


Abstract
Nuclear receptors mediate the hepatic induction of drug-metabolizing enzymes by xenobiotics. Not much is known about enzyme induction in liver tumors. Here, we treated tumor-bearing mice with phenobarbital, an activator of the constitutive androstane receptor (CAR), to analyze the response of chemically induced Ha-ras- and B-raf-mutated mouse liver adenoma to CAR activation in vivo. Both tumor subpopulations possess almost identical gene expression profiles. CAR target gene induction in the tumors was studied at the mRNA and protein levels, and a reverse-phase protein microarray approach was chosen to characterize important signaling cascades. CAR target gene induction was pronounced in B-raf-mutated but not in Ha-ras-mutated tumors. Phosphoproteomic profiling revealed that phosphorylation-activated extracellular signal-regulated kinase (ERK) 1/2 was more abundant in Ha-ras-mutated than in B-raf-mutated tumors. ERK activation in tumor tissue was negatively correlated with CAR target induction. ERK activation is known to inhibit CAR-dependent transcription. In summary, profound differences exist between the two closely related tumor subpopulations with respect to the activation of mitogenic signaling cascades, and these dissimilarities might explain the differences in xenobiotic induction of CAR target genes.
Introduction
Juvenile mice treated with the genotoxin diethylnitrosamine (DEN; N,N-diethylnitrous amide) develop liver tumors with activated mitogen-activated protein kinase (MAPK) signaling, a major driver of proliferation and survival, owing to mutations in Ha-ras or B-raf (Moennikes et al., 2000; Aydinlik et al., 2001; Jaworski et al., 2005). Mouse hepatoma with mutant activated Ha-ras or B-raf exhibit strikingly similar transcriptomic and proteomic profiles; characterized, e.g., by a lack of glutamine synthetase (GS) and drug-metabolizing enzymes from the cytochrome P450 family (Loeppen et al., 2005; Jaworski et al., 2007; Rignall et al., 2009; Unterberger et al., 2014).
The nuclear receptor constitutive androstane receptor (CAR) is activated by phenobarbital (PB; 5-ethyl-5-phenyl-1,3-diazinane-2,4,6-trione), certain polychlorinated biphenyls, pesticides, or other compounds (Hernandez et al., 2009; Oshida et al., 2015; Knebel et al., 2018). CAR activation provokes tumor promotion, transient hepatocyte proliferation, hepatocyte hypertrophy, and transcriptional induction of drug-metabolizing enzymes, especially CYP2B and CYP2C (Wada et al., 2009; Molnár et al., 2013; Elcombe et al., 2014; Kobayashi et al., 2015). Induction of drug metabolism by xenobiotics has a major impact on the pharmaco- and toxicokinetics of foreign compounds (Tannenbaum and Sheehan, 2014).
MAPK activation comprises the activating phosphorylation of extracellular signal-regulated kinase (ERK) downstream of Ha-ras and B-raf (Rubinfeld and Seger, 2005). ERK is the most widely used marker for MAPK activation. This potentially links MAPK activation in tumors and CAR activity: Nuclear translocation of CAR involves a dephosphorylation step mediated by protein phosphatase 2A (Mutoh et al., 2013), and ERK activation in liver cells diminishes the inducibility of CAR downstream targets by retaining the receptor in the cytosol (Koike et al., 2007). Mutational MAPK activation might thus render tumor cells insensitive to exogenous stimulation of drug-metabolizing enzymes via CAR. Here, we report a study with PB treatment of tumor-bearing mice to analyze the responsiveness of Ha-ras- and B-raf-mutated mouse liver tumors to CAR activation in vivo.
Materials and Methods
Animal Experiment.
Mouse strain, treatment, and dosing were chosen on the basis of previous studies demonstrating the induction of MAPK-activated tumors by DEN (Moennikes et al., 2000; Aydinlik et al., 2001; Jaworski et al., 2005). Male mice were selected because of easier tumor induction. Twenty-five C3H/HeN wild-type mice (Charles River, Sulzfeld, Germany) received a single intraperitoneal injection of 10 μg of DEN per gram of body weight (in 0.9% sterile NaCl) at 2 weeks of age. Mice received standard feed (Ssniff, Soest, Germany) and tap water ad libitum. Six months later, mice received 0.05% (w/w) PB via the diet (Ssniff) for 4 weeks (Supplemental Fig. 1). Mice were killed by cervical dislocation between 9 and 11 AM to avoid circadian variation; livers were excised, weighed, and either immediately frozen on dry ice or fixed in Carnoy’s fixative. Animals received humane care and the experimental protocol was approved by a local ethics commission. Tumor samples from non-PB-treated mice, obtained following the identical DEN protocol, were available from previous studies (Moennikes et al., 2000; Jaworski et al., 2005).
