


Abstract
Fluoxetine [±-N-methyl-3-phenyl-3-[(α, α, (-trifluoro-p-tolyl)oxy]-propylamine)] a selective serotonin reuptake inhibitor, is widely used in treating depression and other serotonin-dependent disease conditions. Racemic, (R)- and (S)-fluoxetine are potent reversible inhibitors of CYP2D6, and the racemate has been shown to be a mechanism-based inhibitor of CYP3A4. Racemic fluoxetine also demonstrates time- and concentration-dependent inhibition of CYP2C19 catalytic activity in vitro. In this study, we compared fluoxetine, its (R)- and (S)-enantiomers, ticlopidine, and S-benzylnirvanol as potential time-dependent inhibitors of human liver microsomal CYP2C19. In a reversible inhibition protocol (30 min preincubation with liver microsomes without NADPH), we found (R)-, (S)- and racemic fluoxetine to be moderate inhibitors with IC50 values of 21, 93, and 27 μM, respectively. However, when the preincubation was supplemented with NADPH, IC50 values shifted to 4.0, 3.4, and 3.0 μM, respectively resulting in IC50 shifts of 5.2-, 28-, and 9.3-fold. Ticlopidine showed a 1.8-fold shift in IC50 value, and S-benzylnirvanol shifted right (0.41-fold shift). Follow-up KI and kinact determinations with fluoxetine confirmed time-dependent inhibition [KI values of 6.5, 47, and 14 μM; kinact values of 0.023, 0.085, 0.030 min–1 for (R)-, (S)-, and racemate, respectively]. Although the (S)-isomer exhibits a much lower affinity for CYP2C19 inactivation relative to the (R)-enantiomer, it exhibits a more rapid rate of inactivation. Racemic norfluoxetine exhibited an 11-fold shift (18–1.5 μM) in IC50 value, suggesting that conversion of fluoxetine to this metabolite represents a metabolic pathway leading to time-dependent inhibition. These data provide an improved understanding of the drug-interaction potential of fluoxetine.
Fluoxetine [±-N-methyl-3-phenyl-3-[(α, α, (-trifluoro-p-tolyl)oxy]-propylamine)] is a widely used selective serotonin reuptake inhibitor, with more than 23 million prescriptions filled for the generic drug within the United States in 2006 (http://www.drugtopics.com/Top+200+Drugs). Racemic fluoxetine and its (R)- and (S)-enantiomers are metabolized by N-demethylation to the pharmacologically active metabolite norfluoxetine by CYP2D6 and other cytochrome P450 (P450) isoforms (Margolis et al., 2000; Mandrioli et al., 2006; refer to latter for chemical structure). Fluoxetine also undergoes CYP2C19-mediated O-dealkylation to the p-trifluoromethylphenol metabolite (Liu et al., 2002). In addition, racemic fluoxetine and/or its enantiomers have been shown to be reversible inhibitors of CYP2D6 (Brosen and Skjelbo, 1991; Stevens and Wrighton, 1993), CYP2C19 (Kobayashi et al., 1995; Foti and Wahlstrom, 2008), CYP3A4 (von Moltke et al., 1994; Ring et al., 1995), and CYP2C9 (Schmider et al., 1997; Hemeryck et al., 1999). Fewer studies have been conducted examining the potential for fluoxetine to be a mechanism-based inhibitor of P450. Mayhew et al. (2000) showed fluoxetine to be a mechanism-based inhibitor of CYP3A4, and McGinnity et al. (2006) recently demonstrated time- and concentration-dependent inhibition of CYP3A4 and also CYP2C19 in multiple in vitro systems, including hepatocytes. With heightened awareness of links between mechanism-based inhibitors, covalent binding and idiosyncratic toxicity (Ulrich, 2007), as well as the appearance of regulatory guidance for drug-drug interaction testing (U.S. Food and Drug Administration, http://www.fda.gov/cber/gdlns/interactstud.htm.), many laboratories are establishing or revisiting their procedures for conducting time-dependent P450 inhibition testing. In the process of augmenting our laboratory's CYP2C19 time-dependent inhibition assay, we tested model compounds intended to serve as reference inhibitors. In many laboratories, ticlopidine is used as a positive control inhibitor in this assay (Ha-Duong et al., 2001), but we and others have found it to be only weakly inhibitory (Stresser et al., 2008), and it is therefore unsatisfactory as a potent acting benchmark. In this report, we confirm the findings of McGinnity et al. (2006) with racemic fluoxetine and show that the enantiomers of fluoxetine are effective, but kinetically different, time-dependent inhibitors of CYP2C19.
