


Abstract
With a paradigm shift occurring in health care toward personalized and precision medicine, understanding the numerous environmental factors that impact drug disposition is of paramount importance. The highly diverse and variant nature of the human microbiome is now recognized as a factor driving interindividual variation in therapeutic outcomes. The purpose of this review is to provide a practical guide on methodology that can be applied to study the effects of microbes on the absorption, distribution, metabolism, and excretion of drugs. We also highlight recent examples of how these methods have been successfully applied to help build the basis for researching the intersection of the microbiome and pharmacology. Although in vitro and in vivo preclinical models are highlighted, these methods are also relevant in late-phase drug development or even as a part of routine after-market surveillance. These approaches will aid in filling major knowledge gaps for both current and upcoming therapeutics with the long-term goal of achieving a new type of knowledge-based medicine that integrates data on the host and the microbiome.
Introduction
The promise of precision medicine, wherein drugs are tailored to individuals based on genetic and environmental risk factors, requires a more comprehensive understanding of the complex determinants of interindividual variation in drug metabolism and disposition. Substantial progress has been made by pharmacogenomics research in industry and academia (Relling and Evans, 2015), leading to actionable understandings of interactions between 27 genes and 87 drugs (Whirl-Carrillo et al., 2012). For example, avoiding administration of codeine to ultra-rapid CYP2D6 metabolizers can prevent toxicity and overdose, especially in children (Crews et al., 2012; www.fda.gov/downloads/Drugs/DrugSafety/UCM553814.pdf). However, it is now clear that the interindividual genetic differences in the trillions of microorganisms that colonize the human body (the microbiota) far exceed that of our human genome (Qin et al., 2010). These differences can have meaningful consequences for drug pharmacokinetics and pharmacodynamics due to the direct metabolism of drugs by human gut bacterial enzymes and even more complex host-microbiome interactions (Spanogiannopoulos et al., 2016). This emerging field of study, often referred to as pharmacomicrobiomics (Rizkallah et al., 2010), focuses on how genetic and phenotypic diversity in the microbiome affects therapeutic outcomes and is long overdue for a renaissance given the clear relevance for human health and disease and the broader impacts for our understanding of microbial metabolism.
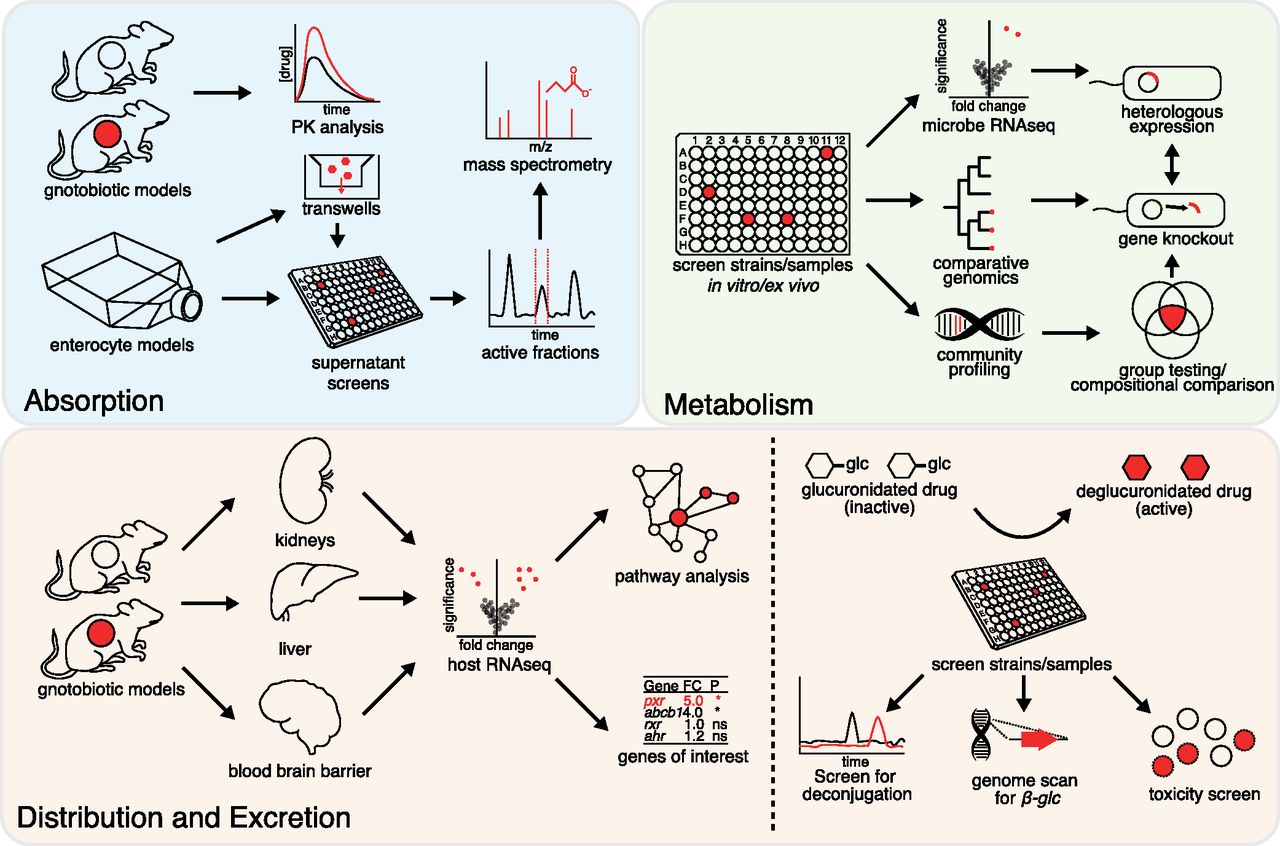