Immunohistochemical Staining.
Formalin-fixed 10-μm cryosections (20-μm for mutation analysis) or 5-μm sections of paraffin-embedded, Carnoy-fixed tissue were stained using standard methods and antibodies against GS (cat. no. G2781; 1:1000 dilution; MilliporeSigma, St. Louis, MO), E-cadherin (CDH1; cat. no. 610181; 1:100; BD Biosciences, San Jose, CA), CYP2B (gift by Dr. R. Wolf, Dundee, UK; 1:300), phosphorylated (pT202/Y204) ERK1/2 (cat. no. 4376; 1:100; Cell Signaling Technology, Danvers, MA), or 5′-bromodeoxyuridine (BrdU; cat. no. M0744; 1:50; Dako/Agilent, Santa Clara, CA) as previously described (Braeuning and Schwarz, 2010; Braeuning et al., 2010). Nuclei were counterstained using hematoxylin. Images were acquired using a Zeiss Axio Imager microscope (Zeiss, Jena, Germany). Grading of ERK1/2 and CYP2B was performed by microscopic examination of stained slices.
Mutation Analysis.
Samples were punched out using a sharpened cannula as previously described (Braeuning et al., 2014). Following proteinase K digestion, mutation analyses of codon 61 of Ha-ras and codon 637 of B-raf were performed using PCR amplification of DNA in combination with restriction fragment–length polymorphism analysis as described recently (Braeuning et al., 2014).
Gene Expression.
Total RNA was extracted using Trizol (Invitrogen/Thermo Fisher Scientific, Karlsruhe, Germany), and cDNA was prepared from 375 ng of RNA by avian myeloblastosis virus reverse transcriptase (Promega, Mannheim, Germany). Real-time gene expression analysis on a capillary-based LightCycler was performed using the Fast Start DNA MasterPLUS SYBR Green I kit (Roche, Mannheim, Germany). Primers:
Cyp2b10 fwd 5′-TACTCCTATTCCATGTCTCCAAA-3′,
Cyp2b10 rev 5′-TCCAGAAGTCTCTTTTCACATGT-3′;
Cyp2c fwd (recognizing multiple Cyp2c isoforms) 5′-CTCCCTCCTGGCCCCAC-3′,
Cyp2c rev 5′-GGAGCACAGCTCAGGATGAA-3′;
Gstm2 fwd 5′-TGGAACCCAAAGTAGGATTACAAA-3′,
Gstm2 rev 5′-TGAGGACCAAGGCAGCACAC-3′;
Gstm3 fwd 5′-GCACTGTGGCTCCCGGT-3′,
Gstm3 rev 5′- AGGCCTGGGGCAGCTCC-3′;
18S rRNA fwd 5′-CGGCTACCACATCCAAGGAA-3′,
18S rRNA rev 5′-GCTGGAATTACCGCGGCT-3′.
Expression was calculated relative to 18S rRNA expression according to Pfaffl (2001). Statistical analysis was performed using Student’s t test; statistical significance was assumed at P < 0.05.
Western Blotting.
Protein extraction and Western blotting from frozen samples was performed as recently described (Braeuning et al., 2011) using antibodies against phosphorylated (pS217/S221) MEK1/2 (cat. no. 9154; 1:1000; Cell Signaling Technology), ERK1/2 (cat. no. 9102; 1:1000; Cell Signaling Technology), phosphorylated (pT202/Y204) ERK1/2 (cat. no. 4376; 1:1000; Cell Signaling Technology), CYP2B and CYP2C (both a gift from Dr. R. Wolf; 1:500), and ACTB (β-actin; cat. no. 1978; 1:8000; loading control; MilliporeSigma). For paraffin-embedded, Carnoy-fixed tissue, 3–5 mg of a sample were heated to 95°C in 40 μl of lysis buffer (26.7 μl of 2xLDS sample buffer, 13.3 μl of sample reducing agent; both from Invitrogen/Thermo Fisher Scientific) for 15 minutes and, after adding 40 μl of additional lysis buffer, for another 10 minutes. Molten paraffin was separated from the aqueous phase by centrifugation. Protein determination was performed using the bicinchoninic acid assay in iodoacetamide-treated aliquots.
Reverse-Phase Protein Microarray.