Materials and Methods
Materials. Pooled human liver microsomes (HLM), S-benzylnirvanol, (S)-mephenytoin, (±)4′-OH-mephenytoin, and stable-isotope–labeled (±)4′-hydroxy mephenytoin-D3, were obtained from BD Biosciences (Woburn, MA). All other chemicals, including (±)-fluoxetine, (R)-fluoxetine, (S)-fluoxetine, (±)-norfluoxetine, and ticlopidine were obtained from Sigma-Aldrich (St. Louis, MO).
S-Mephenytoin 4′-Hydroxylase IC50 Shift Assays. Inhibition by test chemicals was determined using seven concentrations of inhibitor, separated by 0.5 log spacing, in a final volume 0.4 ml. Reactions contained 40 μM S-mephenytoin (approximately the KM), 0.3 mg/ml of pooled HLM, 1.3 mM NADP+, 3.3 mM glucose 6-phosphate, 0.4 U/ml glucose 6-phosphate dehydrogenase, 3.3 mM MgCl2, and 100 mM potassium phosphate buffer, pH 7.4. Reactions containing 5× HLM protein (e.g., 1.5 mg/ml) were incubated for 30 min with or without an NADPH-regenerating system before transfer of an aliquot into a secondary reaction mix containing the S-mephenytoin substrate. Incubations were terminated after 10 min by transferring a 200-μl aliquot to 50 μl of formic acid (0.1%) in acetonitrile containing 0.5 μM stable-labeled isotope internal standard. After stopping the reactions, incubations were subjected to centrifugation at 4000 rpm for 20 min to compress the precipitated protein into a pellet, and the supernatants were retained for high-performance liquid chromatography/mass spectrometry (MS) analysis.
S-Mephenytoin 4′-Hydroxylase KI and kinact Assays. Incubations were performed in 0.1 M potassium phosphate (pH 7.4) with 1.5 mg/ml HLM protein with an NADPH-generating system (as described above). Solvent only and five concentrations of inhibitor (3, 10, 30, 100 and 300 μM) were tested in duplicate. After various incubation times (2, 6, 11, 17, 23, and 30 min), 80 μl was removed and added to 320 μl of a secondary S-mephenytoin assay mixture (0.1 M potassium phosphate with 1.3 mM NADP+, 3.3 mM glucose 6-phosphate, 0.4 U/ml glucose-6-phosphate dehydrogenase, 3.3 mM magnesium chloride, and 200 μM S-mephenytoin). Unless otherwise indicated, after 10 min of incubation, the reaction was stopped and processed for liquid chromatography/MS analysis as described above.
Analytical Methods. The 4′-hydroxy (S)-mephenytoin metabolite was quantified using a 4000 Q TRAP LC/MS/MS System (Applied Biosystems, Foster City, CA) equipped with a dual pump system (Perkin-Elmer, Wellesley, MA) and a LEAP CTC HTS PAL autosampler (LEAP Technologies, Carrboro, NC) as described previously (Perloff et al., 2009). Mass transitions were 235.1 → 150.0 for 4′-hydroxy (S)-mephenytoin metabolite and 238.1→ 150.0 for the internal standard 4-hydroxy-S-mephenytoin-D3.
Data Analysis. The IC50 values were calculated by linear interpolation. IC50 shifts were calculated by dividing the IC50 value in the absence of NADPH by the IC50 values in the presence of NADPH. For KI/kinact assays, the natural logarithm of percentage remaining activity (corrected for decrease in metabolism over time in absence of inhibitor) was plotted against preincubation time for each concentration of inhibitor tested. The slopes of the linear portion of each plot were determined, and the -slope versus inhibitor concentration data set was fitted to a Michaelis-Menten model, k = (kinact × I)/(I + KI), to obtain kinact (maximum rate of inactivation) and KI (inhibitor concentration associated with half-maximal inactivation rate) values. The KI and kinact values were determined by nonlinear regression using SigmaPlot software, version 8.0 (Systat Software, Inc., San Jose, CA), equipped with Enzyme Kinetic module, version 1.1 (SPSS Inc., Chicago, IL). Data for the 300 μM (R)- (S)- and racemic fluoxetine were excluded for analysis because maximal rate of inactivation was consistently found at the 100 μM concentration.