Here, we provide a how-to guide for the current suite of tools that can be leveraged to detect and unravel the mechanistic basis for interactions between host-associated microbial communities and drugs, with the long-term goal of integrating this information into clinical practice. We detail these methods (Fig. 1; Table 1) through their relevance to each classic component of a therapeutic drug’s absorption, distribution, metabolism, and excretion profile. In each section, we describe a relevant methodology that may be applied to discover drug-microbe interactions and understand their mechanistic basis. Although the human microbiome encompasses multiple distinct body habitats, including the oral cavity, genitourinary tract, and skin, we focus predominantly on the gastrointestinal tract (or gut) due to the limited scientific literature regarding the impact of the broader microbiome on pharmacology. Importantly, progress in this area requires more than DNA sequencing, necessitating microbiome researchers to bridge the often-impenetrable divide between biology, chemistry, pharmacology, and computer science.

Methods for studying drug-microbiome interactions. Strengths and limitations of these approaches are discussed in Table 1. Details of each approach is found throughout the main text. FC, fold change; glc, glucuronide; ns, not significant; PK, pharmacokinetics; RNAseq, RNA sequencing.
Comparison of methods for studying drug-microbiome interactions
Mastering Microbial Metabolism
The ability of the gut microbiome to contribute to the metabolism of drugs has been recognized for over 80 years (Fuller, 1937). Although inactive in vitro against most bacteria, the first-ever antibiotic prontosil was effective in animals. Subsequent studies would reveal that microbial cleavage of the azo bond yields the antibiotic sulphanilamide (Fuller, 1937). Not all microbial drug metabolism is beneficial. The coadministration of the antiviral sorivudine alongside fluoropyrimidine chemotherapy results in extreme toxicity due to a microbial-sorivudine metabolite blocking host fluoropyrimidine degradation, causing subsequent toxic buildup of fluoropyrimidine metabolites (Nakayama et al., 1997; Okuda et al., 1998).
At least 50 drugs are known to be metabolized by bacteria, and in most of these cases, neither the microbial species nor genetic determinants responsible for the metabolism are known (Spanogiannopoulos et al., 2016). Furthermore, since no large-scale systematic screens for bacterial metabolism have taken place, the number of drugs metabolized by the microbiome is likely much higher. A recent study screened a collection of >1000 clinically approved drugs, mostly designed for human targets, against 40 representative gut bacterial strains and demonstrated that many of them inhibited bacterial growth in vitro (Maier et al., 2018); however, it was not determined whether metabolism took place. Such systematic screening methodologies coupled with analytical techniques for drug quantification and speciation will help uncover additional interactions between drugs and the gut microbiome. The simplest assays to determine the presence and extent of microbial metabolism involve screening representative strain libraries and/or ex vivo incubations with human stool samples.
