inflammatory diseases such as arthritis. Chronic inflammation caused by infections or autoimmune diseases is clearly associated with an increased cancer risk in a number of instances. For example, it has long been known that patients with persistent hepatitis B, Helicobacter pylori infections, or an immune disorder such as ulcerative colitis have a higher risk for the development of liver or gastrointestinal tract cancer. It has been estimated that chronic inflammation contributes to the development of approximately 15% of malignancies worldwide.16 The connection between inflammation and cancer further supports the concept that anti-inflammatory drugs, NSAIDs, have antineoplastic activity.
COX dependent targets
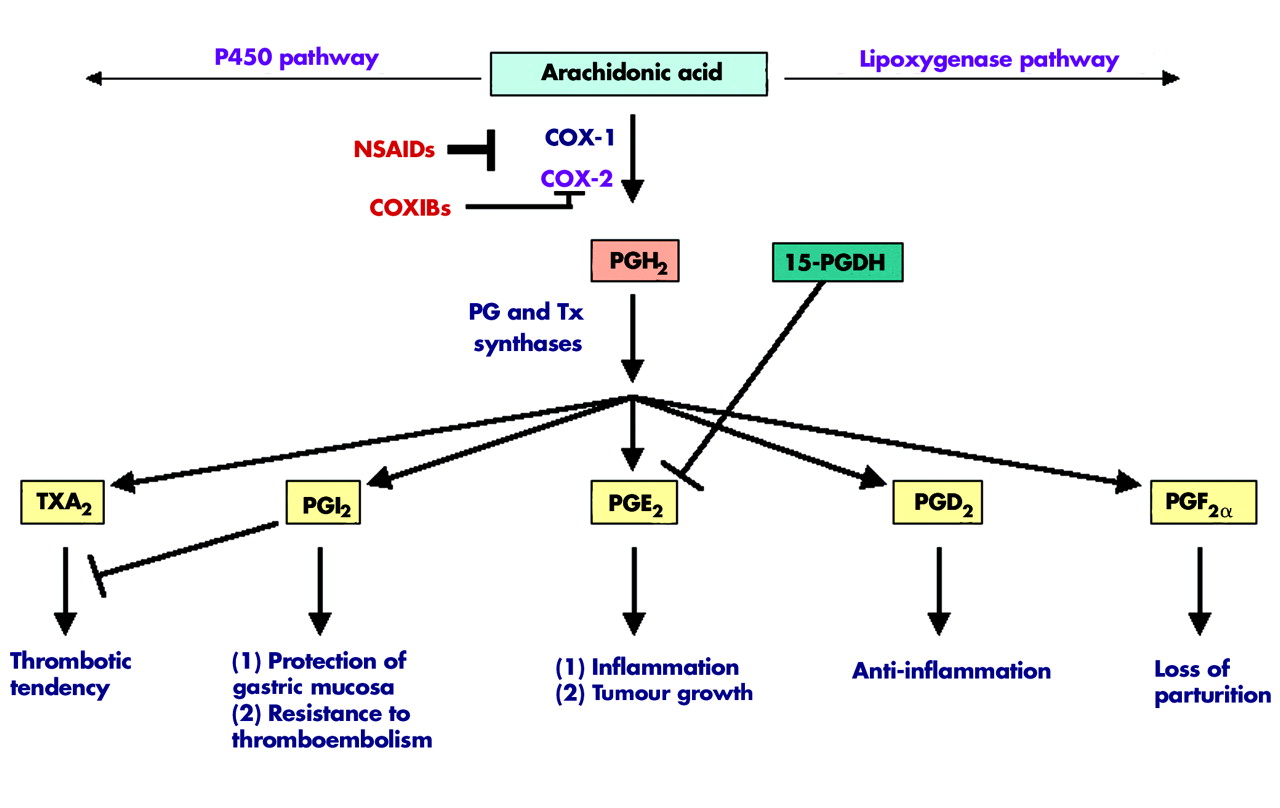
NSAIDs were shown to exert their anti-inflammatory, analgesic, and antipyretic effects mainly by inhibiting COX, a key enzyme responsible for the biosynthesis of PGs from arachidonic acid.6 Aspirin is an irreversible inhibitor of COX through blocking the approach of arachidonic acid, while indomethacin, piroxicam, ibuprofen, and sulindac are competitive inhibitors for substrate binding. When COX converts arachidonic acid to PGs, the key regulatory step in this process is the enzymatic conversion of arachidonate to PGG2, which is then reduced to an unstable endoperoxide intermediate, PGH2. Specific PG synthases in turn metabolise PGH2 to at least five structurally related bioactive lipid molecules, including PGE2, PGD2, PGF2α, PGI2, and thromboxane A2(TxA2)17(fig 1).

