


Abstract
Colon microbiota-based drug metabolism has received little attention thus far in the process of drug development, whereas the role of gut microbiota in clinical safety and efficacy of drugs has become more clear. Many of these studies have been performed using animal studies, but the translational value of these data with respect to drug pharmacokinetics, efficacy, and safety is largely unknown. To investigate human colon microbiota-mediated drug metabolism, we applied a recently developed ex vivo fermentation screening platform, in which human colonic microbiota conditions are simulated. A set of 12 drugs (omeprazole, simvastatin, metronidazole, risperidone, sulfinpyrazone, sulindac, levodopa, dapsone, nizatidine, sulfasalazine, zonisamide, and acetaminophen) was incubated with human colon microbiota under strictly anaerobic conditions, and samples were analyzed using high-performance liquid chromatograph–UV–high-resolution mass spectrometry analysis. The human microbiota in the fermentation assay consisted of bacterial genera regularly encountered in human colon and fecal samples and could be reproducibly cultured in independent experiments over time. In addition, fully anaerobic culture conditions could be maintained for 24 hours of incubation. Five out of the 12 included drugs (sulfasalazine, sulfinpyrazone, sulindac, nizatidine, and risperidone) showed microbiota-based biotransformation after 24 hours of incubation in the ex vivo fermentation assay. We demonstrated that drug metabolites formed by microbial metabolism can be detected in a qualitative manner and that the data are in accordance with those reported earlier for in vivo metabolism. In conclusion, we present a research tool to investigate human colon microbiota-based drug metabolism that may be applied to enable translatability of microbiota-based drug metabolism.
Introduction
Within drug development, drug candidates are extensively tested for efficacy and safety. One of the major concerns for drug efficacy and safety is the metabolism of the drug and rate at which it is eliminated from the body. Whereas the major site of metabolism in the human body is the liver, where drugs and their metabolites are subsequently eliminated from the body through biliary excretion and successive fecal secretion via the gastrointestinal tract, recent insights elucidate the influence of gut microbiota on the metabolism of key therapeutics. Since the colon is identified by its high density and unique collection of bacteria, viruses, and fungi (the microbiota) (Qin et al., 2010), the prospect of microbial interactions with drugs in the colon is rather high. Nevertheless, human colon microbiota-based drug metabolism has received little attention thus far. Previous research with laboratory animals showed the metabolic capacity of the gut microbiome to metabolize various nutrients and drugs (Wallace et al., 2010; Spanogiannopoulos et al., 2016), and a recent paper by Wilkinson et al. (2018) provides a nice overview of clinical examples of the impact of gut microbiota on drug metabolism and therapeutic efficacy.
Although consideration of the role of microbiota in drug metabolism has mostly focused on the conversion of drugs into inactive metabolites, the gut microbiome may also be targeted to enhance clinical response, such as by targeting microbiome-encoded enzymes for activation of prodrugs or alteration of drug metabolism into active metabolites to enhance clinical response. An example of activation of a prodrug by intestinal microbiota is sulfasalazine, designed to be cleaved by gut microbiota into the active metabolites 5-aminosalicylic acid (5-ASA) and sulfapyridine (Peppercorn and Goldman, 1972; Williams et al., 2011). Whereas 5-ASA is responsible for many of the local beneficial effects in patients with inflammatory bowel disease, sulfapyridine appeared to be highly absorbed into the systemic circulation, causing known side effects such as headache, nausea, and fever (Peppercorn and Goldman, 1972). To obtain more insights into the potential consequences of microbiome-based biotransformation of compounds for drug pharmacokinetics, efficacy, and safety, especially for those drugs for which we expect higher dose levels in the colon, it is important to thoroughly investigate human colon microbiota-based drug transformation.
Over the last couple of years, there has been a tendency for newly developed drugs to be characterized by low solubility and/or permeability, thereby belonging to Biopharmaceutics Classification System (BCS) class II drugs or BCS class III or IV drugs, but hardly to BCS class I drugs, which are highly soluble and highly permeable drugs (Ku, 2008). This tendency is mainly caused by the increased structural complexity of newly designed drugs. As a consequence, the solubility and absorption of these drugs is impaired, resulting in prolonged residence in the gastrointestinal tract after oral administration and subsequently resulting in higher drug concentrations in the colon. However, not only can orally administered drugs end up in the colon, but also intravenously administered drugs can reach the intestines and colon via biliary excretion, such as after hepatic glucuronidation (Pellegatti, 2012; Spanogiannopoulos et al., 2016).
Recently, a fermentation screening platform with an anaerobic culturing method simulating colonic microbiota conditions was developed (Ladirat et al., 2013, 2014). In this screening platform, human colon microbiota, retrieved from human stool samples and representing microbiota as found in the colon, are incubated. This platform has been demonstrated to be a very fast and efficient ex vivo animal-free screening platform to study interactions at the level of human colonic microbiota, such as screening for the efficacy of prebiotics and/or xenobiotic-mediated alterations in human microbiota activity or composition. In this paper, we describe the application of this ex vivo fermentation screening platform to study human colon microbiota-based drug metabolism. Human stool samples of various healthy adults were used to create a stable pool of microbiota representing the composition of human colon microbiota (Chen et al., 2012; Gagniere et al., 2016). We focused on the reproducibility of the composition of the microbiota pool in independent experiments over time, verification of the anaerobic culture conditions during experiments, and the incidence of microbiota-based drug metabolism. For the latter, 12 marketed drugs of various BCS classifications and containing chemical substituents amenable to metabolism by anaerobic gut microbiota (Table 1) were incubated in this platform. We screened for colon microbiota-based drug metabolism and investigated whether it was in accordance with metabolites observed in earlier reported in vivo studies.