The veritable tidal wave of human microbiome sequencing data has helped to identify the most prevalent and abundant members of the microbiome (Nayfach et al., 2015). These data have helped guide the systematic isolation of strain collections across multiple research groups (Goodman et al., 2011; Allen-Vercoe, 2013; Lagier et al., 2016). It is now possible to select and screen collections representing broad panels of human-associated strains for drug metabolism via high-throughput culturing. By coincubation of each strain or a collection, referred to as a synthetic community, with each drug, it is possible to observe loss of the substrate and/or the production of new metabolites. With the use of high-density plate-based culturing and robotics, analytical method development for the drug or drugs of interest is a primary bottleneck. This typically involves high-performance liquid chromatography and/or mass spectrometry.
Although high-throughput culture collection screens have the advantage of identifying bacteria capable of drug metabolism with straightforward interpretation, these methods provide only a shallow sampling of microbial genetic diversity. Much of the variation within a species is rendered via horizontal gene transfer: the wholesale movement of blocks of genetic material between cells, which is commonly associated with antibiotic resistance (Riley and Lizotte-Waniewski, 2009). However, many phenotypes, including the ability to inactivate the cardiac drug digoxin, appear sporadically within a species (Koppel et al., 2018). The a priori selection of representative strains is likely to exclude genes of interest due to shallow sampling at the species level. For example, the selection of a single representative strain of Escherichia coli, such as K-12, would sample just 19% of the known genetic diversity in the species: ∼4400 genes out of the 23,107 observed across the E. coli pan-genome (Ding et al., 2018). Nonetheless, this approach has been successful in identifying multiple drug-metabolizing bacterial species (Peppercorn and Goldman, 1972; Caldwell and Hawksworth, 1973; Saha et al., 1983; Strong et al., 1987; Hattori et al., 1988; Shu et al., 1991; Kitamura et al., 1997; Rafii et al., 1997; Shelton et al., 1997).
As an alternative to culture-collection screens, the use of ex vivo incubations of human stool samples may be considered. This method has the advantage of capturing a greater diversity of organisms and has been successfully applied to identify numerous drugs that are susceptible to microbial metabolism (Valerino et al., 1972; Caldwell and Hawksworth, 1973; Smith and Griffiths, 1974; Koch and Goldman, 1979; Powis et al., 1979; Koch et al., 1980; Lindenbaum et al., 1981; Elmer and Remmel, 1984; Harris et al., 1986; Strong et al., 1987; Shu et al., 1991; Kim et al., 1992; Lavrijsen et al., 1995; Watanabe et al., 1995; Kitamura et al., 1997; Sasaki et al., 1997; Basit and Lacey, 2001; Basit et al., 2002; Deng et al., 2011; Li et al., 2012; McCabe et al., 2015). However, when testing a community of microorganisms collectively, interstrain antagonism may mask metabolism. This may be particularly true if the metabolizing organisms are inhibited by the antimicrobial products (e.g., organic acids or bacteriocins) of metabolically inert strains. Additionally, depending on the medium formulation, some organisms may have a strong growth advantage enabling them to dominate the culture. One strategy to combat this is to perform ex vivo incubations across multiple medium formulations with broad selection pressures in place for Gram-positive, Gram-negative, aerobic, strict anaerobic, and spore-forming organisms.
Another approach for studying microbial biochemical transformations from a mixed community is by using a “fecalase” preparation (Tamura et al., 1980), which is a cell-free extract of feces including microbial enzymes (typically starting from a stool sample). Fecalase preparations can be assayed against drugs for metabolism. This method has been used to assay gut microbial enzyme activity toward herbal glycosides generating genotoxic aglycone products (Tamura et al., 1980; Yeo et al., 2012). This technique was also used to demonstrate that gut bacteria can metabolize the cholesterol-lowering drug lovastatin (Yoo et al., 2014). A complicating factor that may lead to false negatives in this approach is the availability of, or competition for, necessary cofactors and membrane-bound coenzymes. These may or may not be present in the fecalase preparation. Also, the release of cytosolic enzymes which may not function in buffer conditions and the lack of continuing turnover of NAD(P)/FAD in the cell may dampen observed metabolic activity. As a result, the sensitivity of this approach can be difficult to determine.