Overview of prostaglandin (PG) synthesis and main functions. Arachidonic acid can be metabolised through three major pathways. In the cyclooxygenase (COX) pathway, each COX-2 derived prostaglandin (PGI2, PGE2, PGD2, PGF2α) or thromboxane A2 (TxA2) has its unique functions. NSAIDs, non-steroidal anti-inflammatory drugs; COXIBs, COX-2 selective inhibitors.
COX exists in two isoforms commonly referred to as COX-1 and COX-2. Although both COX-1 and COX-2 are upregulated in a variety of circumstances, normally COX-1 is constitutively expressed in a broad range of cells and tissues. COX-1 expression remains constant under most physiological or pathological conditions and its constitutive enzymatic activity is linked to renal function, gastric mucosal maintenance, stimulation of platelet aggregation, and vasoconstriction. For example, COX-1 derived prostaglandins play a central role in many normal physiological processes. In the gut, COX-1 derived PGI2(also called prostacyclin) produced by epithelial and stromal cells in subepithelial tissues plays a key role in the cytoprotection of gastric mucosal surfaces and the normal vasculature.18 COX-1 has been shown to play an essential role in gastrulation in zebrafish and a reduction in COX-1 results in posterior mesodermal defects during zebrafish development.19,20 In contrast, COX-2 is an immediate early response gene and its expression is normally absent in most cells and tissues but it is highly induced in response to proinflammatory cytokines, hormones, and tumour promoters.21 Furthermore, COX-2 derived PGE2 is a proinflammatory bioactive lipid and is the major prostaglandin produced in many human solid tumours, including cancer of the colon,22 stomach,23 and breast.24 Recent research has indicated that COX-2 derived PGE2 is a key mediator of acute inflammatory responses,25,26 arthritis,27,28 and inflammatory bowel disease.29,30 Direct evidence supporting the notion that PGE2 promotes tumour growth comes from the following observations. PGE2 reversed NSAID induced adenoma regression in ApcMin mice.31 PGE2 significantly enhanced colon carcinogen induced tumour incidence and multiplicity in rats.32 Furthermore, our group recently reported that PGE2 accelerates intestinal adenoma growth in ApcMin mice.33
Although COX-2 selective inhibitors suppress PGE2 production, the potential inhibition of endothelial cell derived COX-2 activity and subsequent PGI2 production may promote platelet aggregation and lead to an increased risk of coronary thrombosis and stroke.34 As PGI2 antagonises thromboxane produced by platelets, inhibition of PGI2 may shift the homeostatic balance towards more TXA2 effects. In addition, PGI2 appears to be important in protecting cardiomyocytes from oxidative stress.35 Therefore, it will be important to develop chemopreventive agents that do not inhibit production of other prostanoids, such as the antithrombotic PGI2. Given that only PGE2 appears to be procarcinogenic, more selective pharmacological inhibition of PGE2 production downstream of COX-2 may be superior and result in fewer side effects. To achieve this goal, researchers have been investigating precisely how PGE2 promotes tumour growth and its signalling pathways.