Overview of drugs that have been incubated with fermentation screening platform
Material and Methods
Drugs
The following drugs were obtained from Pfizer (Groton, CT): omeprazole, simvastatin, metronidazole, risperidone, sulfinpyrazone, sulindac, levodopa, dapsone, nizatidine, and acetaminophen (Table 1). Zonisamide and sulfasalazine were obtained from Sigma-Aldrich (St. Louis, MO). The drugs were selected based on their BCS classification, chemical structure, widespread prescription, and/or previously reported metabolism detected in human or animal feces. All drugs were diluted in dimethylsulfoxide (DMSO) and were incubated with human colon microbiota using the fermentation screening platform, followed by liquid chromatography–high-resolution mass spectrometry (LC-HRMS) and high-performance liquid chromatography (HPLC) analysis.
Human Colon Microbiota
Stool samples were derived from seven apparently healthy volunteers, as defined by the following criteria: 1) no use of any antibiotics in the past 2 months, 2) no use of pre- or probiotics in the past week, 3) no bowel problems in the past 3 months, 4) no diarrhea, and 5) no use of any medication in the past 2 weeks (excluding vitamin pills). All volunteers were Caucasian individuals, ranged from 25 to 45 years old, and were subject to a European lifestyle and nutrition. As per clinical protocol, subject identities were kept strictly anonymous, so other demographic characteristics, such as sex, age, race, and body weight, were unknown. The stool samples were collected in anaerobic jars by applying self-contained gas-generating systems (AnaeroGen 3.5L; Thermo Scientific, Landsmeer, The Netherlands), establishing an oxygen-depleted environment essential for obligate anaerobic microorganisms. The collected stool samples were anaerobically pooled and fermented for approximately 40 hours in a fed-batch fermenter. This resulted in a homogenized, scaled-up standardized microbiota pool, which could be used repeatedly after subdivision. This pool was stored as human colon microbiota samples as previously described (Minekus et al., 1999; Ladirat et al., 2013).
Ex Vivo Fermentation Screening Platform: Experimental Setup and Sampling
Experiments were performed according to previously published methods with minor adaptations (Ladirat et al., 2013). In brief, the human colon microbiota were precultured overnight (37°C, 300 rpm) under anaerobic conditions in standard ileal efflux medium (SIEM) to stimulate bacteria growth after storage (−80°C, 12% glycerol), as previously published (Minekus et al., 1999). This medium contained 4.5 g/l sodium chloride (NaCl), 2.5 g/l potassium hydrogen phosphate (K2HPO4), 0.45 g/l calcium chloride dihydrate (CaCl2 · 2H2O), 0.5 g/l magnesium sulfate heptahydrate (MgSO4 · 7H2O), 0.005 g/l ferrous sulfate heptahydrate (FeSO4 · 7H2O), 0.05 g/l ox bile, 0.01 g/l hemin, 0.4 g/l cystein, 0.6 g/l pectin, 0.6 g/l xylan, 0.6 g/l arabinogalactan, 0.6 g/l amylopectin, 5 g/l starch, 2 ml/l Tween 80, 3 g/l bactopeptone, and 3 g/l of casein, constituted with 1 ml of a vitamin mixture containing 1 mg/l menadione, 2 mg/l D-biotin, 0.5 mg/l vitamin B-12, 10 mg/l pantothenate, 5 mg/l nicotinamide, 5 mg/l para-aminobenzoic acid, and 4 mg/l thiamine. All components were obtained from Tritium Microbiology (Veldhoven, The Netherlands). After preparation, the SIEM medium was stored for a maximum of 48 hours in an anaerobic jar at 4°C before usage.
Stock concentrations of test compounds (dissolved in DMSO) were further diluted in modified SIEM to final concentrations of 50 and 100 µM in the screening platform (96 2-ml deep-well plates). The final DMSO concentration was 1% in all incubations. The precultured pooled human colon microbiota (0.1%, v/v) were incubated for 24 hours (pH 5.8; 300 rpm) in fully anaerobic chambers (A45 Scientific Anaerobic Workstation; Don Whitley, Bingley, UK) at 37°C. The total samples were split through centrifugation into supernatant containing medium (used for subsequent LC-HRMS or HPLC analysis) and the pellet with microbes [used for subsequent quantitative polymerase chain reaction (qPCR) analysis]. Incubations with only microbiota in SIEM and incubations with only SIEM served as negative background controls for the HPLC and LC-HRMS analyses. Incubations with test compounds in SIEM (without microbiota) served as a negative control for microbiota-based metabolism to determine compound stability or nonspecific degradation of the test compounds due to culture conditions during the assay. Incubations with colon microbiota and the prebiotic fiber inulin served as a positive control for the outgrowth of anaerobic Bifidobacterium as analyzed by qPCR, confirming fully anaerobic culture conditions during incubations. In addition, the log file of the oxygen level within the anaerobe incubator was used as a supplemental indicator for the maintenance of anaerobic conditions during incubations. All experiments and controls were performed in triplicate.
For NMR experiments, a larger-scale incubation was performed in which risperidone was incubated in a volume of 50 ml of SIEM plus microbiota for 24 hours. Follow-up steps were as described earlier. The mixture was processed using a previously described approach (Walker et al., 2014) and used for NMR analysis.
Quantification of Bifidobacterium Levels Using qPCR
To verify whether the anaerobic conditions were maintained during the incubations, the presence and abundance of the anaerobic Bifidobacterium bacteria was analyzed. These species are one of the major genera of bacteria that make up the microbiota of the human colon and are very sensitive to oxygen, since low concentrations of oxygen are already lethal for these bacteria. The outgrowth of Bifidobacterium at different time points was analyzed via qPCR analysis. For qPCR, total DNA was isolated from the microbial pellet samples according to previously described procedures (Crielaard et al., 2011; Ladirat et al., 2013). All Bifidobacterium species were targeted with the following primers: F_allbif_IS (10 µM) (GGG ATG CTG GTG TGG AAG AGA), R_allbif_IS (10 µM) (TGC TCG CGT CCA CTA TCC AGT), and probe MSB-FAM (5 µM) (P_all_bif, TCA AAC CAC CAC GCG CCA) (Haarman and Knol, 2005). DNA isolated from a mix of various species of cultured Bifidobacteria was used as a positive control. For the analysis, 5 µl of DNA sample, 12.5 μL of PCR mixture (2x) (Diagenode, Seraing, Belgium, and 1 µl of the probe, forward primer, and reverse primer were used. The samples were analyzed with the Applied Biosystems (Foster City, CA) 7500 Fast Real-Time PCR system with the settings for a standard run with the amount of cycles adjusted to 45. The ΔCt values were converted into 10Log values to create growth curves.