Depending on the methodology applied for detecting metabolism, a number of complementary approaches may be applied to uncover the molecular mechanisms responsible. If it has already been shown that a specific microbial strain is capable of metabolism, multiomics can be particularly informative. In the bacterial genome, functionally related genes are typically colocalized and transcriptionally regulated as operons. Upon stimulus by a ligand, specific expression of effector genes is induced. It is possible to exploit this functionality to identify candidate effector enzymes and molecules through gene-expression analysis, such as RNA sequencing, by contrasting expression between drug-exposed and vehicle controls. For example, we used RNA sequencing to identify the “cardiac glycoside reductase” operon responsible for the inactivation of digoxin through its >100-fold induction upon incubation with digoxin (Haiser et al., 2013; Koppel et al., 2018). This method has the advantage of requiring only a single metabolizing strain for study and can even be applied to intact human gut microbial communities (Maurice et al., 2013). However, multiple caveats exist. Not all enzymes are under substrate-mediated transcriptional regulation. Furthermore, care needs to be taken to sync the growth phase of control and drug-exposed cultures to avoid false positives due to changes in cellular physiology. Finally, many nonantibiotic drugs can have bacteriostatic or even bacteriocidal effects during in vitro growth (Maier et al., 2018), potentially causing profound changes in gene expression due to stress responses. Once candidate genes are identified, their induction can be readily confirmed by quantitative reverse-transcriptase polymerase chain reaction on independent cultures; however, follow-up studies are critical to make causal links between the identified genes and specific biochemical reactions.
As a complementary approach to expression-based methods, comparative genomics has shown considerable promise in identifying gene candidates while also providing critical information about genetic conservation with important ramifications for the translation of these results. Recently, by screening a collection of 25 Coriobacteriia strains and using our ElenMatchR tool for comparative genomics (Bisanz et al., 2018), we were independently able to identify a single genomic locus conserved in all strains capable of digoxin metabolism (Koppel et al., 2018). Heterologous expression confirmed that a single gene within this locus, the cardiac glycoside reductase 2 gene (cgr2), is sufficient for the reduction of digoxin to its inactive bacterial metabolite dihydrodigoxin (Koppel et al., 2018). In addition to identifying the effectors, we were able to establish that the genes are highly conserved and use this information to design a duplexed assay for the simultaneous detection of the effectors and their species of origin in stool specimens, providing a high-throughput assay for screening patient samples.
Importantly, the bulk of the time investiture for this comparative genomics method is up front, requiring the curation of a collection of strains and genome coding sequences clustered into orthologous groups representing functional clusters. Once established, these collections can be screened multiple times with new substrates and the tools recycled time and time again for new candidate interactions. When cases of convergent functional evolution are present, i.e., two independently evolved effectors with varied amino acid sequence, interpretation can become more difficult; however, machine learning approaches, such as random forests, are particularly adept at uncovering these patterns, as we demonstrated with two divergent ribosomal protection proteins conveying resistance to tetracycline in Eggerthella lenta (Bisanz et al., 2018). Candidate genes can be further prioritized based on biochemical intuition, genomic context, and the scientific literature to match predicted function to biochemical reactions.
A number of genetic screening methodologies have been developed for gut bacteria and have helped identify genetic determinants responsible for various functions from host colonization to nutrient acquisition (Powell et al., 2016; Hibberd et al., 2017). These methods have not yet been widely applied to study drug metabolism from the gut microbiome but should be valuable moving forward. One significant challenge in studying gut bacteria is the lack of tools for genetic manipulations in the vast majority of identified species, limiting which methods can be applied to particular species. In these instances, methods such as functional genomics can be applied, where DNA is expressed using a surrogate organism. This approach is often carried out in E. coli; however, other model gut organisms, such as Bacteroides thetaiotaomicron, have been developed for this purpose (Lam et al., 2018). Gene knockout libraries involve the systematic, or random, creation of inoperative genes across an organism’s genome, either in an arrayed or pooled collection. These libraries are screened for a phenotype, or lack thereof, and then mapped to identify the gene determinant responsible. Concerning gut bacteria, these methods are mostly limited to the model gut bacteria Escherichia and Bacteroides species but have been valuable in understanding microbial interactions with drugs in vitro (Tamae et al., 2008) and in the host (García-González et al., 2017; Scott et al., 2017).