Results and Discussion
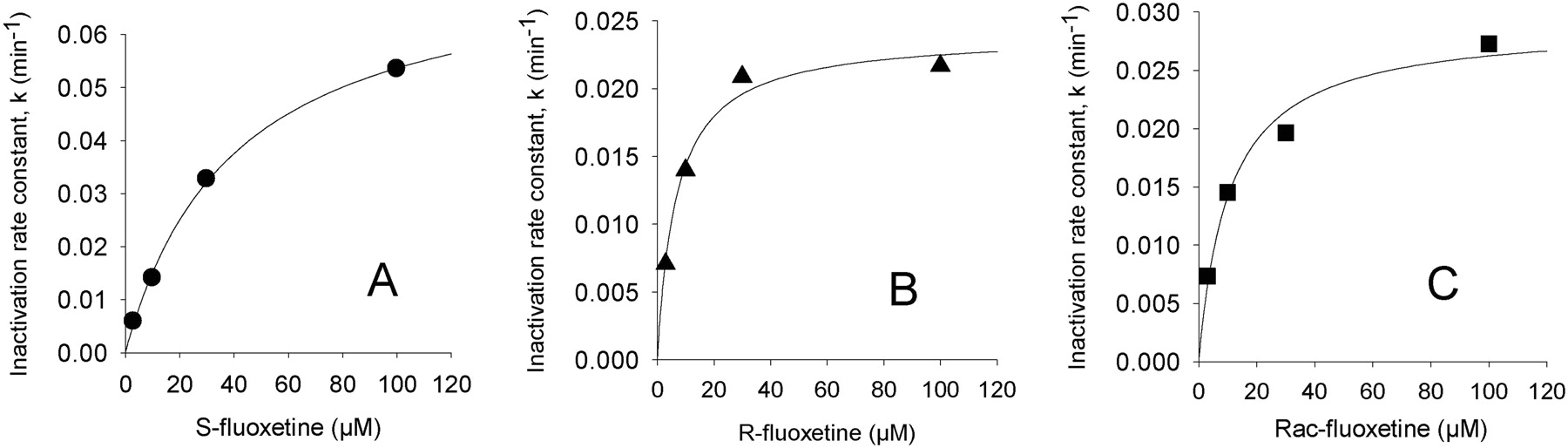
Results for IC50 shifts, kinact, and KI determinations for the inhibition of human liver microsomal CYP2C19-catalyzed S-mephenytoin 4′-hydroxylation are shown Table 1. Obach et al. (2007) demonstrated the utility of IC50 shift assays as an effective and simplified means for assessing the potential for drugs to inactivate P450. Indeed, testing for time- and concentration-dependent inhibition after a preincubation in the presence of NADPH has been advocated in recent U.S. Food and Drug Administration guidance (U.S. Food and Drug Administration, http://www.fda.gov/cber/gdlns/interactstud.htm). In the present study, we found that (S)-fluoxetine exhibited a mean 28-fold IC50 shift, whereas (R)-fluoxetine or racemic fluoxetine exhibited lower shifts of approximately 5- and 9-fold, respectively (Fig. 1). Follow-up studies demonstrated that (S)-, (R)-, and (±)fluoxetine exhibited mean kinact values of 0.085min–1, 0.023min–1, and 0.030min–1, respectively. Mean KI values were 47, 7, and 14 μM for (S)-, (R)-, and (±), respectively. Figure 2 shows representative plots of data used to obtain KI and kinact values. Although the (S)-isomer exhibits an approximately 4-fold more rapid rate of inactivation, the lower affinity for CYP2C19 inactivation makes the (S)-isomer less efficient relative to the (R)-isomer. The kinact and KI values for racemic fluoxetine agree well with the kinact and apparent KI values of 0.03min–1 and 8 μM reported by McGinnity et al. (2006), respectively. Values were not corrected for nonspecific binding, which can be substantial for fluoxetine (Margolis and Obach, 2003). Correcting for unbound fluoxetine, the estimates of KI values would be [10-fold lower (McGinnity et al., 2006)] and are within steady-state total plasma levels of fluoxetine (ranging from 0.15 to 1.5 μM) found after therapeutic dosing of fluoxetine (Orsulak et al., 1988). Obach et al. (2007) has suggested a relationship between the magnitude of drug interactions, I/KI and kdeg/kinact, where kdeg is the in vivo degradation rate of the P450 under investigation. Using an estimate of kdeg of 0.0008 min–1 (Mayhew et al., 2000), we would estimate kinact/kdeg ratios of 25 to 150 and I/KI ratios ranging from approximately 0.1 to 1 for fluoxetine and its isomers. These data indicate the likelihood of drug interactions of fluoxetine and drug cleared predominantly by CYP2C19 may be significant based on the contour plot model suggested by Obach et al. (2007). Indeed, there have been several reports of drugs interactions and/or inhibition by fluoxetine of CYP2C19-mediated metabolism in vivo (Flockhart, 1995; Dingemanse et al., 1998; Harvey and Preskorn, 2001). Consistent with CYP2C19 inactivation is the previous finding that both (R)- and (S)-isomers of fluoxetine are substrates for this enzyme (as well as CYP3A4, CYP2D6, and CYP2C9), with (S)-isomer having marginally higher intrinsic clearance (Margolis et al., 2000).
Summary of CYP2C19 IC50 shift and Ki and kinact values for ticlopidine, (S)-benzylnirvanol, racemic, (S)- and (R)-fluoxetine, and racemic norfluoxetine in human liver microsomes
Values shown are results from experiments conducted in duplicate on independent days.