Reverse-phase protein microarray (RPPA) profiling of tumors was performed as previously described (Braeuning et al., 2011). Frozen liver tissue (50–80 mg) was ground under liquid nitrogen and lysed with CLB1 lysis buffer (Bayer Technology Services, Leverkusen, Germany). Protein concentration of the lysate was determined by the Bradford assay and adjusted to 0.4 mg/ml. RPPAs were printed as described by Pirnia et al. (2009). Detection of proteins was performed using a two-step immunoassay (antibodies listed in Supplemental Table 1) and Alexa647-labeled secondary antibodies (Invitrogen/Thermo Fisher Scientific). Images of the microarrays were taken and analyzed using the ZeptoREADER microarray imager (Bayer Technology Services) and ZeptoVIEW Pro 3.0 software. The weighted mean of replicate sample spots was used for statistical analysis; S.D. was calculated according to S.E. propagation rules from the S.D. of raw and blank signals. For statistical analysis and graphical representation of the data, MEV 4.8.1 software (Saeed et al., 2006) was used. Hierarchical clustering (Euclidian distance, complete linkage) of median-centered and log2-transformed data were performed to visualize the differences.
Results
As expected (Aydinlik et al., 2001; Jaworski et al., 2005; Braeuning et al., 2014), DEN treatment of mice yielded GS-negative, basophilic and CDH1-positive hepatocellular adenoma, indicative of MAPK activation by Ha-ras or B-raf mutations (data not shown). The CAR activator PB was administered to the tumor-bearing mice to test for the responsiveness of tumor tissue to stimulation by an exogenous signal.
Surprisingly, some tumors from PB-treated animals exhibited pronounced immunoreactivity for CYP2B and CYP2C, but others did not. Ha-ras and B-raf mutation analyses revealed that PB-treated tumors with activated Ha-ras expressed lower levels of the CAR target enzyme CYP2B than tumors with activated B-raf (Fig. 1A). This was observed likewise for the CAR target mRNAs Cyp2b10, Cyp2c, Gstm2, and Gstm3 (Fig. 1B) and confirmed at the protein level (Supplemental Fig. 2). CAR mRNA levels were likewise reduced in both tumor types (Supplemental Fig. 3).

Expression of marker proteins in mouse liver tumors following treatment with the CAR activator PB. (A) Tumors with activated Ha-ras and B-raf lack GS and overexpress CDH1. The model CAR target CYP2B is high in B-raf-mutated but not in Ha-ras-mutated hepatomas following exposure to PB. Representative images are shown. (B) Levels of CAR target mRNAs in normal liver (NL) or tumor tissue with or without PB treatment, as determined by real-time reverse-transcription–polymerase chain reaction (n ≥ 3 per group). Statistical significance: *P < 0.05; **P < 0.01; Student’s t test.
Phosphorylation/dephosphorylation plays a substantial role in the regulation of CAR activity (Mutoh et al., 2013). We therefore conducted an RPPA analysis of Ha-ras- and B-raf-mutated tumors to analyze the phosphorylation of kinases involved in MAPK signaling and/or CAR regulation. Cluster analysis well separated normal liver from the tumors, whereas separation of tumor genotypes was not consistent (Fig. 2). Both tumor types were likewise characterized by their previously known lack of GS and Rh family, B glycoprotein (Jaworski et al., 2007) and elevated CDH1 levels (Hailfinger et al., 2006). The phosphorylated isoforms of numerous proliferation- and/or survival-related kinases were altered in the tumors generally showing hyperphosphorylation (Fig. 2A). Certain phosphorylated, active kinases were present at especially high levels in Ha-ras-mutated tumors, whereas the levels in B-raf-mutated tumors were lower (Fig. 2). Clustering, whose only bases are the active, phosphorylated versions of ERK1/2, ribosomal S6 kinase (RSK) 1, MAPK kinase (MEK) 1/2, and Jun N-terminal kinase (JNK) 1/2, separated the vast majority of tumors with activating Ha-ras mutations from normal liver samples and B-raf-mutated tumors by their high levels of these phospho-proteins (Fig. 2B). Preferential kinase activation in Ha-ras-mutated tumors was verified by immunohistochemistry (Fig. 2C) and Western blotting (Fig. 2D; Supplemental Fig. 2). Immunohistochemically stained tumors were classified into tumors positive or negative for phosphorylated active ERK1/2 and correlated to CYP2B levels. Phospho-ERK-positive tumors generally exhibited weaker staining for CYP2B than their phospho-ERK-negative or only weakly phospho-ERK-positive counterparts (Fig. 2, E and F).