Analysis of Colon Microbiota Composition
Microbial composition of the fecal samples collected from the seven individuals (before and after pooling, and before and after the ex vivo fermentation screen for 24 hours) was analyzed by sequencing using MiSeq (Illumina, EindhoveThe Netherlands), as previously published (Arnoldussen et al., 2017).
Analysis by Ultra-High-Performance Liquid Chromatography, HPLC, and NMR
Ultra-High-Performance Liquid Chromatography–UV–HRMS.
Acetonitrile (2.5 ml) was added to an aliquot of terminated incubation samples (0.5 ml), and the mixture was spun in a centrifuge at 1700g for 5 minutes. The supernatant was transferred to a 15-ml conical glass tube and subject to vacuum centrifugation. To the dried residue, 0.1 ml of water (omeprazole, nizatidine), 1% aqueous formic acid (metronidazole, sulindac, sulfasalazine, zonisamide, levodopa, dapsone, acetaminophen), 1% formic acid in 10% acetonitrile in water (risperidone, sulfinpyrazone), or 20% acetonitrile in water (simvastatin) was added for analysis by ultra-high-performance liquid chromatography (UHPLC)–UV–HRMS. Samples (0.01 ml) were injected into a Thermo Orbitrap Elite equipped with an Accela UHPLC system (Thermo Fisher Scientific, Waltham, MA, USA) composed of a quaternary pump, photodiode array detector, and CTC Analytics autoinjector (CTC Analytics, Zwingen, Switzerland). Separations were effected using conditions listed in Supplemental Table S1–S7.
HPLC-UV.
Sulfasalazine was used as a quality reference drug in all experiments to screen for anaerobic metabolism. Sulfasalazine is metabolized into 5-ASA and sulfapyridine by azoreductases of gut microbiota (Peppercorn and Goldman, 1972). The concentration of sulfasalazine and its metabolite sulfapyridine was determined based on the UV absorption using HPLC, with a UV detector, an autosampler and a Xbridge BEH C18 column (4.6 × 150 mm; Waters, Milford, MA). The absorption peaks were identified and quantified using Laura software (Lablogic, South Yorkshire, UK). The incubated samples were filtered (10 minutes, 1690g, 45 µm), diluted with acetonitrile (10%), and stored at −80°C until further use. The samples were diluted with acetonitrile (30%) and injected (10 µl) into the HPLC column. Acetonitrile with 0.1% formic acid was used as a mobile phase with a flow rate of 0.6 ml/min−1 at 10°C.
NMR.
All samples were dissolved in DMSO-d6 “100%” and placed in a 1.7-mm NMR tube (0.04 ml) under a dry argon atmosphere. 1H and 13C spectra were referenced using residual DMSO-d6 (1H δ = 2.50 ppm relative to tetramethylsilane, δ = 0.00; 13C δ = 39.50 ppm relative to tetramethylsilane, δ = 0.00). NMR spectra were recorded on a Bruker Avance 600 MHz (Bruker BioSpin Corporation, Billerica, MA) controlled by Topspin V3.2 (Bruker, Billerica, MA); TCI CryoProbe (Bruker). One-dimensional (1D) spectra were recorded using an approximate sweep width of 8400 Hz and a total recycle time of approximately 7 seconds. Two-dimensional (2D) data were recorded using the standard pulse sequences provided by Bruker. Postacquisition data processing was performed with either Topspin V3.2 or MestReNova V9.1. Quantitation of NMR isolates was performed by external calibration against the 1H NMR spectrum of a 5-mM benzoic acid standard using the ERETIC2 function within Topspin V3.2 (Mestrelab Research, Escondido, CA).
Results
Microbial Composition and Stability.
Microbiota composition was analyzed in the stool samples collected from seven healthy adult volunteers, before and after pooling (Fig. 1), showing the presence of all microbial phyla regularly encountered in the human microbiome (Firmicutes, Bacteroidetes, Actinobacteria, Proteobacteria, and Verrucomicrobia) in the inoculation material. These data showed that despite the rather large interindividual variation in the microbiota composition of all prefermented stool samples of the seven individuals, the microbial diversity of the pooled sample was representative of the individual samples and of the human in vivo situation (Fig. 1, A–H). Pooled stool samples were fermented to increase the sample sizes, aliquoted, and stored at <−70°C upon usage. Although the fermentation and storage process inevitably affected the colon microbiota composition, the overall diversity of the microbiota composition of the pooled sample was still maintained, demonstrated by the microbial composition of the pooled sample at the beginning and end of the ex vivo fermentation screen (Fig. 1, I and J). Limited intra- and interexperimental variation in microbial composition was observed, demonstrating the robustness of the assay under highly controlled laboratory conditions (Supplemental Fig. S1).

Microbial composition diversity of a pooled sample of seven different individual fecal samples. (A–H) Microbial composition of individual and pooled fecal samples obtained from seven healthy adult volunteers analyzed by sequencing, differentiating between the five main microbial phyla present in the human microbiota (Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, and Verrucomicrobia). (I and J) Microbial composition of pooled human fecal samples at the start of the ex vivo fermentation assay (t = 0) and after 24 hours of incubation in the assay (t = 24). Data are presented as the mean relative abundance of the individual species (n = 3).
Maintenance of Anaerobic Culture Conditions.
Maintenance of strict anaerobic culture conditions during incubations was verified by analyzing the outgrowth of the strictly anaerobic Bifidobacteria over 24 hours in the absence and presence of the probiotic fiber inulin. Proper growth curves were observed for all experiments (Supplemental Fig. S2), showing an outgrowth of Bifidobacteria of more than 1 log, indicating that anaerobic conditions allowing for active growth of strictly anaerobic bacteria were indeed maintained throughout all incubations. These data were confirmed by the data log of the oxygen concentrations within the incubator, showing no deviations during the experiments.