The mechanism of time-dependent inhibition was investigated by assessing the IC50 shift of a major fluoxetine metabolite, norfluoxetine racemate. We found an 11-fold shift, suggesting that metabolic conversion to this metabolite represents one pathway leading to time-dependent inhibition. Alternate metabolites may be responsible for the time-dependent inhibition found here. Indeed, a recent report suggests that the primary fluoxetine metabolites (S)- and (R)-N-hydroxyfluoxetine exhibit time-dependent inhibition in CYP2C19 Supersomes and are capable of forming a metabolite intermediate complex (VandenBrink et al., 2008).
In our hands, the CYP2C19 mechanism-based inhibitor ticlopidine exhibited a relatively weak but reproducible IC50 shift of 1.8 ± 0.53 and was consistent with previous reports (Obach et al., 2007). In the IC50 shift assay, compounds that are metabolically depleted or cause very rapid inactivation may have little or no shift or even shift in a reverse direction. The latter occurred when we tested the competitive inhibitor (S)-benzylnirvanol (0.41-fold shift) and was probably due to metabolic depletion.
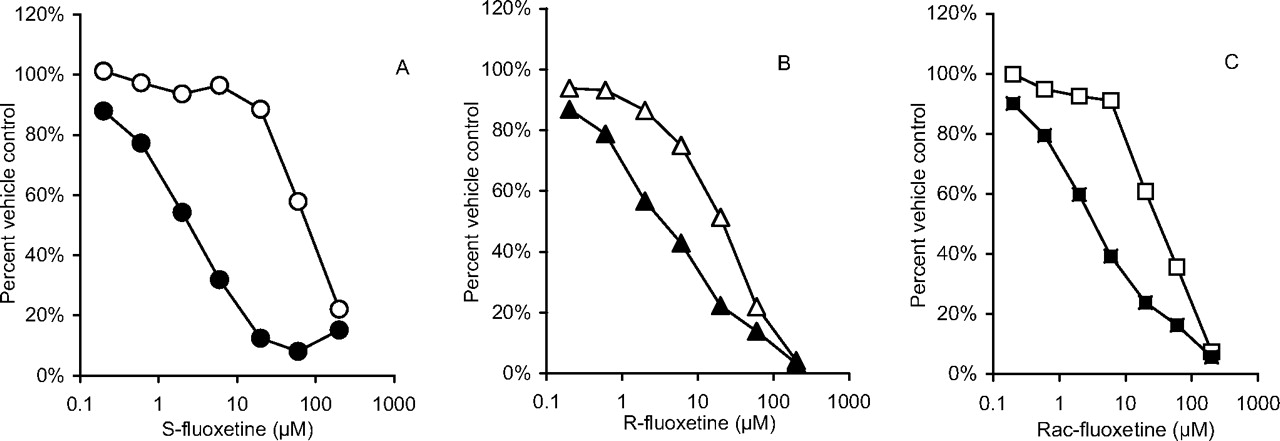
Representative plots of IC50 curves for S-fluoxetine (A), R-fluoxetine (B), and racemic fluoxetine (C). Data points represent means of duplicate incubations. Calculated IC50 shifts for each plot were 33, 5.5, and 9.4, respectively. Open symbols represent data generated after preincubation for 30 min without NADPH, whereas filled symbols represent preincubation for 30 min with NADPH. After the preincubation period, a portion of the incubation was combined with substrate S-mephenytoin and supplemental NADPH and the reaction was allowed to proceed as described under Materials and Methods.

Representative plots of inactivation rate constant (k) and inhibitor concentration for S-fluoxetine (A), R-fluoxetine (B), and racemic fluoxetine (C). The data were generated in pooled human liver microsomes by monitoring the natural log of percentage S-mephenytoin 4′-hydroxylase activity remaining after preincubation times of 2, 6, 11, 17, 23, and 30 min and correction for fluoxetine-independent loss of enzyme activity. Nonlinear regression analysis using the equation k = (kinact × I)/(I + KI) was applied to determine values of kinact and KI.
In summary, we have shown that enantiomers of fluoxetine exhibit time-dependent inhibition of human liver microsomal CYP2C19 and differ in both affinity and rate of inactivation. These data should help in our understanding of potential drug-drug interactions elicited by fluoxetine. From a practical viewpoint, due to its large IC50 shift value, (S)-fluoxetine seems to be a highly robust reference inhibitor for use in the routine measurement of time-dependent inhibition of liver microsomal CYP2C19.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.108.025726.
-
ABBREVIATIONS: P450, cytochrome P450; HLM, human liver microsomes; MS, mass spectrometry; KI, inhibition constant; kinact, maximal inactivation rate constant.
- Received November 19, 2008.
- Accepted January 12, 2009.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}