Where ex vivo incubations have been applied, either 16S ribosomal RNA gene amplicon sequencing or metagenomic shotgun sequencing may be used to associate microbial abundance and gene content with metabolic capability. To further the utility of this approach, a combinatorial group testing approach can be used wherein rationally designed pools of samples and/or isolates are tested for activity. By deconvoluting which microbes and genes are associated with activity across the pools, a greater sampling of diversity can be obtained with less analytical cost (Dorfman, 1943).
Alternative Alterations to Absorption
The bioavailability and absorption of an orally administered drug is a major barrier to clinical efficacy, with ∼40% of candidate drugs abandoned due to poor bioavailability and/or pharmacokinetics in the 1990s (Kola and Landis, 2004). The parameters governing absorption typically lie in the chemical properties of the drug itself and how the host may actively import, or export, the molecule. Early microbiome studies hinted that drug transport may be influenced by the microbiome in a composition-dependent manner (Hooper et al., 2001), and multiple animal studies have demonstrated that the microbiome modulates drug absorption (Kim et al., 2018; Subramanian et al., 2018). In this section, we highlight methods for examining the influence of microbes on drug transport.
Cell-based drug-absorption assays are among the most common methods of in vitro screening of absorption parameters (Artursson et al., 2001) and are readily adopted for high-throughput screening (Larson et al., 2012). The Caco-2 cell line (ATCC HTB-37; American Type Culture Collection, Manassas, VA) is the preferred model for these types of assay because of its ability to differentiate into a monolayer resembling the enterocytes of the small intestines with characteristic expression of transporters including P-glycoprotein and organic anion transporting polypeptide A/B. Furthermore, parameters generated in Caco-2 cells have been shown to correlate with human studies (Artursson et al., 2001). Concerns of interlaboratory variation in Caco-2 lines have also led to the generation of clonal derivatives (Peterson and Mooseker, 1992). When grown on a semipermeable membrane, as in Transwells (Corning, NY), a polarized monolayer is formed with an apical and basolateral chamber functionally equivalent to the intestinal lumen and underlying circulation, respectively. The apical to basolateral and basolateral to apical transport are measured for a candidate drug or appropriate surrogate compound, such as P-glycoprotein model substrates digoxin, calcein-AM, or rhodamine 123. More complex models of Caco-2 cells, such as the “gut-on-a-chip” and HuMiX models, allow for the development of three-dimensional structures including villi and divergent cell types with concomitant enhancement of CYP3A4 activity while facilitating coculture with bacterial cultures (Kim and Ingber, 2013; Shah et al., 2016).
To test the effect of microbiome-drug interactions in these models, microbes, or preparations thereof, can be spiked into the Transwell system in the apical compartment and the effect on transport measured. Live bacteria may be used with caution, but this is not advised as their metabolic activity may have unintended consequences, such as extreme acidification of the medium by viable lactic acid bacteria. Transepithelial electrical resistance and/or indicators of cytotoxicity, such as l-lactate dehydrogenase release, should be monitored to rule out effects due to cell death. Heat-killed or gamma-irradiated microbes may be a preferable treatment to retain the presence of cell-associated structures including the microbe-associated molecular patterns that have known interactions with transporters (Frank et al., 2015). Cell-free supernatants spiked into cell culture medium represent the most attractive preparation when prolonged incubation is desired and/or microbial metabolites are suspected to be the causative agent. Organoids, a form of three-dimensional cell culture, offer additional promise for studying the effects of transport. Models using the fluorescent model P-glycoprotein substrate rhodamine 123 offer the advantage of allowing real-time tracking and kinetic analysis (Mizutani et al., 2012). Alternatively, nonimmortalized primary human cells may be seeded on Transwells (Bhatt et al., 2018). Although a highly relevant model, growth medium costs and availability of human surgical samples may limit the availability of this approach for many groups.