COX independent targets
Another explanation for the antitumour effects of NSAIDs recently emerged, based on studies showing that high doses of NSAIDs inhibit tumour cell growth and induce apoptosis through COX independent mechanisms by regulating several different targets,36 such as 15-LOX-1,37 a proapoptotic gene Par-4,38 antiapoptotic gene Bcl-XL,39 and nuclear factor κB (NFκB) signalling.40,41 In the above studies, higher NSAID concentrations may be impossible to achieve in vivo without significant toxicity. For example, the proapoptotic effects of NSAIDs seen at concentrations above 50 μM are most likely modulated through COX-2 independent pathways. However, the best characterised biochemical target of NSAIDs at therapeutic concentrations remains the COX enzymes. It is likely that many of the chemopreventive effects of NSAIDs are carried out via inhibition of COX-2.
PGE2 AND CANCER
PGE2 and its receptors play a predominant role in promoting cancer progression. The only other COX-2 derived prostaglandin implicated in oncogenesis is TxA2, which was reported to promote angiogenesis.42 NSAIDs have been shown to inhibit PGE2 mediated processes that play essential roles in tumour progression, such as tumour cell proliferation, invasion, angiogenesis, and immunosuppression. Therefore, we will focus on modulation of PGE2 and its downstream targets that control these processes.
PGE2 regulation and its receptors
Steady state cellular levels of PGE2 depend on the relative rates of COX-2/PGE synthase dependent biosynthesis and 15-hydroxyprostaglandin dehydrogenase (15-PGDH) dependent degradation (fig 1). For example, cytosolic or microsomal PGE2 synthases can convert PGH2 to PGE2. Two cytosolic PGE2 synthases called cytosolic glutathione transferases (GSTM2-2 and GSTM3-3) catalyse the conversion of PGH2 to PGE2 in the human brain.43 The two microsomal PGE2 synthases characterised to date are mPGES1 and mPGES2. mPGES1 exhibits a higher catalytic activity than other PGES isomerases, indicating that it probably plays a key role in the synthesis of PGE2 from PGH2.44,45
15-PGDH, a prostaglandin degrading enzyme, catalyses oxidisation of the 15(S)-hydroxyl group of PGE2 to yield an inactive 15-keto PGE2.46 Genetic deletion of 15-Pgdh in mice leads to increased tissue levels of PGE2.47 Although 15-PGDH may promote certain androgen sensitive prostate cancers,48 we and others recently reported that loss of expression of 15-PGDH correlates with tumour formation, including colorectal cancer,49,50 lung,51 and transitional bladder cancer.52 Interestingly, NSAIDs have been shown to upregulate 15-PGDH expression in colorectal and medullary thyroid cancers.49,53 Taken together, these studies suggest that loss expression of 15-PGDH may contribute to tumour progression. The functional role of 15-PDGH in promoting tumour growth is currently under investigation in several laboratories.
PGE2 exerts its cellular effects by binding to its cognate receptors (EP1-4) that belong to the family of seven transmembrane G protein coupled rhodopsin-type receptors. The central role of PGE2 in tumorigenesis has been further confirmed through homozygous deletion of its receptors. Mice with homozygous deletions in EP1 and EP4 receptors, but not EP3, were partially resistant to colon carcinogen mediated induction of aberrant crypt foci.54,55 EP2 disruption decreases the number and size of intestinal polyps in APCΔ716 knockout mice.56 Moreover, in carcinogen treated wild-type mice, an EP1 receptor antagonist also decreased the incidence of aberrant crypt foci whereas ApcMin mice treated with the same EP1 receptor antagonist and an EP4 receptor antagonist developed 57% and 69% fewer intestinal polyps, respectively, than untreated mice.54,55 In addition to colorectal cancer, it has been shown that EP1, 2, and 4 receptors were elevated whereas EP3 receptor levels were decreased in mammary tumours in COX-2-MMTV mice.57 Furthermore, an EP1 receptor antagonist was shown to reduce tumour burden in a carcinogen induced rat mammary model.58 However, Amano and colleagues recently reported that EP3 receptor activation is required for lung tumour associated angiogenesis and tumour growth.59 In the future, it will be important to carefully determine the EP receptor profile in human cancers and to examine whether NSAIDs can modulate PGE2 receptor expression. Taken together, these findings may provide a rationale for the development of EP receptor antagonists which may offer an alternative to COX-2 selective inhibitors.