Microbiome-Mediated Biotransformation of Drugs.
To study the significance of gut microbiota as a determinant of drug pharmacokinetics and metabolism (and, accordingly, therapeutic response), the developed platform was applied to incubate a subset of 12 drugs (Table 1) with pooled human adult colon microbiota. Five of the 12 included drugs showed microbiota-based biotransformation after 24 hours of incubation (nizatidine, risperidone, sulfasalazine, sulfinpyrazone, and sulindac), whereas 8 hours after incubation, no major biotransformation of these drugs was observed based on LC-HRMS or HPLC data (Fig. 2; Table 2). After 24 hours of incubation with human colon microbiota in the fermentation assay, the prodrug sulfasalazine was completely reduced to sulfapyridine, whereas the cleavage product, 5-ASA, could not be detected (Fig. 3). Sulindac and sulfinpyrazone displayed a sulfoxide reduction to the thioethers due to colon microbial metabolism (Figs. 4 and 5). Sulindac was completely converted by the colon microbiota within 24 hours into deoxysulindac. Nizatidine was also reduced by human colon microbiota, resulting in a nitroso-deoxygenated metabolite (Fig. 6). The benzisoxazole N-O bond of risperidone was reduced and the resulting imine metabolite converted to the ketone, either spontaneously or possibly by the microbiota (Fig. 7). After 24 hours of incubation with colon microbiota, the reduction of risperidone was complete. These five drugs appeared to be stable during incubation in the assay, as no degradation or metabolite formation was detected when incubated without microbiota in the assay (Figs. 3–7). For acetaminophen, levodopa, metronidazole, dapsone, omeprazole, simvastatin, and zonisamide, no specific metabolites were detected after incubation with human colon microbiota for 6 or 24 hours. Although a decline of the parent drug was detected for levodopa and metronidazole after 24-hour incubation with human colon microbiota, no specific metabolites were found.
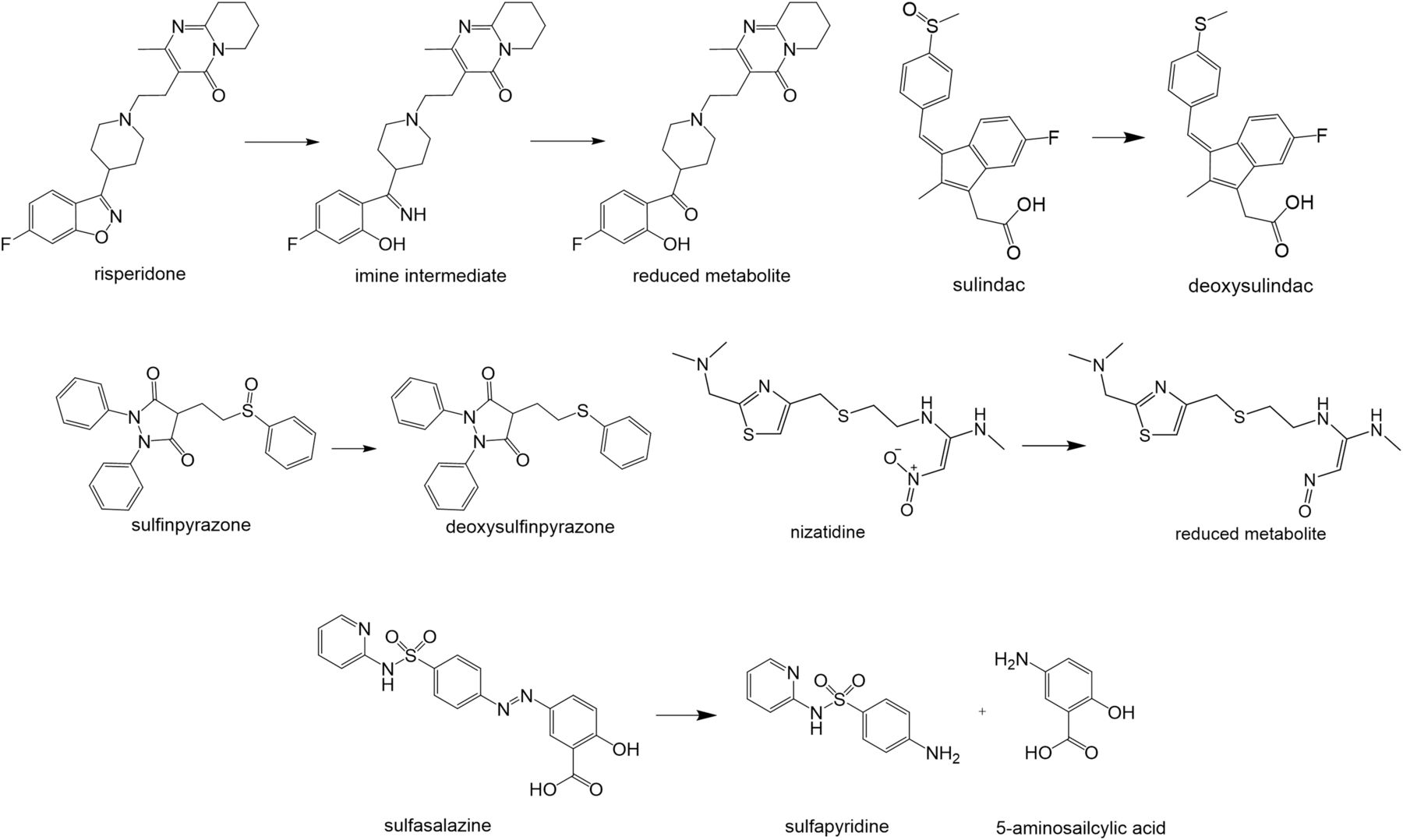
Summary of metabolic reactions observed in the fermentation screening platform. Schematic representation of microbiome-induced metabolic reactions observed in the fermentation screening platform for risperidone, sulindac, nizatidine, sulfinpyrazone, and sulfasalazine.