Although in vitro models are an attractive option, there is still considerable need for animal models which encompass a level of complexity not possible in cell culture. In particular, pharmacokinetic analyses of gnotobiotic mice and other animals allow isolation of the in vivo impact of single microorganisms or complex consortia. Gnotobiotic animals are raised in germ-free conditions and can subsequently be colonized with an arbitrary combination of microbes capable of host colonization (Williams, 2014). These models are the microbiome scientist’s equivalent of transgenic animals wherein bacteria can be knocked in, or out, of communities by design. Pre-existing mouse lines are routinely “rederived” in a germ-free state by surgically removing the offspring, sterilizing their external surface, and transferring them into isolators where everything the animal will come into contact with has been sterilized. Although mice are by far the most frequently used gnotobiotic model, rats and even pigs can also be made germ-free (Miniats and Jol, 1978). Pigs offer the advantage of having a gastrointestinal tract physiologically more similar to humans (Zhang et al., 2013) but require considerably more resources and may not be an attractive model for early-stage research. Despite gnotobiotic facilities increasing in number globally, other options can be used where gnotobiotic models are unavailable or prohibitively expensive. Antibiotic knockdown of endogenous microbes in conventional animals is an acceptable alternative; however, issues involving reproducibility and off-target effects on host cells represent barriers to the use and interpretation of this approach (Lundberg et al., 2016). In addition, the native rodent microbiome shares little resemblance to the human microbiome in strain and gene content (Xiao et al., 2015), and it can prove difficult or impossible to fully supplant endogenous communities with human-associated microbes in any long-term capacity (Staley et al., 2017). Nonetheless, the use of antibiotic-knockdown models has successfully demonstrated effects on drug transport in the case of lovastatin (Yoo et al., 2014).
Multiple mechanisms through which the microbiome controls host transport have been proposed, but broadly speaking, they involve regulation of host gene expression, substrate competition, or binding of microbial products acting as inhibitors or allosteric regulators. Examination of host expression is possible in a targeted manner using quantitative reverse-transcription polymerase chain reaction panels, or globally via RNA sequencing or similar approaches. Assessment of codifferentially regulated genes may shine light on the pathways through which modulation occurs. P-glycoprotein, for example, is under the influence of pregnane X receptor (PXR), farnesoid X receptor, nuclear factor κ-light-chain-enhancer of activated B cells, and activator protein 1 pathways (Miller, 2010), but examination of coregulated transcripts using pathway analysis tools may help determine the specific pathway that is active (Krämer et al., 2014). It has been demonstrated that microbial-derived compounds including indole metabolites can function as PXR ligands (Venkatesh et al., 2014). The intestinal environment also contains a variety of cell types, many of which do not have absorptive function. To target the absorptive enterocytes, laser capture microdissection or single-cell RNA sequencing could be applied to limit differential expression analyses to target cell types of interest. This may be particularly important in the case of genes which have functions in other cell types, such is the case with P-glycoprotein expressed in phagocytes and T cells (Ramesh et al., 2014; Cory et al., 2016).
To disentangle expression and functional inhibition, constitutive expression cell lines can obtained or created. As an example, the Madin-Darby canine kidney cell model (MDCK) was modified for this purpose through the constitutive expression of P-glycoprotein through a retroviral vector (Pastan et al., 1988). Use of these models can establish the direct modulation of transporter activity. In addition to the methods previously identified, fractionation of microbial preparations based on size and/or chemical properties can be used to narrow down candidate modulators. The use of high-throughput cell-culture assays facilitates the rapid screening of fractions for activity, and then analytical approaches can be further applied to identify effectors.
When studying interactions between microbes and models of the small intestine, it should be noted that fecal samples do not necessarily represent the composition or absolute abundances of the small intestinal community, and modeling these differences should be incorporated where possible (Zoetendal et al., 2012).
Demonstrating Diversity in Distribution
Whereas many drugs can passively distribute through the body (e.g., ethanol), some require facilitated movement via transporters. Even drugs that readily diffuse through membranes are often impeded at barriers, such as the blood-brain barrier (BBB), blood–cerebrospinal fluid barrier, placental barrier, and blood-testes barrier. Interindividual variability in transporter and barrier function alters the extent of drug distribution past these sites and can be attributed to genetic polymorphisms or physiologic changes, such as pregnancy or disease, that affect the function or expression levels of transporters and proteins at target sites and barriers (Hoffmeyer et al., 2000; Hebert et al., 2008; Hammarlund-Udenaes et al., 2013).
Given the previously mentioned potential of bacterial metabolites to regulate expression patterns of transporters and enzymes, exploration of barrier modulation by bacteria requires further study. Typical in vitro cell assays, such as BeWo trophoblastic cell lines of the placental barrier and the hCMEC/D3 cell monolayer model of the BBB, can be augmented by spiking the system with cell-free supernatants to determine induction or inhibition of key enzymes and transporters at these barriers. Recent studies have explored modulation of cell barriers by exposing cell lines to known bacterial metabolites of interest, such as short-chain fatty acids (Hoyles et al., 2018). These findings could be readily extended to include experiments with supernatant preparations, thereby allowing exploration of novel bacterial modulators.