PGE2 signalling pathways and its downstream targets
An increasingly large body of evidence indicates that PGE2 promotes tumour growth by stimulating EP receptor signalling with subsequent enhancement of cellular proliferation, promotion of angiogenesis, inhibition of apoptosis, stimulation of invasion/motility, and suppression of immune responses. These findings prompted research to elucidate PGE2 signalling pathways and identify PGE2 downstream targets that are involved in promoting tumour growth (fig 2).
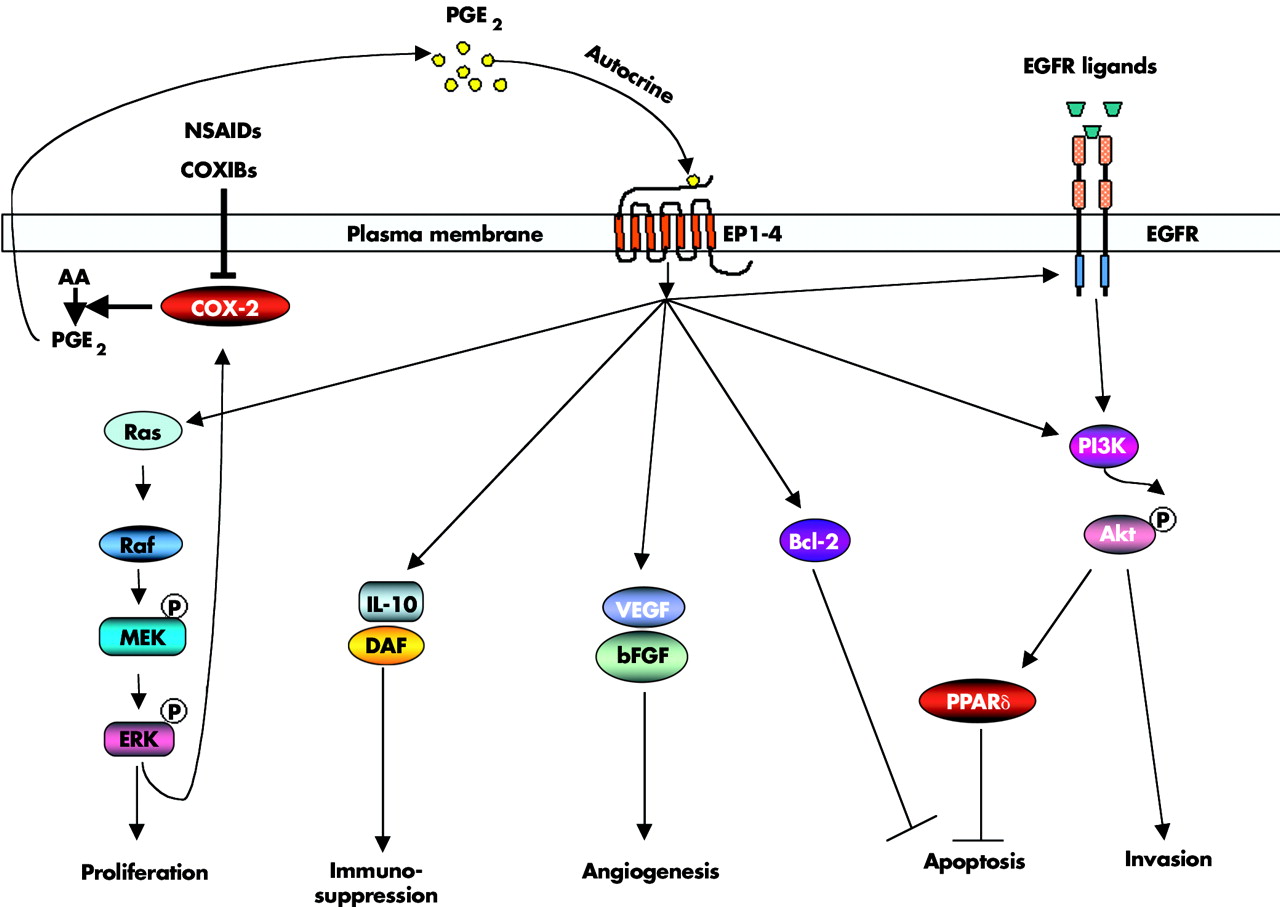
{kind=link}
{kind=link}
Prostaglandin (PG) E2 in carcinogenesis. PGE2 promotes tumour growth by stimulating EP receptor downstream signalling and subsequent enhancement of cellular proliferation, promotion of angiogenesis, inhibition of apoptosis, stimulation of invasion/motility, and suppression of immune responses. NSAIDs, non-steroidal anti-inflammatory drugs; COXIBs, COX-2 selective inhibitors; AA, arachidonic acid; EGFR, epidermal growth factor receptor; IL-10, interleukin 10; DAF, decay accelerating factor; VEGF, vascular endothelial growth factor; bFGF, basic fibroblast growth factor; PPARδ, peroxisome proliferator activated receptor δ.