Overview of metabolic reactions of drugs observed in fermentation screening platform

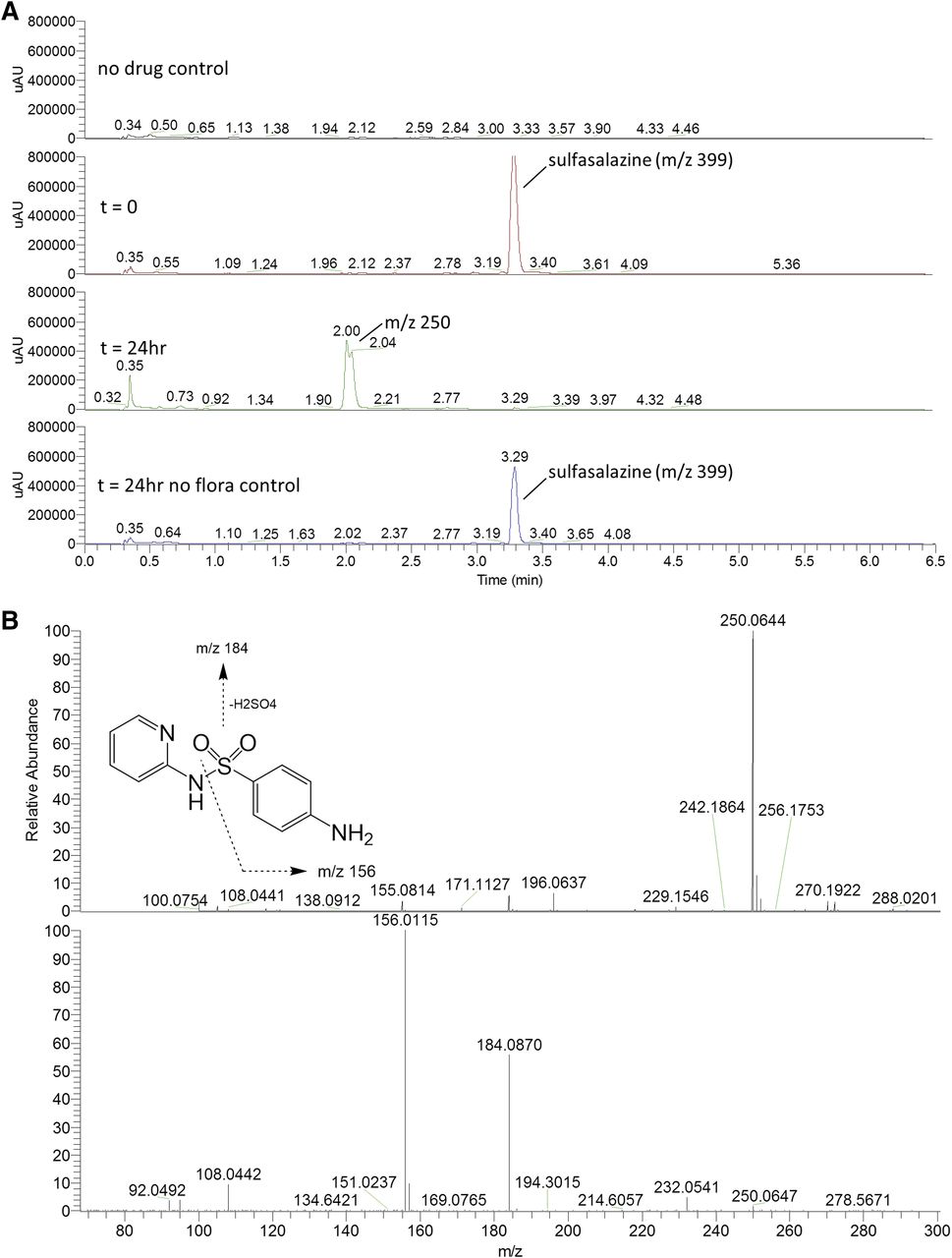
(A) HPLC chromatograms of sulfasalazine (50 µM) incubations in the fermentation screening platform. The appearance of a peak representing reductive cleavage of the azo bond hydrolysis was observed (m/z 250.0644; −0.3 ppm; Rt = 2.0 minutes). (B) High-resolution mass spectra for reductively cleaved metabolite of sulfasalazine in the fermentation screening platform. The top panel shows the protonated molecular ion at m/z 4250.0644 (−0.3 ppm), and the bottom panel is the CID-induced fragmentation pattern.

(A) HPLC chromatograms of sulfinpyrazone (50 µM) incubations in the fermentation screening platform. The appearance of a peak represented deoxy metabolite eluted at 3.9 minutes (λ = 260 nm). (B) High-resolution mass spectra of deoxysulfinpyrazone metabolite generated in the fermentation screening platform. The top panel shows the protonated molecular ion at m/z 389.1322 (1 ppm), and the bottom panel is the CID-induced fragmentation pattern.

(A) HPLC chromatograms of sulindac (50 µM) incubations in the fermentation screening platform. The appearance of a peak represented deoxy metabolite eluted at 7.19 minutes (λ = 328 nm). (B) High-resolution mass spectra of deoxysulindac metabolite generated in the fermentation screening platform. The top panel shows the protonated molecular ion at m/z 341.1005 (1 ppm), and the bottom panel is the CID-induced fragmentation pattern.