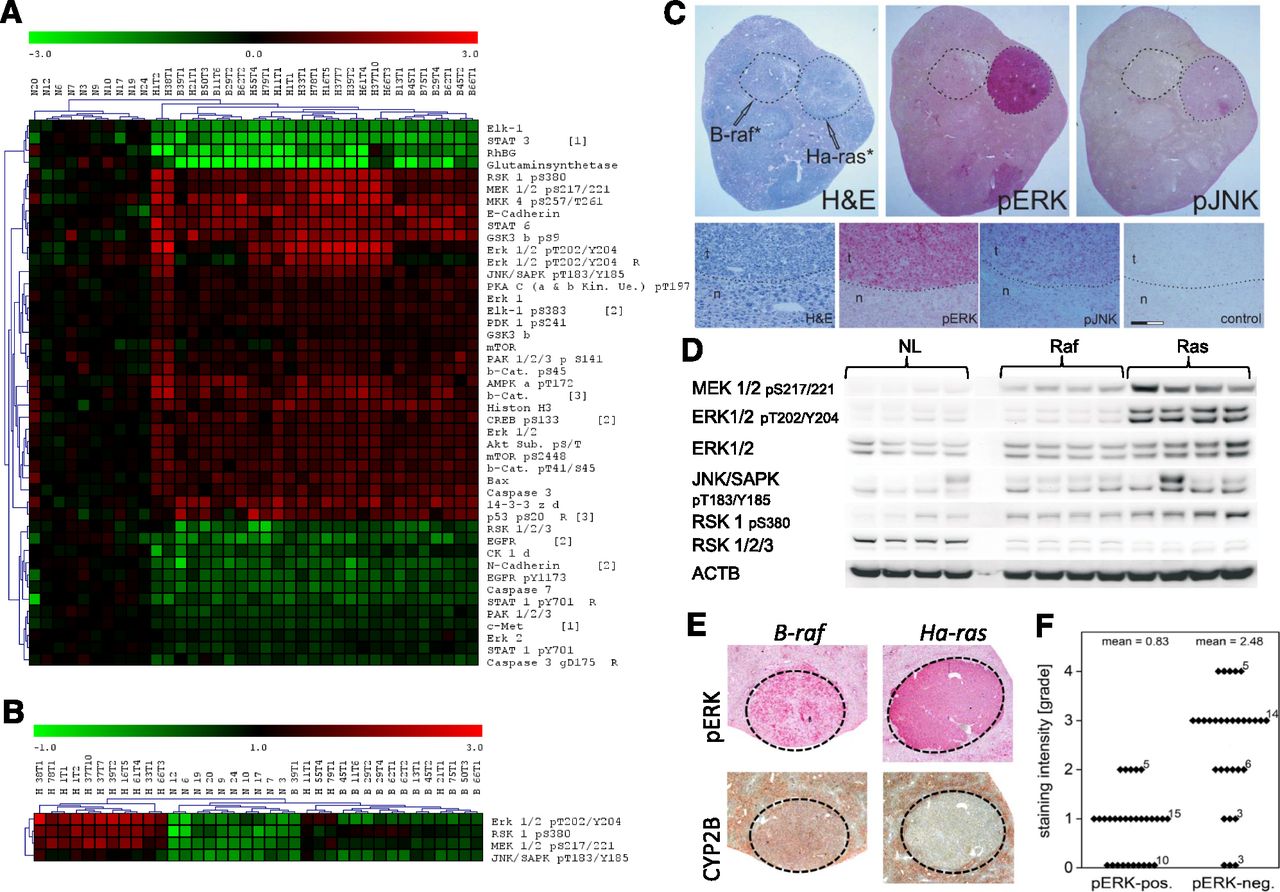
CAR-dependent protein expression in mouse liver tumors. (A) Phospho-proteomic profiling by RPPA, visualized as clustered heat-map. (B) Reclustering on the basis of the activated phospho-forms of selected kinases. Sample designations: N, normal liver; H, Ha-ras-mutated tumor; B, B-raf-mutated tumor. Up- and downregulation are shown in red and green, respectively. (C) Verification of different levels of the phosphorylated active kinases in Ha-ras- and B-raf-mutated liver tumors. Hematoxylin/eosin staining (H&E), staining for ERK 1/2 pT202/Y204 (pERK), and JNK/SAPK pT183/Y185 (pJNK) is shown. Identified gene mutations are indicated. Enhanced display details from the Ha-ras-mutated liver tumor from (C) are also presented. Control staining was performed without the primary antibody. n, normal tissue; t, tumor. Scale bar, 50 μm. (D) Western blotting of tumor tissue protein extracts. Representative blots for the different phosphorylated kinases are comparatively shown for normal liver (NL) and liver tumors harboring activating mutations in either Ha-ras (Ras) or B-raf (Raf). (E) Correlation of PB-induced CYP2B expression, ERK 1/2 phosphorylation status, and mutation status in mouse liver tumors: immunohistochemical staining demonstrating the linkage between ERK 1/2 phosphorylation at T202/Y204 (pERK) and CYP2B expression. Representative images are shown. (F) Correlation of ERK 1/2 phosphorylation and CYP2B expression in tumors, as determined by visual grading of staining intensity of CYP2B-immunostained tissue slices, demonstrating the lack of CYP2B induction by PB in tumors with strong phospho-ERK immunoreactivity. Numbers of investigated tumors are indicated in the graph.