EGFR pathway
Both COX-2 and epidermal growth factor receptor (EGFR) pathways are activated in most human cancers.60 The observation that forced expression of COX-2 in human colorectal cancer (CRC) cells stimulates cellular proliferation through induction of EGFR61 indicated the likelihood of crosstalk between these two pathways. We and others have demonstrated that PGE2 can transactivate EGFR, which results in stimulation of cell migration through increased PI3K-Akt signalling in CRC cells.62–64 As expression of EGFR directly correlates with the ability of human CRC cells to metastasise to the liver,65 it is possible that inhibiting both the EGFR tyrosine kinase and COX-2 at lower doses could yield additive effects, blocking the spread of metastatic disease. Moreover, preclinical studies support the notion that combined treatment of NSAIDs plus EGFR tyrosine kinase inhibitors is more effective than either single agent alone in several models. In colorectal carcinoma cells, blocking both COX-2 and the HER-2/neu pathways synergistically reduced tumour growth.66 In soft agar and xenograft assays, the combination of a COX-2 selective inhibitor, an EGFR tyrosine kinase inhibitor, and a protein kinase A antisense construct markedly reduced proliferation and angiogenesis of human colon and breast cancer cells.67 Similarly, combined treatment with inhibitors of both pathways significantly decreases polyp formation in ApcMin mice,68 as Apc deficiency is associated with increased EGFR activity in the intestinal enterocytes.69 Hence we feel that it will be essential to examine the use of COX-2 selective inhibitors as potential agents in combination with EGFR tyrosine kinase inhibitors in clinical trials.
Nuclear receptor pathways
Peroxisome proliferator activated receptors δ(PPARδ, also referred to as PPARβ) is a ligand activated nuclear transcription factor that is a member of the nuclear hormone receptor superfamily. Published data indicate that the PPARδ/β is important for regulating fat metabolism,70 inhibiting obesity induced by either genetic or high-fat-diet,70 and decreasing weight gain and insulin resistance in mice fed high fat diets.71 However, PPARδ has been identified as a direct transcriptional target of the APC/β-catenin/Tcf pathway.72 Our recent findings show that PGE2 transactivates PPARδ which in turn promotes tumour cell survival.33 PGE2 activates PPARδ via a PI3K-Akt pathway. Most importantly, we demonstrated that PGE2 promotes intestinal epithelial cell survival and colorectal adenoma growth in Apcmin mice, but not in PPARδ-/-/Apcmin mice,33 indicating that PPARδ is a critical downstream mediator in PGE2 stimulated tumour growth. Consistent with this result, a selective PPARδ agonist also accelerates intestinal polyp growth in Apcmin mice via inhibition of tumour cell apoptosis.73 These results support the rationale for considering the development of PPARδ antagonists for use in cancer prevention and/or treatment and raise caution for developing PPARδ agonists for clinical use to treat dyslipidaemia, obesity, and insulin resistance.