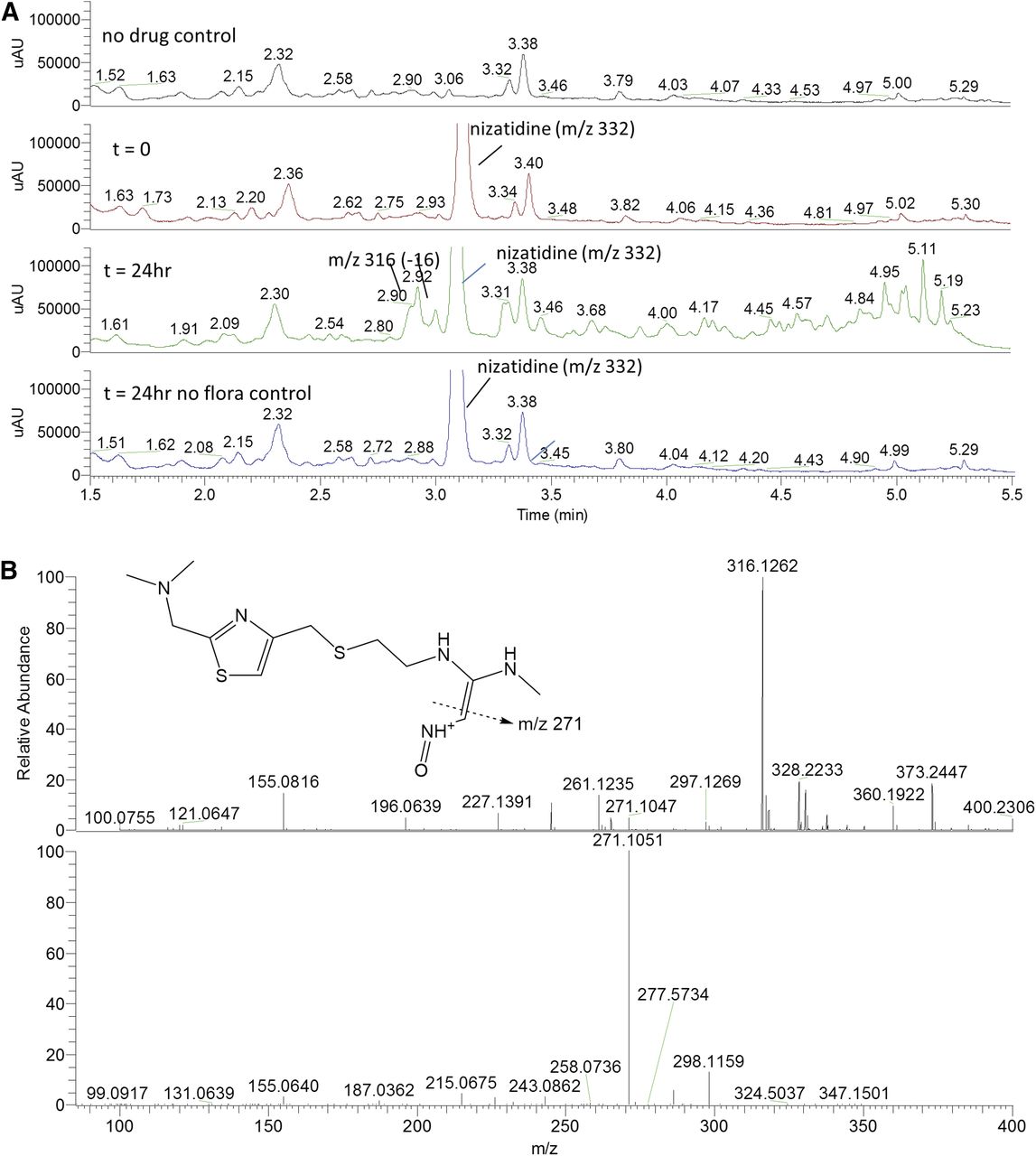
(A) HPLC chromatograms of nizatidine (50 µM) incubations in the fermentation screening platform. The appearance of peaks represented deoxy metabolites eluted between 2.9 and 3.0 minutes (λ = 275 nm). (B) High-resolution mass spectra for deoxynizatidine metabolites generated in the fermentation screening platform. The top panel shows the protonated molecular ion at m/z 316.1262 (0.5 ppm), and the bottom is the CID-induced fragmentation pattern.

(A) HPLC chromatograms of risperidone (50 µM) incubations in the fermentation screening platform. The appearance of a peak represented reduction of the benzisoxazole N-O bond, and subsequent hydrolysis of the intermediate imine was observed (m/z 414.2184; −0.8 ppm). Parent drug and metabolite coeluted at Rt = 4.20 minutes, but distinguished by different masses. (B) High-resolution mass spectra for reduced risperidone metabolite generated in the fermentation screening platform. The top panel shows the protonated molecular ion at m/z 414.2184 (−0.8 ppm), and the bottom panel is the CID-induced fragmentation pattern.
Although HPLC-MS data are frequently obtained in drug-metabolism experiments, in many cases NMR spectral data are needed to verify structures of metabolites. In the current study, we used microbiota-mediated metabolism of risperidone as an example to demonstrate that the fermentation assay could be scaled up to produce enough metabolite for NMR analysis. This was successful, and 1D proton and 2D homonuclear and heteronuclear experiments confirmed the risperidone metabolite structure as the previously described ketone (Fig. 8).