In vivo assessments of microbiome-induced barrier modulation can likewise be conducted via use of gnotobiotic (i.e., microbial knockouts) or antibiotic depletion (i.e., microbial knockdown) rodent models. As mentioned previously, antibiotics have been useful in assessments of distribution, but when possible, gnotobiotic models should also be used to mitigate off-target effects of the antibiotics on host tissues, such as PXR activation by rifampicin (Cheng et al., 2009). In both gnotobiotic and antibiotic knockdown models, there is mounting evidence of a microbiome modulation of the BBB. Compared with conventional mice, germ-free and antibiotic-treated mice exhibit increased permeability of the BBB and decreased expression of occludin and claudin-5 tight junction proteins (Fröhlich et al., 2016), which is reversible upon introduction of sodium butyrate or by monocolonization of butyrate-producing bacteria (Braniste et al., 2014). An important point to keep in mind when conducting distribution assays in rodents is the interspecies variation in PXR ligands; typical rodent models do not accurately recapitulate the range of ligand activation possible for human PXR. As a result, future exploration of nuclear hormone receptor modulation by the microbiome would benefit from the use of “humanized” rodent models when possible (Ma et al., 2007).
Beyond transporters, enzymes, and barrier proteins, the extent of drug distribution is also governed by the rate of fluid (blood, interstitial fluid, cerebrospinal fluid) exchange between compartments and the fraction unbound of drug in the plasma. Studies using ultracentrifugation, ultrafiltration, or equilibrium dialysis can be conducted to determine the binding affinity of microbial metabolites for common plasma proteins, such as albumin and α1-acid glycoprotein, which bind acidic and basic therapeutics, respectively (Liu et al., 1999). Microbial metabolites displaying high affinity for plasma proteins have the potential to displace drug from plasma proteins, thereby increasing both the fraction unbound and, by extension, the apparent volume of distribution.
Et Vous Excrétion?
Drug detoxification and excretion, key components of drug disposition, are primarily driven by hepatocytes for most medications. The liver and gut are close neighbors, connected by the biliary tract and portal vein, both of which allow for a ready exchange of metabolites (host and microbially derived) and other compounds. Multiple lines of evidence point to a gut-liver axis in which both the microbiome and liver influence each other through biliary excretion and recycling, signaling, and regulation of gene expression (Tripathi et al., 2018). In addition, increasing evidence points to the importance of the gut-liver axis in drug excretion, such as the anticancer drug irinotecan (Wallace et al., 2010) and the Alzheimer’s drug tacrine (Yip et al., 2018), both of which are discussed later.
The most commonly used methodologies to study microbial mediation of hepatic metabolism and transport are gene expression based, frequently involving gnotobiotic or antibiotic-depleted rodent models. For drugs impacted by a known, specific signaling pathway, hepatic transporter, or enzyme, liver expression studies with and without the drug of interest may illuminate the microbiome’s impact on metabolism and clearance. Similar to bacteriological gene-expression experiments, great care must be taken to avoid spurious results. A multitude of xenobiotic metabolism pathways are known to be under the influence of circadian rhythm (DeBruyne et al., 2014), and as such, if the germ-free group is sacrificed in the morning and the colonized group after lunch, investigators could falsely conclude that differential expression was the result of the microbiome. Animal sacrifices should be carefully and reproducibly timed, and animals should be interleaved between colonization groups to prevent these artifacts from skewing results. In addition to gene-expression analysis, metabolomic techniques may be valuable for the detection of microbial mediators of modulation.
A recent study demonstrated differential expression of xenobiotic metabolism in livers of germ-free or conventionally raised mice, demonstrating that microbial metabolites of tryptophan act as aryl hydrocarbon receptor activators (Selwyn et al., 2015). Additionally, the colonization of germ-free mice can result in increased hepatic expression of Cyp3a11 and Cyp2c29 (Claus et al., 2011). As well as acting as paradigms, these studies offer the potential for meta-analysis and recycling of data to determine effects on a particular investigator’s favorite gene. Indeed, some of these data sets are available with user-friendly interfaces (http://microbiota.wall.gu.se/) (Larsson et al., 2012). Researchers with a particular gene of interest can perform a preliminary analysis with these tools prior to designing their own experiments.