Ras-MAPK pathway
Ras is an oncogene and its activation is found in a wide variety of human malignancies. Ras induces cell transformation, proliferation, and survival by triggering downstream signalling pathways such as the Raf/MEK/ERKs and PI3K/Akt pathways. The Ras-MAP kinase cascade is one of the major intracellular signalling pathways responsible for cell proliferation. Forced expression of constitutively active Ras (mutant Ras) or MEK upregulates COX-2 expression and enhances cell proliferation in a variety of cell culture models, respectively.74–77 Non-selective NSAIDs and COX-2 selective inhibitors inhibit cell proliferation and transformation by blocking the Ras-MAPK signalling pathway.78–80 Our group recently reported that PGE2 activates a Ras-MAPK pathway which in turn upregulates COX-2 expression and stimulates colorectal cancer cell proliferation.81 This finding supports a novel mechanism by which COX-2 derived PGE2 promotes human cancer cell growth by autoregulation of COX-2 expression, which depends primarily on PGE2 induced activation of the Ras-MAPK pathway.
PGE2 downstream targets: angiogenic factors
Angiogenic factors are important for the growth and survival of endothelial cells and to stimulate vascular endothelial cell migration and capillary formation.82 Overexpression of COX-2 in CRC cells induces the production of angiogenic factors such as vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF),83 and NSAIDs block the production of these angiogenic factors leading to inhibition of proliferation, migration, and vascular tube formation.83–86 The observations that PGE2 can reverse the antiangiogenic activity of NSAIDs87 and homozygous deletion of EP2 completely abrogated induction of VEGF in APCΔ716 mouse polyps56 support the concept that PGE2 mediates the major role of COX-2 in inducing expression of proangiogenic factors. Several reports demonstrated that PGE2 upregulates VEGF in cultured human fibroblasts88 and increases VEGF and bFGF expression through stimulation of ERK2/JNK1 signalling pathways in endothelial cells.89 EP2/EP4 mediate PGE2 induction of VEGF in ovarian cancer cells90 and human airway smooth muscle cells.91 Interestingly, VEGF and bFGF induce COX-2 and subsequent PG production in endothelial cells, suggesting that the effects of PGE2 on regulation of VEGF and bFGF are likely amplified through a positive feedback loop.92 However, PGE2 induction of VEGF may provide another explanation for the undesired side effect of COX-2 selective inhibitors on cardiovascular complications. VEGF is implicated in cardiovascular homeostasis. For example, treatment with Avastin (bevacizumab), a humanised anti-VEGF antibody, has only marginal improvement in survival for colorectal cancer patients and is also associated with an increased risk of hypertension. Therefore, it is important to identify new molecular targets that drive colorectal tumour associated angiogenesis. Preliminary data from our laboratory indicate that PGE2 induces a proangiogenic chemokine expression in CRC cells (unpublished data).
PGE2 downstream targets: antiapoptotic factors
Overexpression of COX-2 in rat intestinal epithelial cells increased their resistance to undergo apoptosis and resulted in increased Bcl-2 protein expression.93 The role of COX-2 in preventing apoptosis is likely mediated by COX-2 derived PGE2, which attenuates cell death induced by the COX-2 selective inhibitor SC-58125.94 These findings have stimulated great interest in identifying PGE2 downstream targets responsible for modulating apoptosis. PGE2 induces antiapoptotic protein expression such as Bcl-294 and increases NFκB transcriptional activity,95 which is a key antiapoptotic mediator. As chemotherapeutic agents and radiation therapy enhance COX-2 protein expression as well as PGE2 synthesis in human cancer cells, elevated PGE2 production may increase resistance to therapy by giving cells a survival advantage. It will be important to determine whether patients treated with combinations of chemotherapy or/and radiation therapy with NSAIDs respond better than those not treated with NSAIDs. Preliminary study suggests that COX-2 selective inhibitors may increase the beneficial effects of radiotherapy.96
PGE2 downstream targets: chemokines and their receptors
Although chemokines play a crucial role in immune and inflammatory reactions, recent studies indicate that they have an equally important role in the cancer.97 PGE2 has been shown to inhibit production of CC chemokines, macrophage inflammatory protein (MIP)-1α and MIP-1β, in dendritic cells via binding to the EP2 receptor,98,99 and suppress CC chemokine RANTES (regulated upon activation normal T cell expressed and secreted) production in human macrophages through the EP4 receptor.100 These CC chemokines are crucial for macrophage and lymphocyte infiltration in human breast, cervix, pancreas, and gliomas cancers.101,102 Moreover, a recent study showed that PGE2 mediates VEGF and bFGF induced CXCR4 dependent neovessel assembly in vivo.103 The important role of CXCR4 for cancer pharmacology is based on findings that activation of CXCR4 is involved in stimulating cancer cell migration and increasing invasion in breast, prostate, bladder, and pancreatic cancers.104–107 These preclinical data indicate that chemokines and their receptors are potential drug targets of PGE2 downstream signalling for cancer prevention and treatment.