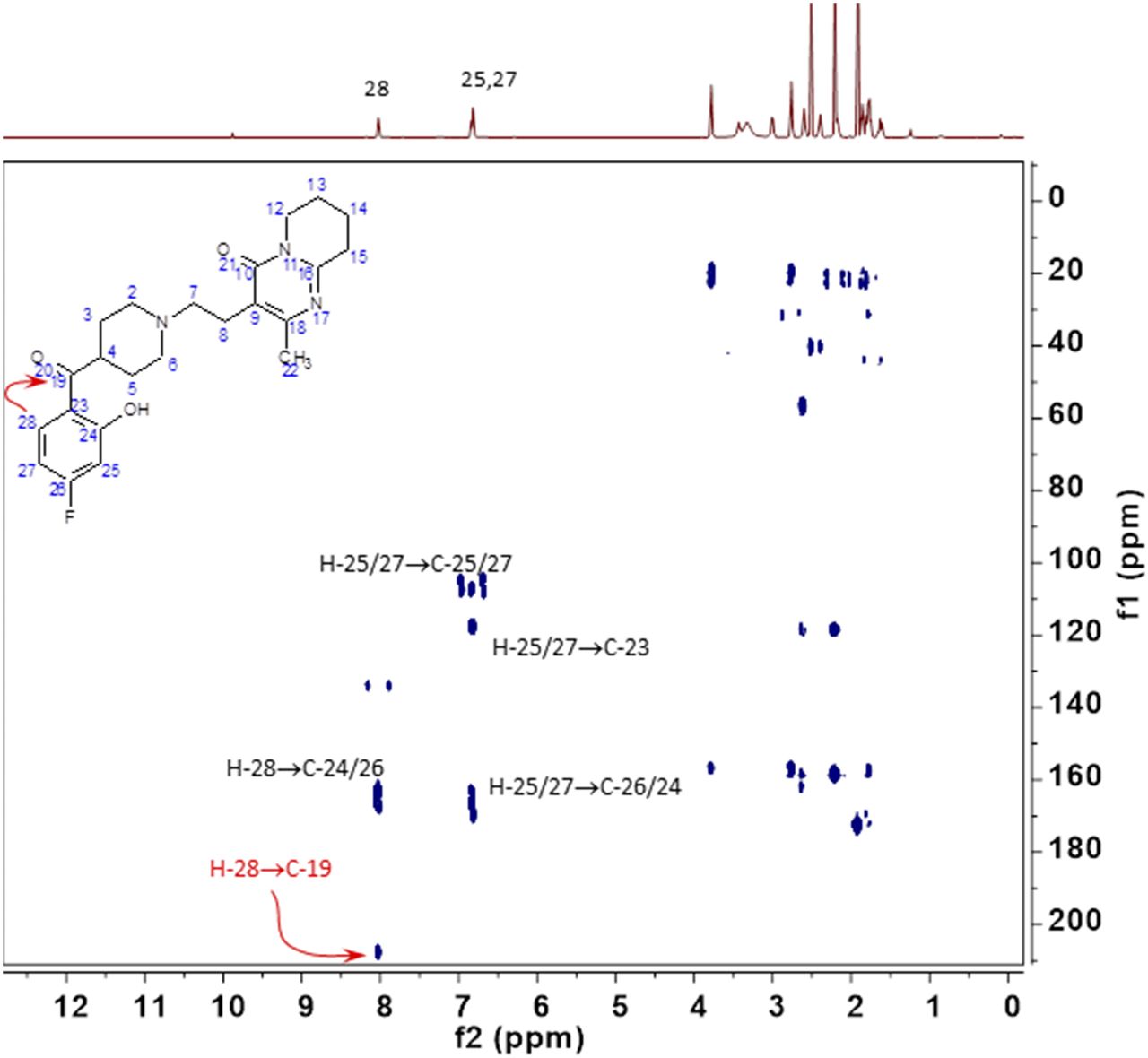
NMR of reduced risperidone metabolite 1H and 1H-13C heteronuclear multiple bond correlation. 1H chemical shift and coupling patterns established the 8.02 ppm resonance as H28. Within the 1H-13C heteronuclear multiple bond correlation data set, there is a cross-peak from the 1H 8.02 ppm resonance to a 13C resonance at 207.5 ppm, which is assigned as the C19 carbon of the ketone. All other observed resonances and cross-peaks in the isolated metabolite are unchanged from those observed in similarly acquired data from risperidone.
Discussion
In the current study, we applied a previously developed microbial fermentation assay (Ladirat et al., 2013) for studying human colon microbiota-mediated biotransformation of drugs. For this, microbiota from stool samples, which were collected from seven healthy adult volunteers, were homogenized, pooled, and scaled up to create a standardized microbiota pool representing human colon microbiota. Despite the rather large interindividual variation in the microbiota composition of all prefermented stool samples of the seven individuals, the microbial diversity of the pooled sample was representative of the individual samples and of the human in vivo situation, as has previously been published (Ladirat et al., 2013). In this latter publication, the representativeness of the human colon microbiota samples was studied by determining the presence and abundance of more than 200 bacterial targets, of which Bacteroides, Bifidobacteria, Enterobacteria, Clostridia, and Lactobacillus are examples of prominent bacteria species present in the human gut (Eckburg et al., 2005; Zoetendal et al., 2006). These data showed that the microbiota composition of the pooled prefermented samples was representative of the composition of human colon microbiota since key species as found in the human colon were present and the microbial diversity of the pooled sample was representative of the individual samples. During fermentation, storage, and assay incubation, the overall diversity of the human microbiota composition of the pooled sample was still maintained, demonstrated by the microbial composition of the pooled sample at the beginning and end of the ex vivo fermentation screen and the comparison of microbial composition at the end of experiments performed over time. The use of a pool in this manner is similar in practice to the use of pooled human hepatocytes or liver microsomes to study drug-metabolism rates and pathways. Understanding interindividual variability is lost in such an approach, but once the data are generated using a pooled sample, follow-up experiments to address interindividual variability can be pursued as warranted.
Maintenance of strict anaerobic conditions during the ex vivo fermentation assay is important for reliability of the obtained results since, in the colon, the microbiota thrive in an anaerobic environment. Stringent anaerobic conditions were verified by proper outgrowth of the strictly anaerobic Bifidobacterium over 24 hours in the assay in the presence of the probiotic fiber inulin and by data log of the oxygen concentrations within the incubator.
To study the potential significance of gut microbiota as a determinant of drug pharmacokinetics and metabolism, the developed platform was applied to incubate a subset of 12 drugs with pooled human adult colon microbiota. The drugs were selected based on their widespread prescription, BCS classification, and potential for microbial metabolism as has previously been reported for some in the literature (Table 1). Five of the 12 included drugs showed microbiota-based biotransformation after 24 hours of incubation in the ex vivo fermentation assay: sulfasalazine, sulfinpyrazone, sulindac, nizatidine, and risperidone. Colonic transit time was previously determined to be 20–40 hours in healthy adult people (Zaslavsky et al., 1998), so the 24-hour incubation used in our experiments may offer a reasonable reflection of residence time of drugs and drug metabolites in the colon. Results were compared with the literature to determine whether the metabolic reactions that were detected corresponded to clinical data; a summary of the results is presented in Table 2. In the ex vivo microbial fermentation assay, sulfasalazine was completely reduced to sulfapyridine within 24 hours of incubation, whereas the cleavage product 5-ASA could not be detected. In the clinic, sulfasalazine is provided as a prodrug, designed to reach the colon to be metabolized into sulfapyridine and 5-ASA by azoreductases of gut microbiota (Peppercorn and Goldman, 1972; Williams et al., 2011). Due to cleavage, the active metabolite 5-ASA is formed in the colon, where it has its therapeutic effect. The other metabolite, sulfapyridine, has been shown to have toxic effects in humans (Das et al., 1973; Pullar and Capell, 1986; Davila and Ranganathan, 2011). The bacteria species Bacteroides, Bifidobacterium, Lactobacillus, and Enterococcus have been linked to the metabolism of sulfasalazine (Peppercorn and Goldman, 1972). The reason that 5-ASA could not be detected during analysis might be that 5-ASA is extensively further metabolized after cleavage from sulfasalazine, which could complicate detection of 5-ASA, since it has been shown that both 5-ASA and sulfapyridine can be acetylated by gut microbiota in animals and humans (Dull et al., 1987).