Discussion
We have shown that mouse liver adenoma with activating mutations in Ha-ras or B-raf differ in their activation of kinases from the MAPK pathway. This was unexpected since previous analyses did not reveal remarkable differences between the gene or protein expression profiles of the two tumor types (Jaworski et al., 2007; Rignall et al., 2009). Ha-ras and B-raf activation are believed to activate downstream MAPK signaling in a similar manner, including the kinases found to be differentially activated (Rubinfeld and Seger, 2005). The data altogether suggest that the downstream consequences of MAPK activation must be very similar in these tumors, despite considerable differences in the phosphorylation state of the engaged kinases. These results question the frequent use of ERK1/2 phosphorylation as a surrogate marker for the biologic activity of the MAPK pathway.
In addition to differences in kinase signaling, our study revealed a differential response of tumors with either Ha-ras or B-raf mutations to stimulation with a CAR activator: B-raf-mutated tumors responded with pronounced induction of CAR target genes, as occurs in normal tissue, whereas induction was weak in Ha-ras-mutated tumors. This was also unexpected given the known similarity of the two tumor types (Jaworski et al., 2007; Rignall et al., 2009). Interestingly, kinase activation data provide a possible explanation for the differential behavior of the two tumor subpopulations: ERK1/2 activation inhibits CAR-dependent transcription by retaining the receptor in the cytosol (Koike et al., 2007). Thus, the strong ERK phosphorylation in Ha-ras-mutated tumors may explain the diminished response to CAR activation. Unfortunately, commercially available CAR antibodies did not allow for sufficiently specific immunostaining to verify this hypothesis by demonstrating preferential CAR translocation in B-raf-mutated tumors. It appears improbable that variations in the levels of CAR are the cause of the observed differences: Both tumor types show CAR mRNA levels likewise reduced, whereas B-raf-mutated tumors still exhibit substantial CAR target gene induction comparable to normal tissue. Even though no data are available for other CAR agonists or for other species, one might speculate that similar results would be produced with different CAR activators also in other strains or species.
Of note, PB inhibits the outgrowth of MAPK-activated hepatocytes to manifest tumors when DEN injection is followed by chronic PB treatment (Lee, 2000; Moennikes et al., 2000). We assessed tumor multiplicity, tumor volume, and BrdU incorporation, as a surrogate for cell proliferation, to determine whether the administration of PB at a later time point would still have a similar inhibitory effect. However, PB treatment did not affect the aforementioned parameters, compared with non-PB-treated tumors obtained under otherwise identical experimental conditions (our unpublished data). This indicates that PB might not inhibit the growth of manifest mouse liver adenoma with activated MAPK signaling.
In summary, the present study improves our knowledge on the state of kinase signaling in chemically induced mouse liver tumors and furthermore demonstrates that otherwise very similar tumor subpopulations might react strikingly differently when the tumor cells are exposed to a xenobiotic activator of the nuclear receptor CAR.
Acknowledgments
Technical assistance by Johanna Mahr and Elke Zabinsky is acknowledged. We thank Dr. R. Wolf (Dundee, UK) for the gift of cytochrome P450 antibodies.
Authorship Contributions
Participated in research design: Braeuning, Templin, Schwarz.
Conducted experiments: Braeuning, Kollotzek, Knorpp.
Performed data analysis: Braeuning, Zeller, Knorpp, Templin.
Wrote or contributed to the writing of the manuscript: Braeuning, Schwarz.
Footnotes
- Received June 14, 2018.
- Accepted August 14, 2018.
This work was supported by the Medical Faculty of the University of Tübingen [fortüne program].
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- CAR
- constitutive androstane receptor
- DEN
- N,N-diethylnitrous amide (diethylnitrosamine)
- ERK
- extracellular signal-regulated kinase
- GS
- glutamine synthetase
- JNK
- Jun N-terminal kinase
- MAPK
- mitogen-activated protein kinase
- PB
- 5-ethyl-5-phenyl-1,3-diazinane-2,4,6-trione (phenobarbital)
- RPPA
- reverse-phase protein microarray
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}