PGE2 downstream targets: immunosuppressive mediators
The tumour microenvironment is predominantly shifted from a Th1 to a Th2 dominant response (immunosuppressive immune responses). COX-2 selective inhibitors restore the tumour induced imbalance between Th1 and Th2 and promote antineoplastic responses in lung cancer108 and metastatic spread of colorectal cancer.109 These findings led to extensive efforts to understand how PGE2 can regulate immunosuppression. PGE2 has been shown to downregulate Th1 cytokines (tumour necrosis factor α, interferon γ, and interleukin (IL)-2)110 and upregulate Th2 cytokines such as IL-4, IL-10, and IL-6.111–113 IL-10 is an immunosuppressive cytokine. Moreover, PGE2 can modulate immune function through inhibiting dendritic cell differentiation and T cell proliferation and suppressing the antitumour activity of natural killer cells and macrophages.114,115 In addition, our group showed that PGE2 upregulates the complement regulatory protein decay accelerating factor which results in blocking the complement C3 into two active compounds, C3a and C3b in CRC cells.116 Thus the effects of PGE2 on the immune system may allow neoplastic cells to evade attack.
CONCLUSIONS
Chemoprevention is being carefully evaluated on several fronts as an effective measure to insure cancer control. NSAIDs and COX-2 selective inhibitors have been touted for their possible use as chemopreventive agents. However, concerns over the safety of COX-2 selective inhibitors have prompted researchers to develop more effective chemopreventive agents with minimal toxicity. Understanding the molecular mechanisms of COX-2 and its downstream targets will help to identify specific molecular targets for developing more safe agents which target this pathway. Preclinical studies provide evidence to demonstrate that non-selective NSAIDs and COX-2 selective inhibitors decrease the risk of colorectal cancer as well as other cancers through reduction of COX-2 derived PGE2 synthesis. Significant progress has been made in the elucidation of PGE2 downstream signalling pathways which mediate the chemopreventive effect of NSAIDs. These cumulative data indicate that the development of agents that lower cellular levels of PGE2 or that specifically inhibit the PGE2 downstream signalling pathway might be useful for cancer prevention. PGE synthases, 15-PDGH, and/or PGE2 receptors may also serve as rational targets for lowering cellular levels of PGE2 and EGFR, MAPK, and chemokines, and chemokine receptors are molecular targets for specific inhibition of PGE2 downstream signalling pathways. Moreover, another approach for decreasing the undesired side effects of COXIBs may be to lower the drug dose used. The combined use of multiple agents may allow for a lower dose of drug to be used. Taken together, efforts to develop novel chemopreventive agents with minimal toxicity and to design strategies for combinations of different agents targeting multiple pathways may yield significant benefits for cancer patients.
Acknowledgments
We acknowledge support from the United States Public Health Services Grants RO1-DK-62112 and PO1-CA77839. RND is the Hortense B Ingram Professor of Molecular Oncology and the recipient of an NIH MERIT award (R37-DK47297). We also thank the TJ Martell Foundation and the National Colorectal Cancer Research Alliance (NCCRA) for generous support.
REFERENCES
Footnotes
-
Conflict of interest: None declared.