The observed microbiome-mediated sulfoxide reduction of sulindac and sulfinpyrazone was in line with previous studies in which reduced metabolites were detected in human and rabbit feces (Strong et al., 1987). Nizatidine was also reduced by human colon microbiota in the assay, resulting in a nitroso-deoxygenated metabolite, conforming to previously published results with an alternative in vitro fermentation system with human feces (Basit et al., 2002). The benzisoxazole N-O bond of risperidone was reduced and the resulting imine metabolite converted to the ketone, either spontaneously or possibly by the microbiota. This metabolic pathway of risperidone has previously been reported in studies using rat, dog, and human feces (Mannens et al., 1993; Meuldermans et al., 1994; Jourova et al., 2016). After 24-hour incubation with colon microbiota, risperidone was completely reduced to the ketone, which was structurally confirmed by 1D proton and 2D homonuclear and heteronuclear experiments. This also demonstrated that these fermentations can be conducted at a larger scale such that quantities of metabolites that can be generated are high enough for NMR spectroscopy.
For acetaminophen, levodopa, metronidazole, dapsone, omeprazole, simvastatin, and zonisamide, no specific metabolites were detected after incubation with human colon microbiota. However, for levodopa and metronidazole, a decline of the parent drug was detected after 24-hour incubation, but no specific metabolites were found, whereas the other drugs were found to be stable during incubation with human colon microbiota. Previous studies showed gut microbiota-mediated reduction of C-14–labeled metronidazole into N-(2-hydroxyethyl)-oxamic acid and acetamide in rat feces (Koch and Goldman, 1979; Koch et al., 1979). Levodopa undergoes p-hydroxylation by which m-hydroxyphenylacetic acid is formed in humans, but this was inhibited upon treatment with the antibiotic neomycin (Sandler et al., 1969). We did not confirm these data for metronidazole and levodopa or for the other drugs using human colon microbiota, which may be due to potential species-specific differences in microbial enzymatic activity or due to the absence of certain microbial enzymatic activity in the used pool of human microbiota. Also, the UHPLC-MS analysis method used was relatively generic to drug metabolism, whereas for some of these drugs, specific sample work-up and analytical methods may be required for the detection of their metabolites. Small amounts of small hydrophilic metabolites would need to be detected within a very complex matrix (Duda-Chodak et al., 2015). Another possibility is that the metabolites are unstable in the sample work-up method. Generally speaking, the use of radiolabeled drugs would facilitate the process of quantification and identification of metabolism in this type of ex vivo screening platform. Nevertheless, this was outside the scope of the current research.
In conclusion, this study shows that the previously developed fermentation screening platform is a valid technique to screen for microbiota-based drug metabolism. The anaerobic culture conditions were maintained throughout the incubations. The composition of the human colon microbiota in the assay was representative of microbiota as found in the colon, and the diversity of the microbial composition was maintained during incubations and was reproducible between experiments. Twelve drugs with varying BCS classification were incubated and analyzed for metabolism by human colon microbiota and we were able to demonstrate metabolism in a qualitative manner. Among the drugs, those in BCS class II are more likely to reach the colon and microbiota since they are not as readily absorbed. Our data showed that these drugs can be metabolized by the gut microbiota in the in vitro system (Table 1), indicating that they can penetrate the cells despite being less absorbed in the intestine. The detected metabolic transformations were comparable to reactions as previously described in studies with human or animal feces, demonstrating the utility of this assay (Nayak and Turnbaugh, 2016; Wilson and Nicholson, 2017). Future research will be aimed toward determining whether this tool could be useful to understand the reversal of metabolism back to parent drugs (e.g., glucuronide hydrolysis, N-oxide reduction). In addition, we will determine if data obtained with this in vitro system can be more quantitatively translated to human drug disposition, and we will aim to use the assay for precision medicine by applying personalized microbiota to determine interindividual variation.
Acknowledgments
We thank L. Bouw and M. Ossendrijver for their excellent scientific and technical assistance.
Authorship Contributions
Participated in research design: van de Steeg, Schuren, Obach, Nooijen, Woudenbergh, Vaes.
Conducted experiments: Obach, Woudenbergh, Walker.
Performed data analysis: Obach, Nooijen, Woudenbergh, Walker, Heerikhuisen.
Wrote or contributed to the writing of the manuscript: van de Steeg, Schuren, Obach, Woudenbergh, Nooijen, Vaes.
Footnotes
- Received February 22, 2018.
- Accepted August 1, 2018.
↵1 E.v.d.S. and F.H.J.S. contributed equally to the manuscript.
The work described was partly funded by the Dutch Government, Ministry of Economic Affairs.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- 5-ASA
- 5-aminosalicylic acid
- BCS
- Biopharmaceutics Classification System
- 1D
- one-dimensional
- 2D
- two-dimensional
- DMSO
- dimethylsulfoxide
- HPLC
- high-performance liquid chromatography
- LC-HRMS
- liquid chromatography–high-resolution mass spectrometry
- qPCR
- quantitative polymerase chain reaction
- SIEM
- standard ileal efflux medium
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}