Although less used, human hepatic cell assays in tandem with microbial metabolites provide a less-resource-intensive, in vitro approach for understanding the impact of the microbiome on host gene expression. For example, Hubbard et al. (2015) assayed human immortalized hepatocytes (HepG2) and demonstrated varied gene aryl hydrocarbon receptor expression with the microbial tryptophan metabolite indole.
The most well characterized impact of the gut microbiome on drug excretion is through bacterial deglucuronidation. Host glucuronidation of xenobiotics contributes to elimination from the host through renal or biliary excretion (Wells et al., 2004). Ex vivo incubations may be a good first step towards evaluating if hepatic glucuronidation and subsequent gut microbial metabolism following reintroduction to the intestinal tract may predict adverse effects. A seminal example of this approach comes from the study of the side effects presented in a subset of patients taking irinotecan, a widely used chemotherapeutic for colon cancer. Its metabolite, compound SN-38, undergoes glucuronidation in the liver, resulting in SN-38G (SN-38 glucuronide). Following excretion to the intestine SN-38G is acted upon by β-glucuronidases that revert the metabolite to SN-38 and, as a result, cause severe diarrhea (Wallace et al., 2010). The link between patient variation in this side effect and the gut microbiome was hypothesized and recently quantified by Guthrie et al. (2017). Utilizing healthy stool sample incubations with irinotecan, they identified highly efficient or non–drug metabolizers based on quantities of irinotecan derived from the glucuronated metabolite, then used a metagenomics approach to identify β-glucuronidases gene abundance as the driving distinction between these two groups (Guthrie et al., 2017).
The approaches discussed here require a priori knowledge of the pathway for drug excretion and its potential link to the microbiome. Alternatively, unbiased “systems biology” approaches can be used to quantify drug pharmacokinetics, host genetics, and microbial community structure and function. This approach has helped to link specific microbial metabolites to drug excretion (Clayton et al., 2009). Recently, this approach was used to elucidate the source of variations in hepatotoxicity observed in patients taking tacrine, a cholinesterase inhibitor approved for treating Alzheimer’s disease. Similar to irinotecan, following hepatic glucuronidation of tacrine, the gut bacterial β-glucuronidases contribute to enteric reactivation of tacrine, increasing liver toxicity (Yip et al., 2018).
Fomenting Future Pharma
Importantly, many of the approaches described here can be readily applied to human cohorts, enabling the adoption of this strategy in late-phase drug development or even as a part of routine after-market surveillance. Ideally, these data would be paired with in-depth and publicly accessible data from in vitro and in vivo models generated during the preclinical efficacy and toxicity testing of each new medication. A more difficult challenge will be to fill the gaps in our knowledge for the current set of Food and Drug Administration–approved drugs, requiring new efforts to establish partnerships between academia and industry built upon mutual respect and devotion. These efforts will have a transformative impact on our understanding of drug disposition, helping to answer long-standing questions about the source of variation in drug response and side effect profiles between patients. However, the challenges ahead are unprecedented, requiring nothing less than a reconsideration of the way in which we treat human disease and a new type of physician scientist with deep knowledge of both human physiology and the microbiome. Indeed, the patient of the future may look back on our primitive “microbiome-naive” medicine just like we now view the use of pharmaceuticals without respect for polymorphisms in the human genome.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Bisanz, Spanogiannopoulos, Pieper, Bustion, Turnbaugh.
Footnotes
- Received June 28, 2018.
- Accepted August 13, 2018.
This work was supported by the National Institutes of Health [R01HL122593] and the Searle Scholars Program [SSP-2016-1352]. P.J.T. is a Chan Zuckerberg Biohub investigator and a Nadia’s Gift Foundation Innovator supported, in part, by the Damon Runyon Cancer Research Foundation [DRR-42-16]. P.S. is supported by a Canadian Institutes of Health Research fellowship. J.E.B. was the recipient of a Natural Sciences and Engineering Council of Canada fellowship.
Abbreviations
- BBB
- blood-brain barrier
- PXR
- pregnane X receptor
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}